
The Development and Study of Some Composite Membranes Based on Polyurethanes and Iron Oxide Nanoparticles †


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
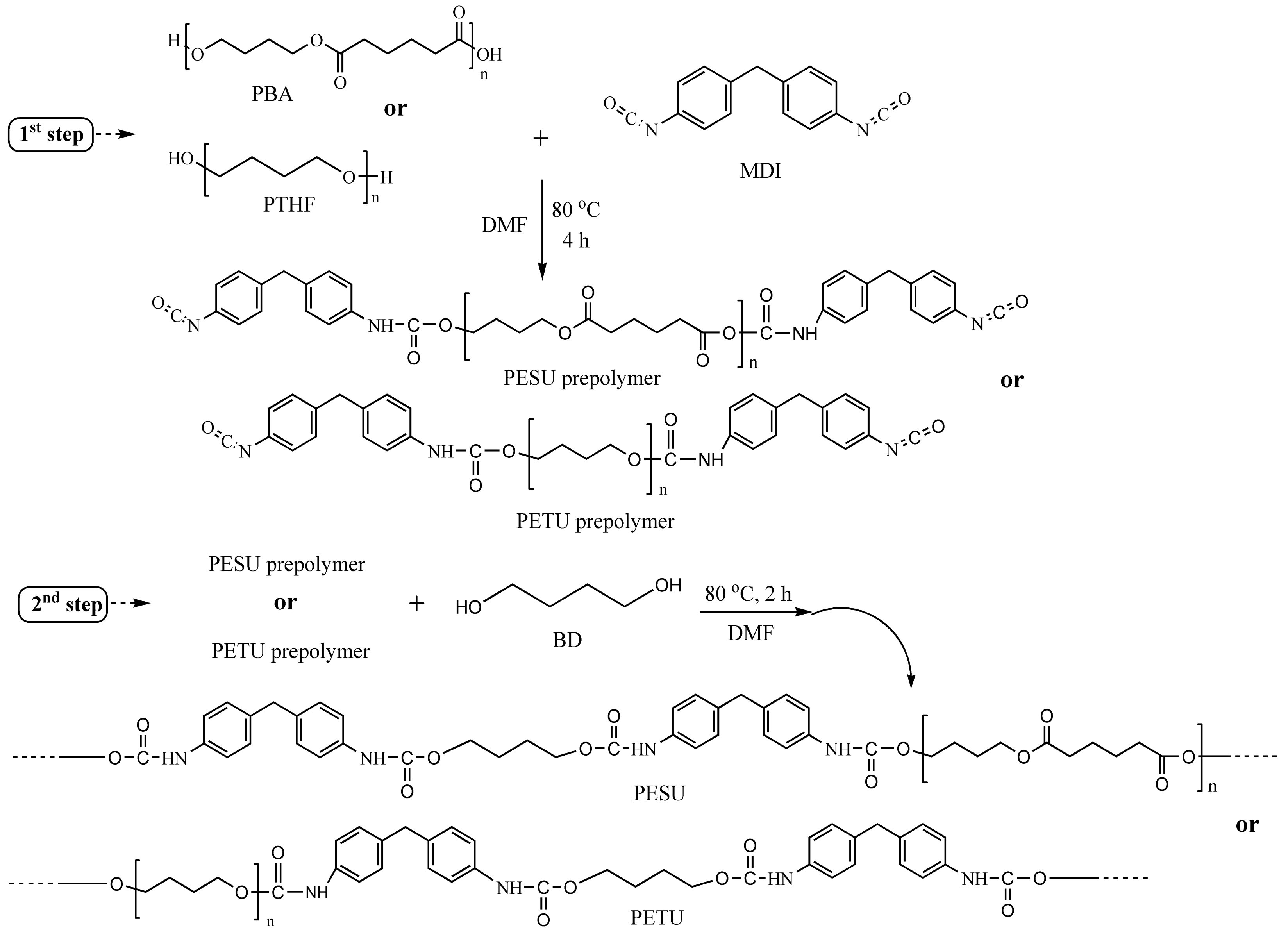
2.2. Synthesis of Polyurethanes
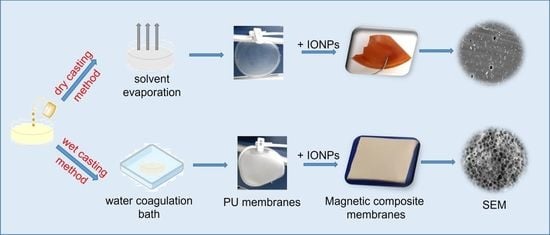
