
Mitochondria Isolated from Hearts Subjected to Ischemia/Reperfusion Benefit from Adenine Nucleotide Translocase 1 Overexpression

 , ,
, ,  and
and
Abstract
: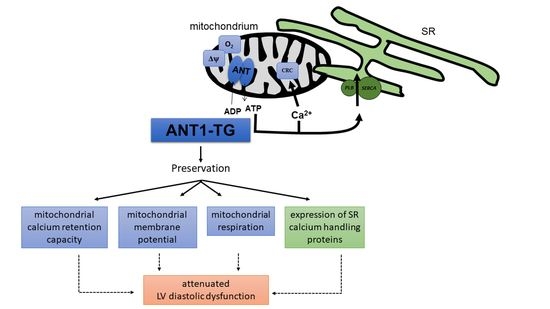
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Langendorff Perfusion (Volume-Constant Mode)
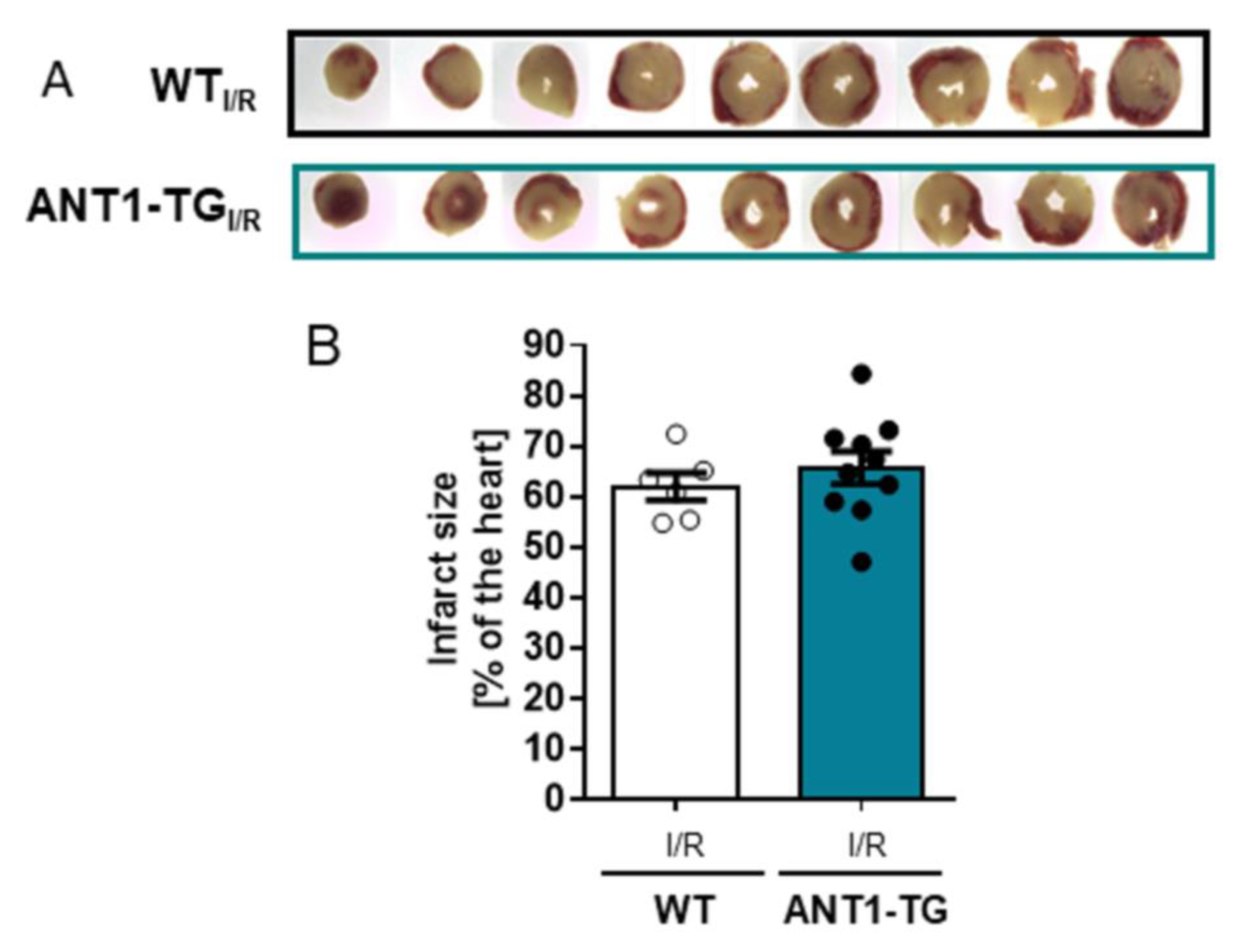
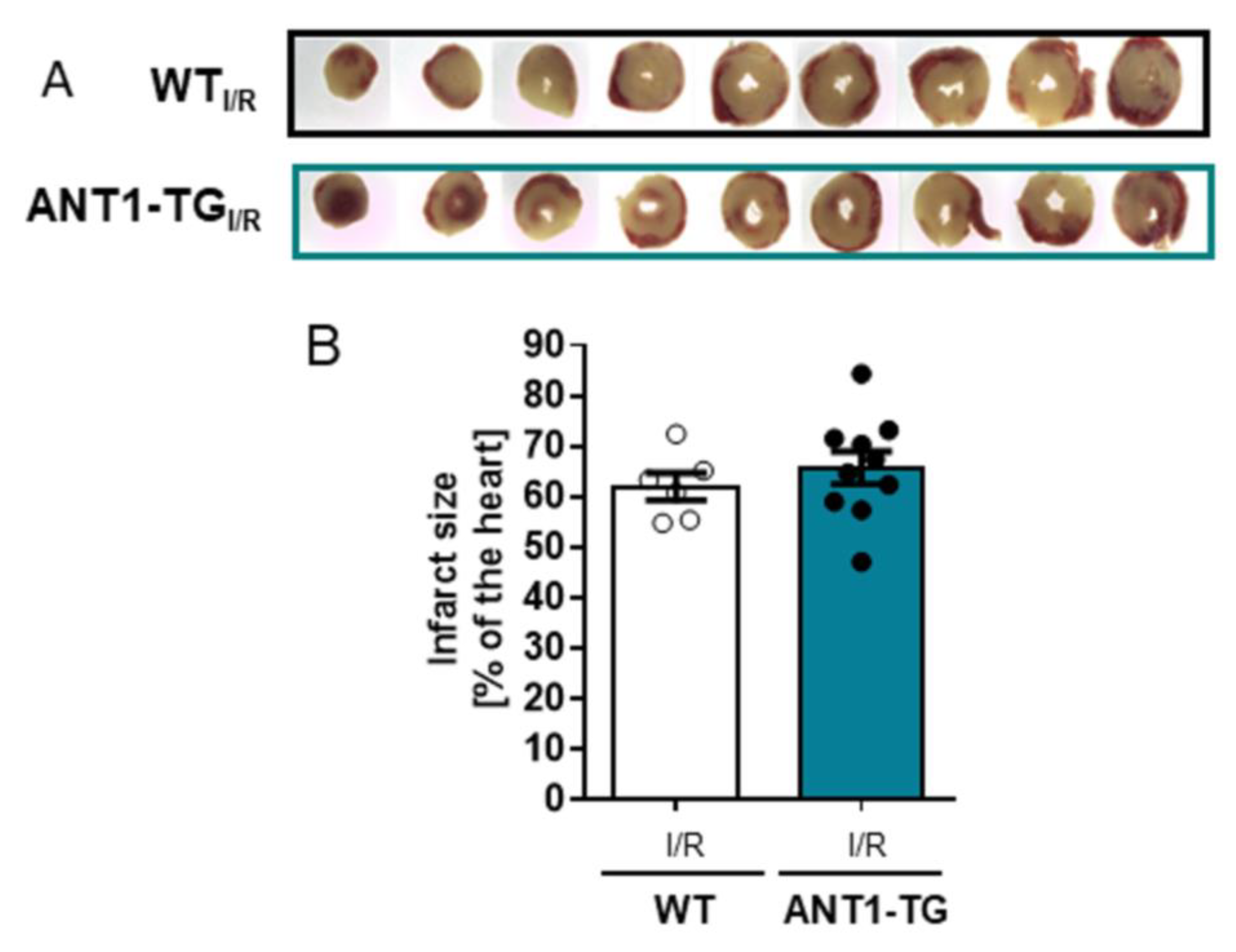
2.3. Infarct Size Measurement
2.4. Real-Time RT-PCR Analysis
2.5. Western Blot Analysis
2.6. Mitochondria Isolation
2.7. Calcium Retention Assay
2.8. Membrane Potential (Δψm)
2.9. Oxygen Consumption
2.10. Statistical Analysis
3. Results
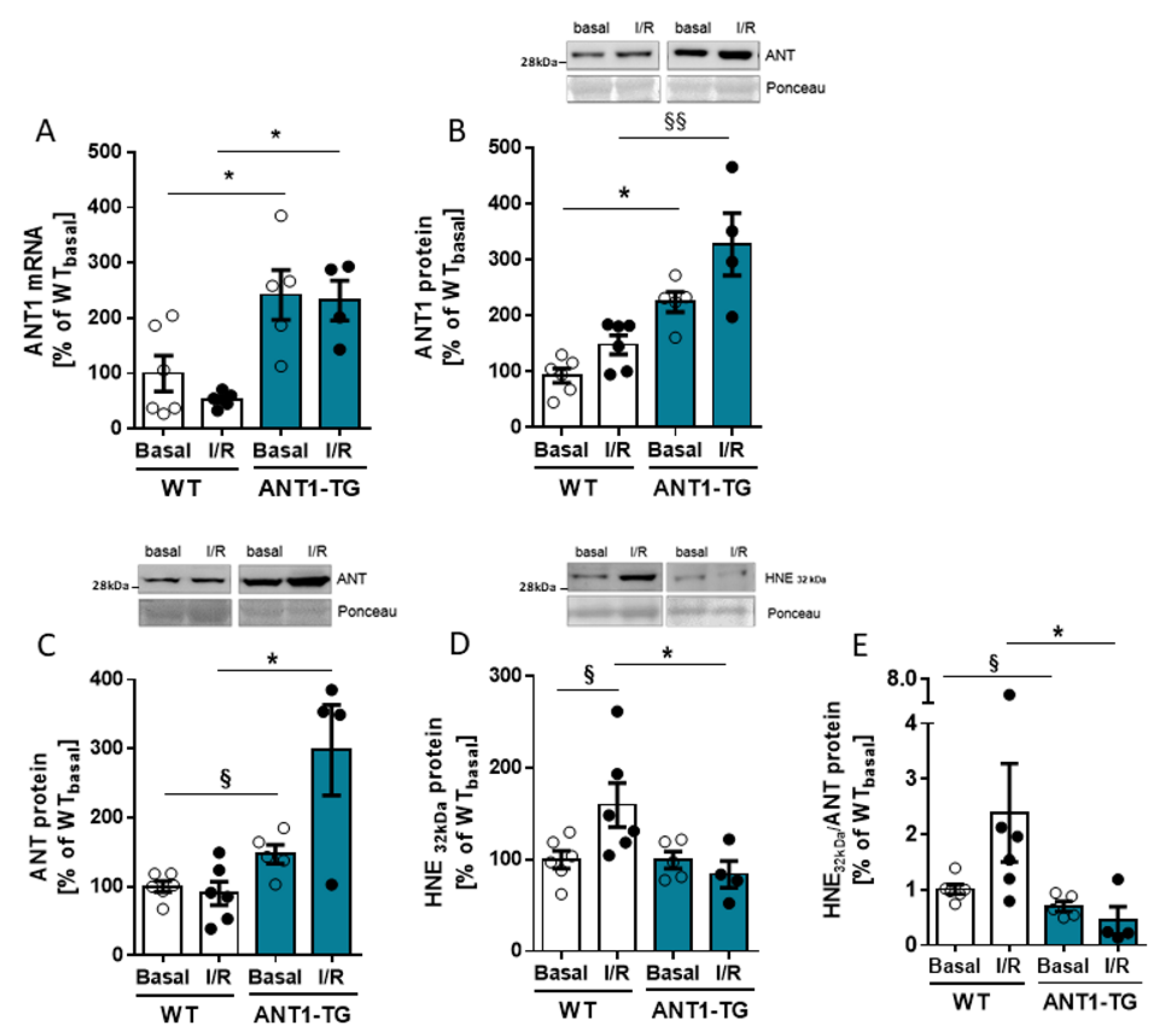
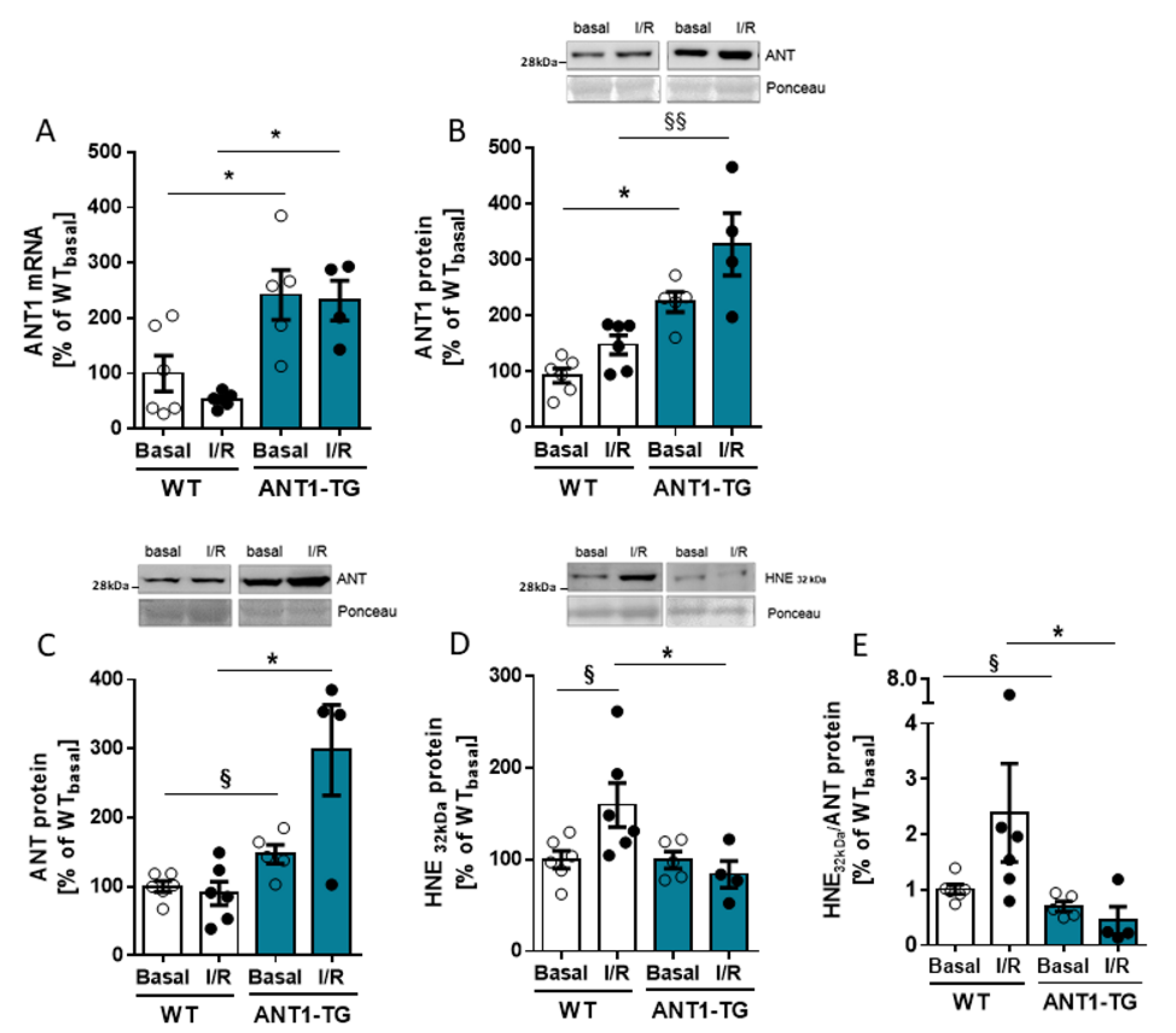
3.1. ANT Expression and Modification in I/R-Treated Hearts
3.2. ANT1 Overexpression Stabilizes Mitochondrial Calcium Retention Capacity and Membrane Potential after I/R
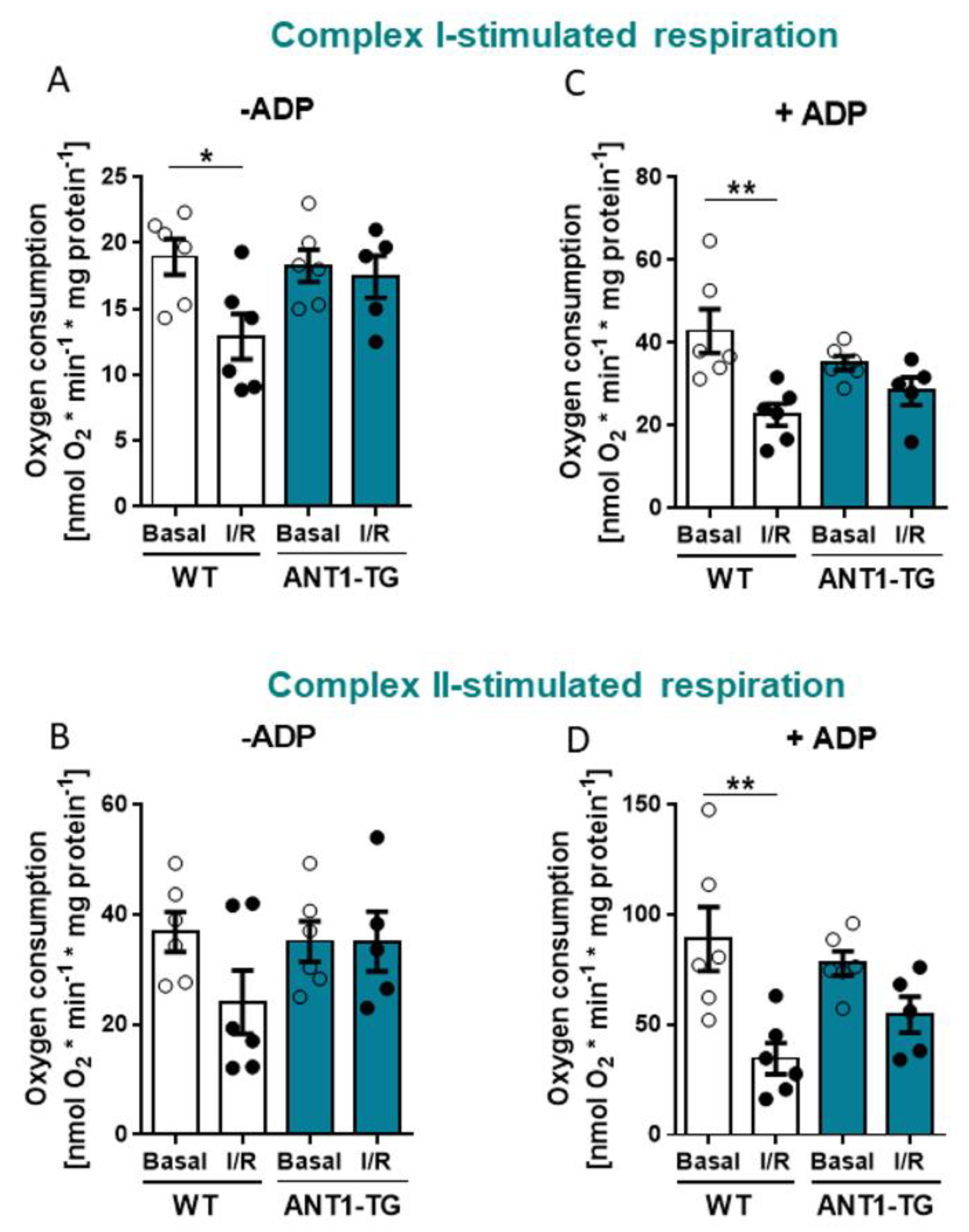
3.3. ANT1 Overexpression Stabilizes Mitochondrial Oxygen Consumption after I/R
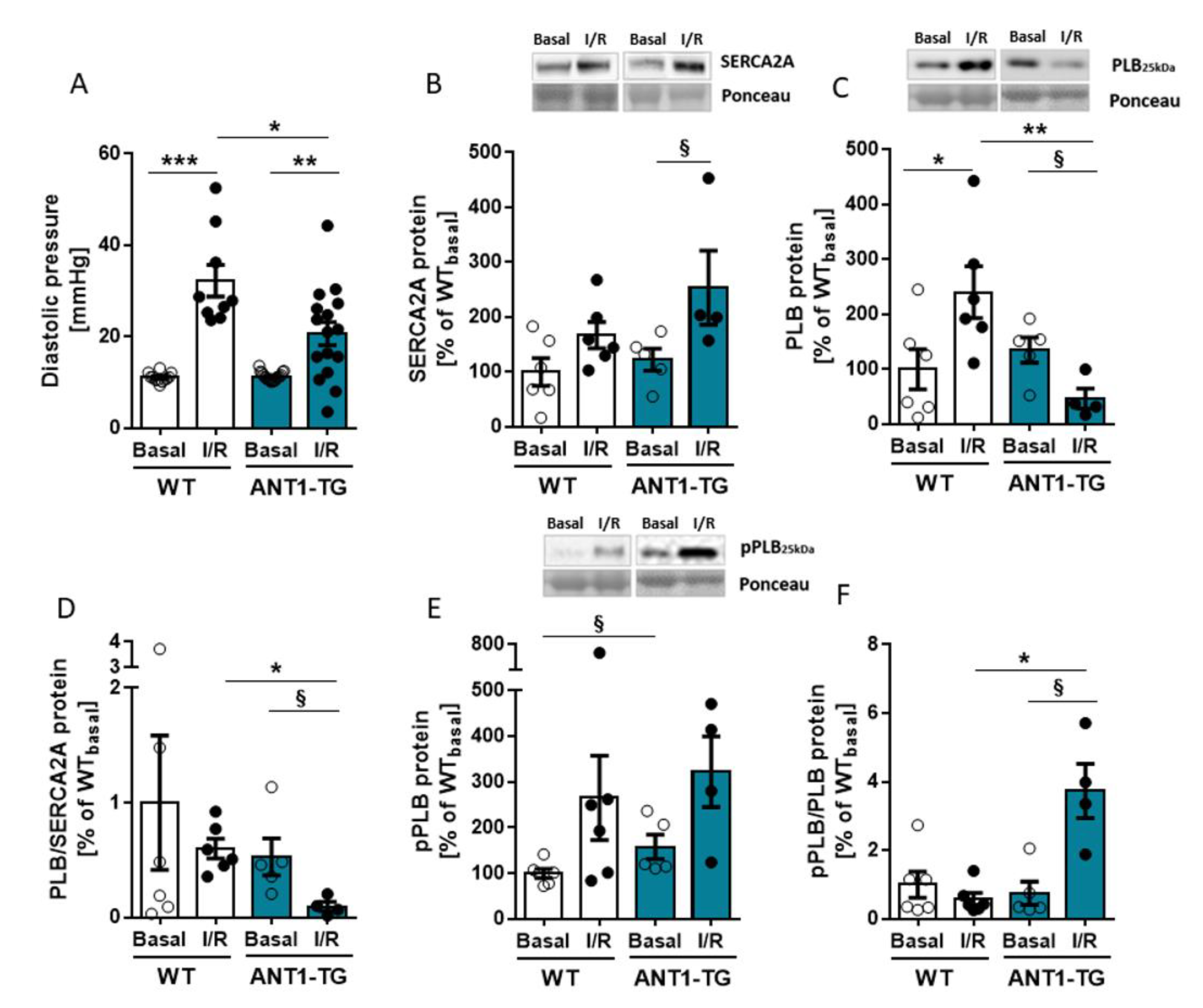
3.4. ANT1 Overexpression Reduces the PLB/SERCA2a Ratio and Stabilizes a Beneficial pPLB/PLB Ratio Leading to Reduced Diastolic Pressure after I/R
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Detect. Mitochondrial Dis. 1997, 174, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Valverde, C.A.; Kornyeyev, D.; Ferreiro, M.; Petrosky, A.D.; Mattiazzi, A.; Escobar, A.L. Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc. Res. 2010, 85, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Ladilov, Y.; Efe, O.; Schäfer, C.; Rother, B.; Kasseckert, S.; Abdallah, Y.; Meuter, K.; Schlüter, K.D.; Piper, H.M. Reoxygenation-induced rigor-type contracture. J. Mol. Cell. Cardiol. 2003, 35, 1481–1490. [Google Scholar] [CrossRef]
- Iwanaga, Y.; Hoshijima, M.; Gu, Y.; Iwatate, M.; Dieterle, T.; Ikeda, Y.; Date, M.O.; Chrast, J.; Matsuzaki, M.; Peterson, K.L.; et al. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J. Clin. Investig. 2004, 113, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Carraro, M.; Carrer, A.; Urbani, A.; Bernardi, P. Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection. J. Mol. Cell. Cardiol. 2020, 144, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Salinas, K.; Zuo, X.; Kucejova, B.; Chen, X.J. Dominant membrane uncoupling by mutant adenine nucleotide translocase in mitochondrial diseases. Hum. Mol. Genet. 2008, 17, 4036–4044. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Chen, X.J. Misfolding of mutant adenine nucleotide translocase in yeast supports a novel mechanism of Ant1-induced muscle diseases. Mol. Biol. Cell 2015, 26, 1985–1994. [Google Scholar] [CrossRef]
- Dörner, A.; Schultheiss, H.P. Adenine Nucleotide Translocase in the Focus of Cardiovascular Diseases. Trends Cardiovasc. Med. 2007, 17, 284–290. [Google Scholar] [CrossRef]
- Walther, T.; Tschöpe, C.; Sterner-Kock, A.; Westermann, D.; Heringer-Walther, S.; Riad, A.; Nikolic, A.; Wang, Y.; Ebermann, L.; Siems, W.E.; et al. Accelerated mitochondrial adenosine diphosphate/adenosine triphosphate transport improves hypertension-induced heart disease. Circulation 2007, 115, 333–344. [Google Scholar] [CrossRef]
- Heger, J.; Abdallah, Y.; Shahzad, T.; Klumpe, I.; Piper, H.M.; Schultheiss, H.P.; Schlüter, K.D.; Schulz, R.; Euler, G.; Dörner, A. Transgenic overexpression of the adenine nucleotide translocase 1 protects cardiomyocytes against TGFbeta(1)-induced apoptosis by stabilization of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 2012, 53, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Hammer, E.; Heger, J.; Schultheiss, H.P.; Rauch, U.; Landmesser, U.; Dörner, A. Adenine Nucleotide Translocase 1 Expression is Coupled to the HSP27-Mediated TLR4 Signaling in Cardiomyocytes. Cells 2019, 8, 1588. [Google Scholar] [CrossRef] [Green Version]
- Klumpe, I.; Savvatis, K.; Westermann, D.; Tschöpe, C.; Rauch, U.; Landmesser, U.; Schultheiss, H.P.; Dörner, A. Transgenic overexpression of adenine nucleotide translocase 1 protects ischemic hearts against oxidative stress. J. Mol. Med. 2016, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, M.; Frodermann, T.; Böning, A.; Schreckenberg, R.; Schlüter, K.D. Citrulline Improves Early Post-Ischemic Recovery or Rat Hearts In Vitro by Shifting Arginine Metabolism from Polyamine to Nitric Oxide Formation. Clin. Med. Insights Cardiol. 