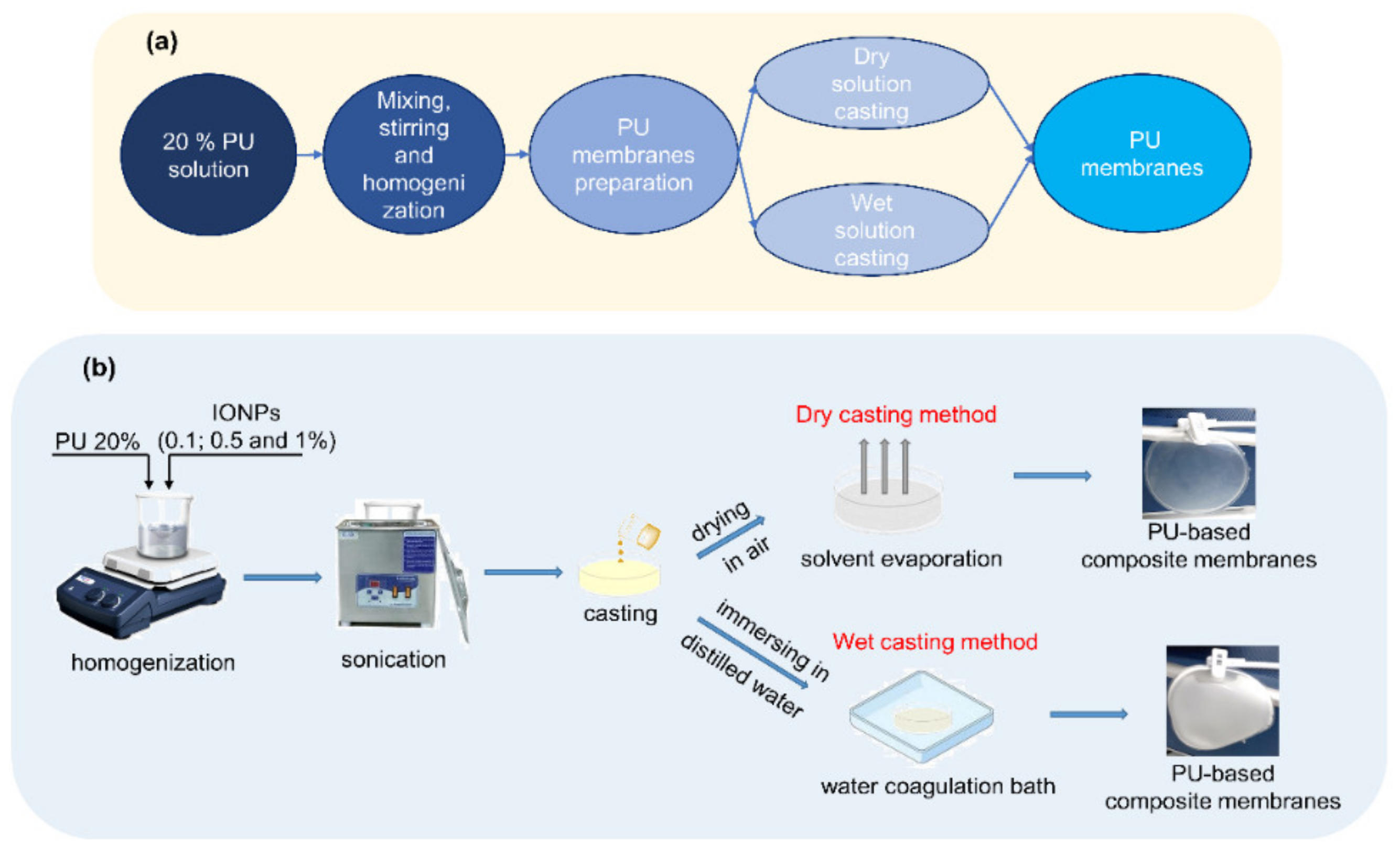
2.3. Preparation of Polyurethane-Based Membranes
2.4. Characterization Methods
3. Results and Discussion