2018, 12, 1179546818771908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.; Schreckenberg, R.; Schlüter, K.D. Effect of preischemic beta-adrenoceptor stimulation on postischemic contractile dysfunction. Life Sci. 2009, 84, 437–443. [Google Scholar] [CrossRef]
- Boengler, K.; Bulic, M.; Schreckenberg, R.; Schlüter, K.D.; Schulz, R. The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. Br. J. Pharmacol. 2017, 174, 2060–2073. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Boengler, K.; Bencsik, P.; Palóczi, J.; Kiss, K.; Pipicz, M.; Pipis, J.; Ferdinandy, P.; Schlüter, K.D.; Schulz, R.L. Lack of Contribution of p66shc and Its Mitochondrial Translocation to Ischemia-Reperfusion Injury and Cardioprotection by Ischemic Preconditioning. Front. Physiol. 2017, 8, 733. [Google Scholar] [CrossRef] [Green Version]
- Hirschhäuser, C.; Bornbaum, J.; Reis, A.; Böhme, S.; Kaludercic, N.; Menabò, R.; Di Lisa, F.; Boengler, K.; Shah, A.M.; Schulz, R.; et al. NOX4 in Mitochondria: Yeast Two-Hybrid-Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid. Redox Signal. 2015, 23, 1106–1112. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. USA 1998, 95, 12896–12901. [Google Scholar] [CrossRef] [Green Version]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panel, M.; Ahmed-Belkacem, A.; Ruiz, I.; Guichou, J.F.; Pawlotsky, J.M.; Ghaleh, B.; Morin, D.A. Phenyl-Pyrrolidine Derivative Reveals a Dual Inhibition Mechanism of Myocardial Mitochondrial Permeability Transition Pore, Which Is Limited by Its Myocardial Distribution. J. Pharmacol. Exp. Ther. 2021, 376, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Solaini, G.; Harris, D.A. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem. J. 2005, 390, 377–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borutaite, V.; Mildaziene, V.; Katiliutt, Z.; Kholodenko, B.; Toleikis, A. The function of ATP/ADP translocator in the regulation of mitochondrial respiration during development of heart ischemic injury. Biochim. Biophys. Acta 1993, 1142, 175–180. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–1044. [Google Scholar] [CrossRef]
- Heusch, G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: Benefit from selective bradycardic agents. Br. J. Pharmacol. 2008, 153, 1589–1601. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, C.; Blanchard, D. Diastolic Heart Failure: Challenges of Diagnosis and Treatment. Am. Fam. Physician 2004, 69, 2609–2616. [Google Scholar] [PubMed]
- Bowers, K.C.; Allshire, A.P.; Cobbold, P.H. Bioluminescent measurement in single cardiomyocytes of sudden cytosolic ATP depletion coincident with rigor. J. Mol. Cell. Cardiol. 1992, 24, 213–218. [Google Scholar] [CrossRef]
- Tran, K.; Loiselle, D.S.; Crampin, E.J. Regulation of cardiac cellular bioenergetics: Mechanisms and consequences. Physiol. Rep. 2015, 3, e12464. [Google Scholar] [CrossRef]