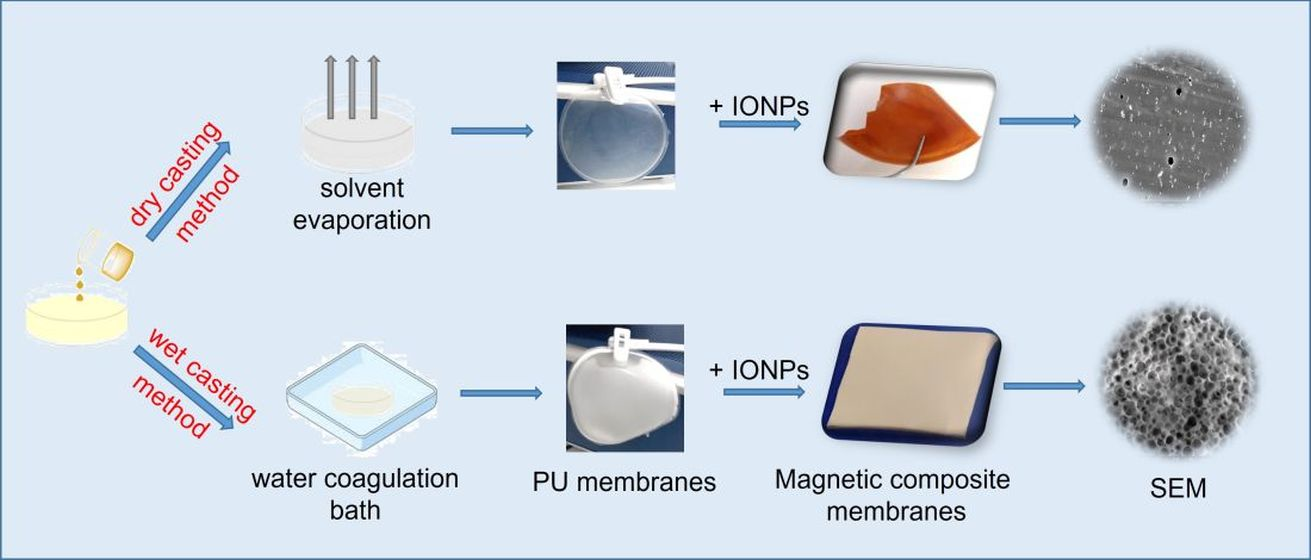
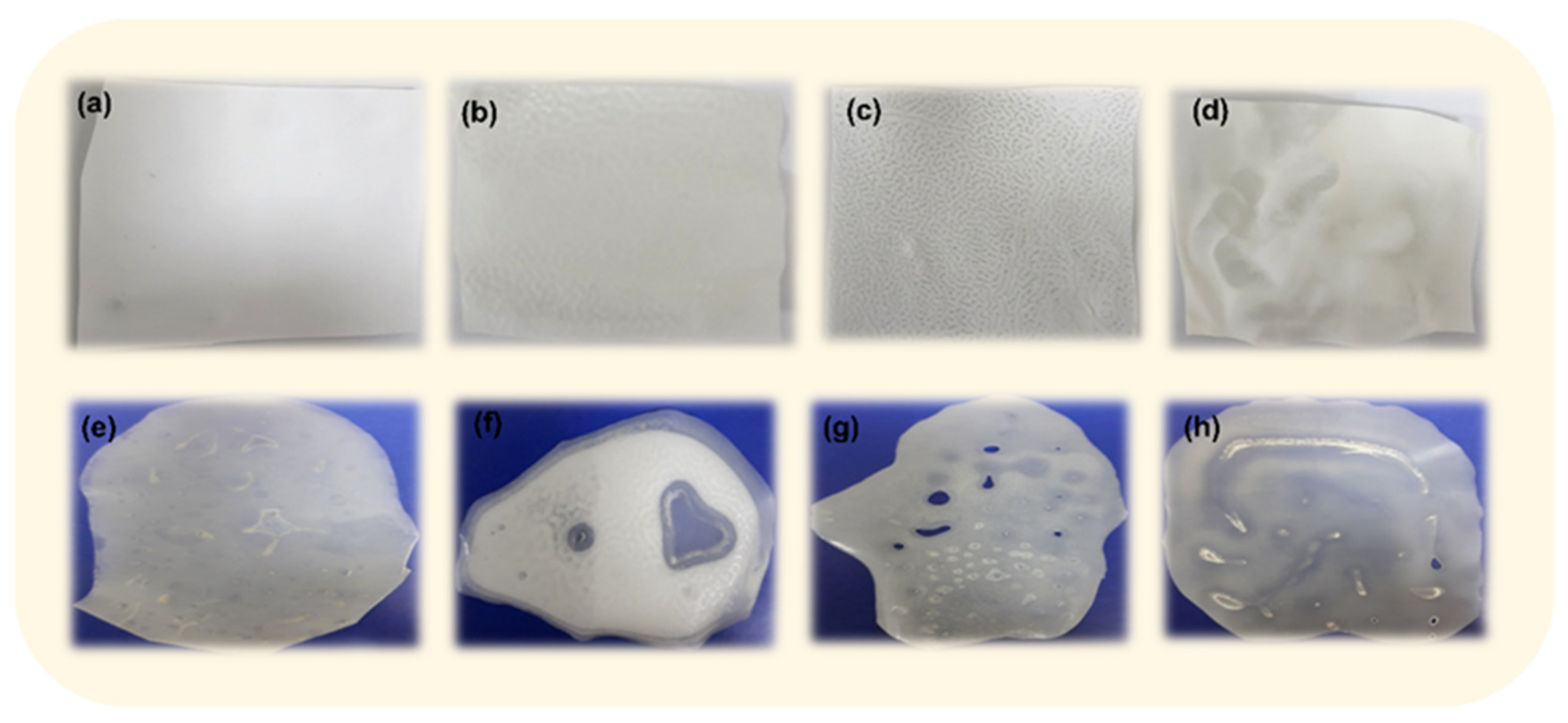
3.1. Preliminary Studies for the Selection of Conditions for Membranes’ Preparation