- Piper, H.M.; Garcia-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, B.; Schlüter, K.D.; Piper, H.M. Calcium and the oxygen paradox. Cardiovasc. Res. 1993, 27, 1778–1783. [Google Scholar] [CrossRef]
- Calderón-Sánchez, E.M.; Domínguez-Rodríguez, A.; López-Haldón, J.; Jiménez-Navarro, M.F.; Gómez, A.M.; Smani, T.; Ordóñez, A. Cardioprotective Effect of Ranolazine in the Process of Ischemia-reperfusion in Adult Rat Cardiomyocytes. Revista Española de Cardiología 2016, 69, 45–53. [Google Scholar] [CrossRef]
- Dibb, K.M.; Graham, H.K.; Venetucci, L.A.; Eisner, D.A.; Trafford, A.W. Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium 2007, 42, 503–512. [Google Scholar] [CrossRef]
- Antoons, G. Regulation of Ca2+ Release from the Sarcoplasmic Reticulum in the Normal and Failing Heart, 1st ed.Leuven University Press: Leuven, Belgium, 2003; ISBN 13-978-9058673121. [Google Scholar]
- Vogelpohl, I.; Vetter, R.; Heger, J.; Ebermann, L.; Euler, G.; Schultheiss, H.P.; Dörner, A. Transgenic overexpression of heart-specific adenine nucleotide translocase 1 positively affects contractile function in cardiomyocytes. Cell. Physiol. Biochem. 2011, 27, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Vittone, L.; Mundina-Weilenmann, C.; Mattiazzi, A. Phospholamban phosphorylation by CaMKII under pathophysiological conditions. Front. Biosci. 2008, 13, 5988–6005. [Google Scholar] [CrossRef] [Green Version]
- Batulan, Z.; Pulakazhi Venu, V.K.; Li, Y.; Koumbadinga, G.; Alvarez-Olmedo, D.G.; Shi, C.; O’Brien, E.R. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front. Immunol. 2016, 7, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Long, B.; Li, N.; Li, L.; Liu, C.Y.; Dong, Y.H.; Gao, J.N.; Zhou, L.Y.; Wang, C.Q.; Li, P.F. MicroRNA-2861 regulates programmed necrosis in cardiomyocyte by impairing adenine nucleotide translocase 1 expression. Free Radic. Biol. Med. 2016, 91, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Szibor, M.; Schreckenberg, R.; Gizatullina, Z.; Dufour, E.; Wiesnet, M.; Dhandapani, P.K.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Nyman, T.A.; et al. Respiratory chain signalling is essential for adaptive remodelling following cardiac ischaemia. J. Cell. Mol. Med. 2020, 24, 3534–3548. [Google Scholar] [CrossRef] [Green Version]
- Yabe, K.; Nasa, Y.; Sato, M.; Lijima, R.; Takeo, S. Preconditioning preserves mitochondrial function and glycolytic flux during an early period of reperfusion in perfused rat hearts. Cardiovasc. Res. 1997, 33, 677–685. [Google Scholar] [CrossRef]
- Takeo, S.; Nasa, Y. Role of energy metabolism in the preconditioned heart—A possible contribution of mitochondria. Cardiovasc. Res. 1999, 43, 32–43. [Google Scholar] [CrossRef] [Green Version]