3.2. Characterization of Magnetic Polyurethane-Based Composite Membranes
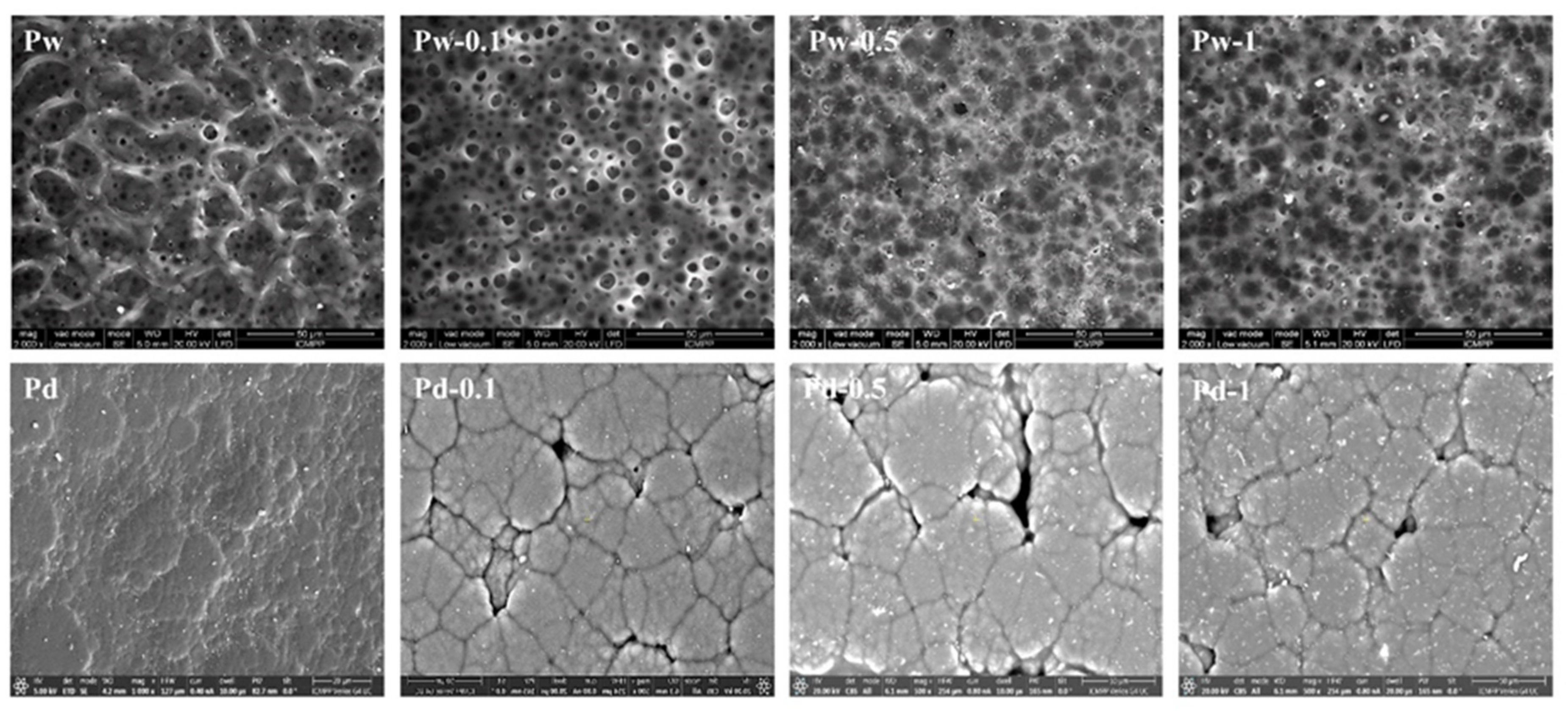