- McLeod, C.J.; Jeyabalan, A.P.; Minners, J.O.; Clevenger, R.; Hoyt, R.F., Jr.; Sack, M.N. Delayed ischemic preconditioning activates nuclear-encoded electron-transfer-chain gene expression in parallel with enhanced postanoxic mitochondrial respiratory recovery. Circulation 2004, 110, 534–539. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörner, A.; Lynetskiy, O.; Euler, G.; Landmesser, U.; Schlüter, K.-D.; Heger, J. Mitochondria Isolated from Hearts Subjected to Ischemia/Reperfusion Benefit from Adenine Nucleotide Translocase 1 Overexpression. Membranes 2021, 11, 836. https://doi.org/10.3390/membranes11110836
Dörner A, Lynetskiy O, Euler G, Landmesser U, Schlüter K-D, Heger J. Mitochondria Isolated from Hearts Subjected to Ischemia/Reperfusion Benefit from Adenine Nucleotide Translocase 1 Overexpression. Membranes. 2021; 11(11):836. https://doi.org/10.3390/membranes11110836
Chicago/Turabian StyleDörner, Andrea, Oleg Lynetskiy, Gerhild Euler, Ulf Landmesser, Klaus-Dieter Schlüter, and Jacqueline Heger. 2021. "Mitochondria Isolated from Hearts Subjected to Ischemia/Reperfusion Benefit from Adenine Nucleotide Translocase 1 Overexpression" Membranes 11, no. 11: 836. https://doi.org/10.3390/membranes11110836
APA StyleDörner, A., Lynetskiy, O., Euler, G., Landmesser, U., Schlüter, K.-D., & Heger, J. (2021). Mitochondria Isolated from Hearts Subjected to Ischemia/Reperfusion Benefit from Adenine Nucleotide Translocase 1 Overexpression. Membranes, 11(11), 836. https://doi.org/10.3390/membranes11110836