3.2.1. Surface Morphology Investigations
SEM Analysis of the Surfaces
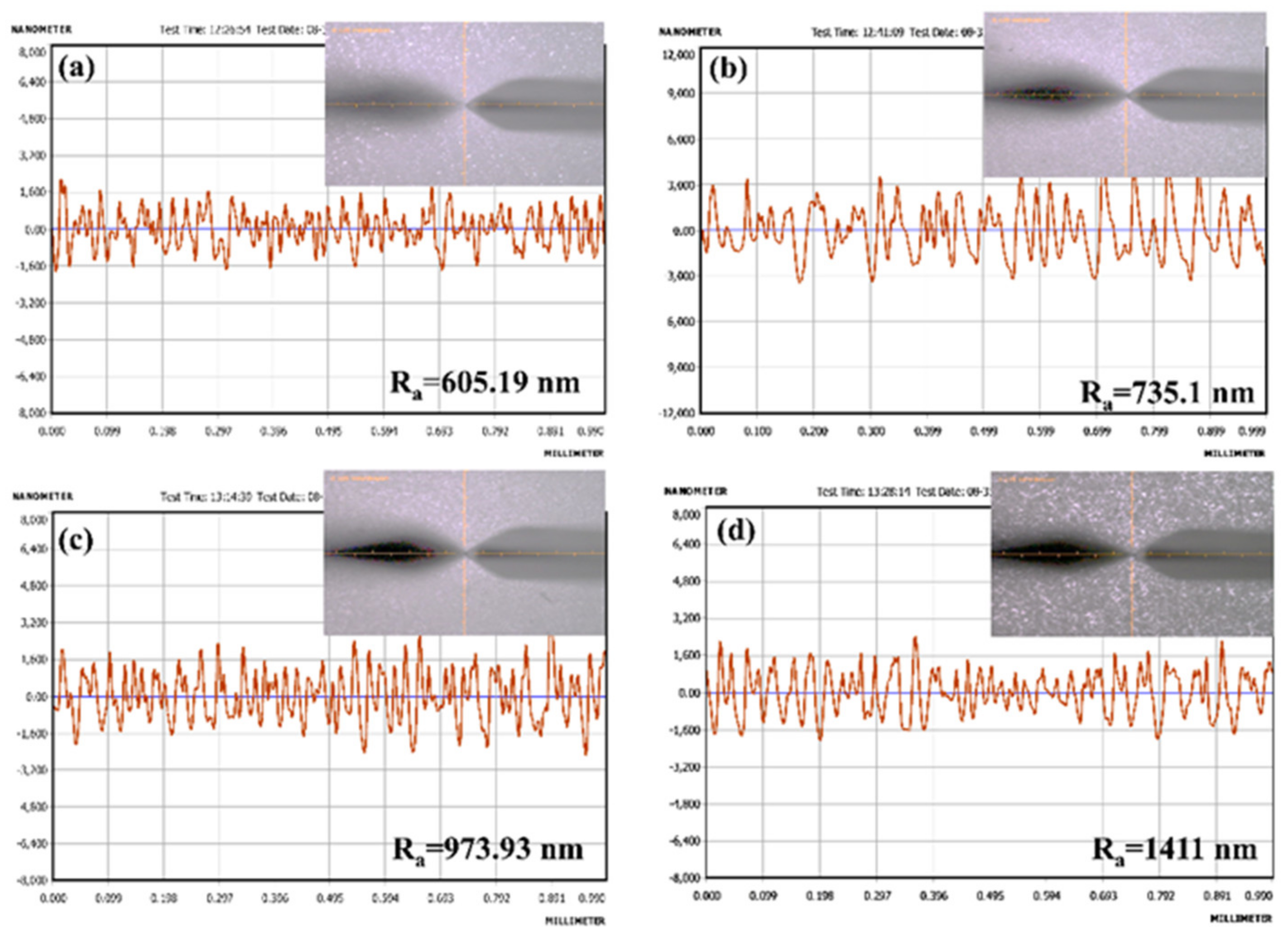
Surface Roughness
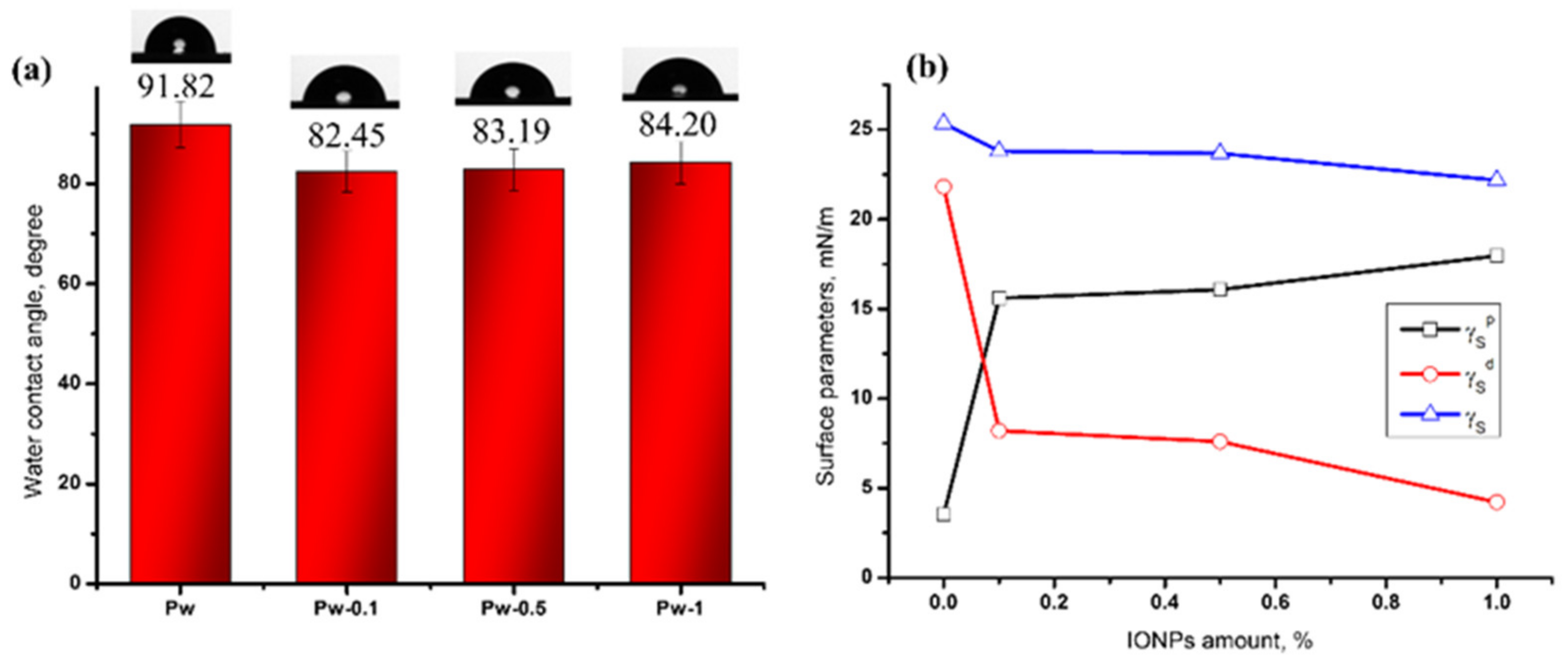
Surface Hydrophilicity and Surface Energy
Water Vapor Permeability
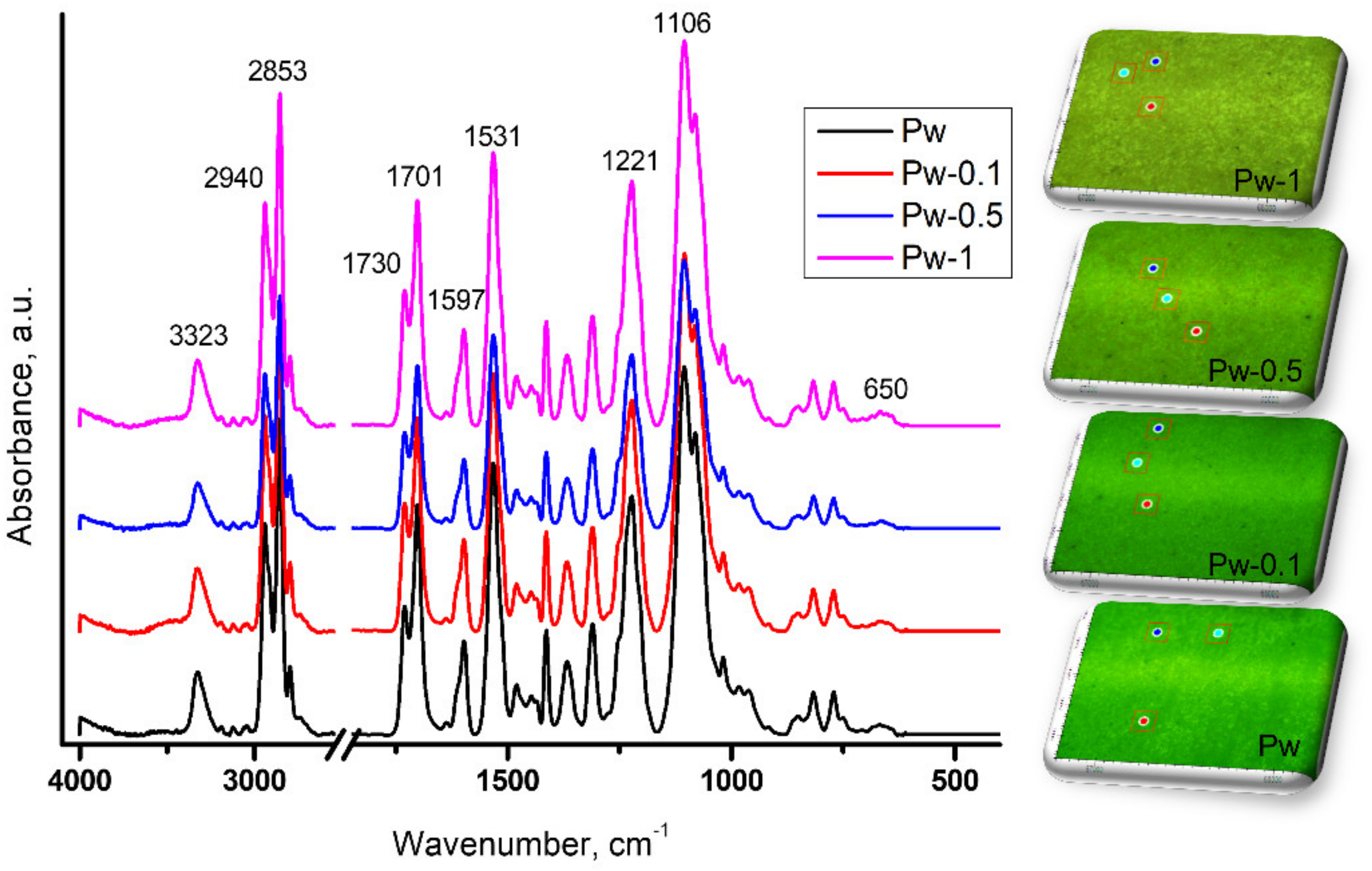
3.2.2. ATR-FTIR Investigations
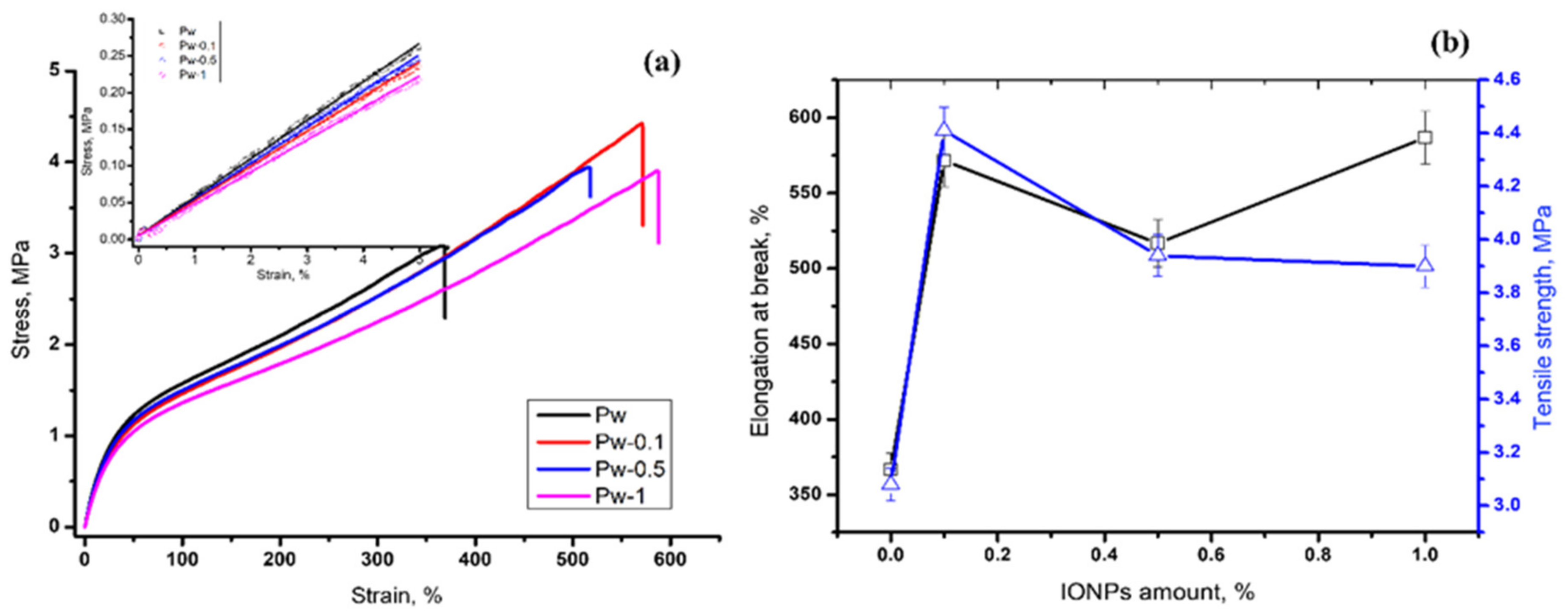
3.2.3. Tensile Tests
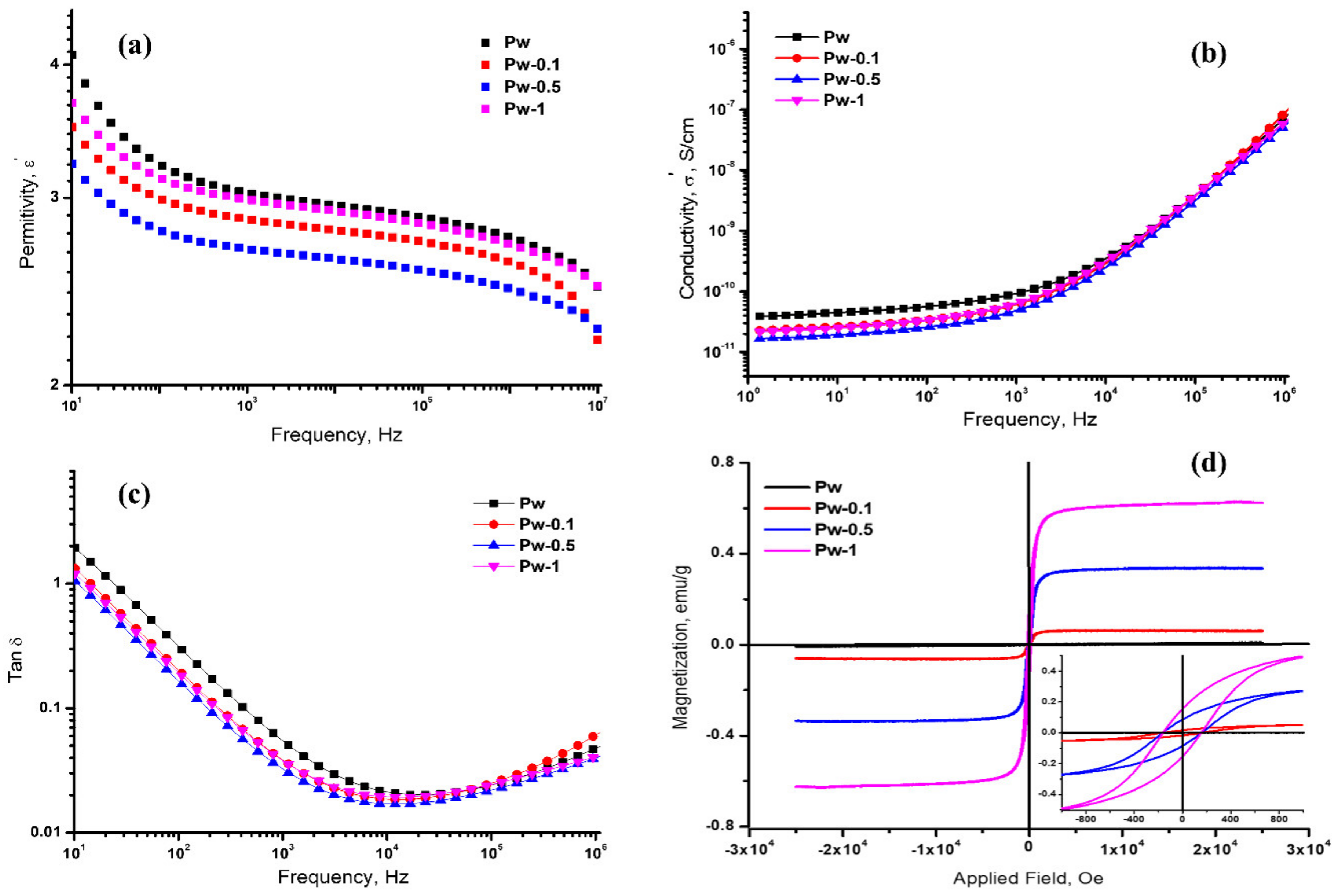
3.2.4. Dielectric and Magnetic Properties
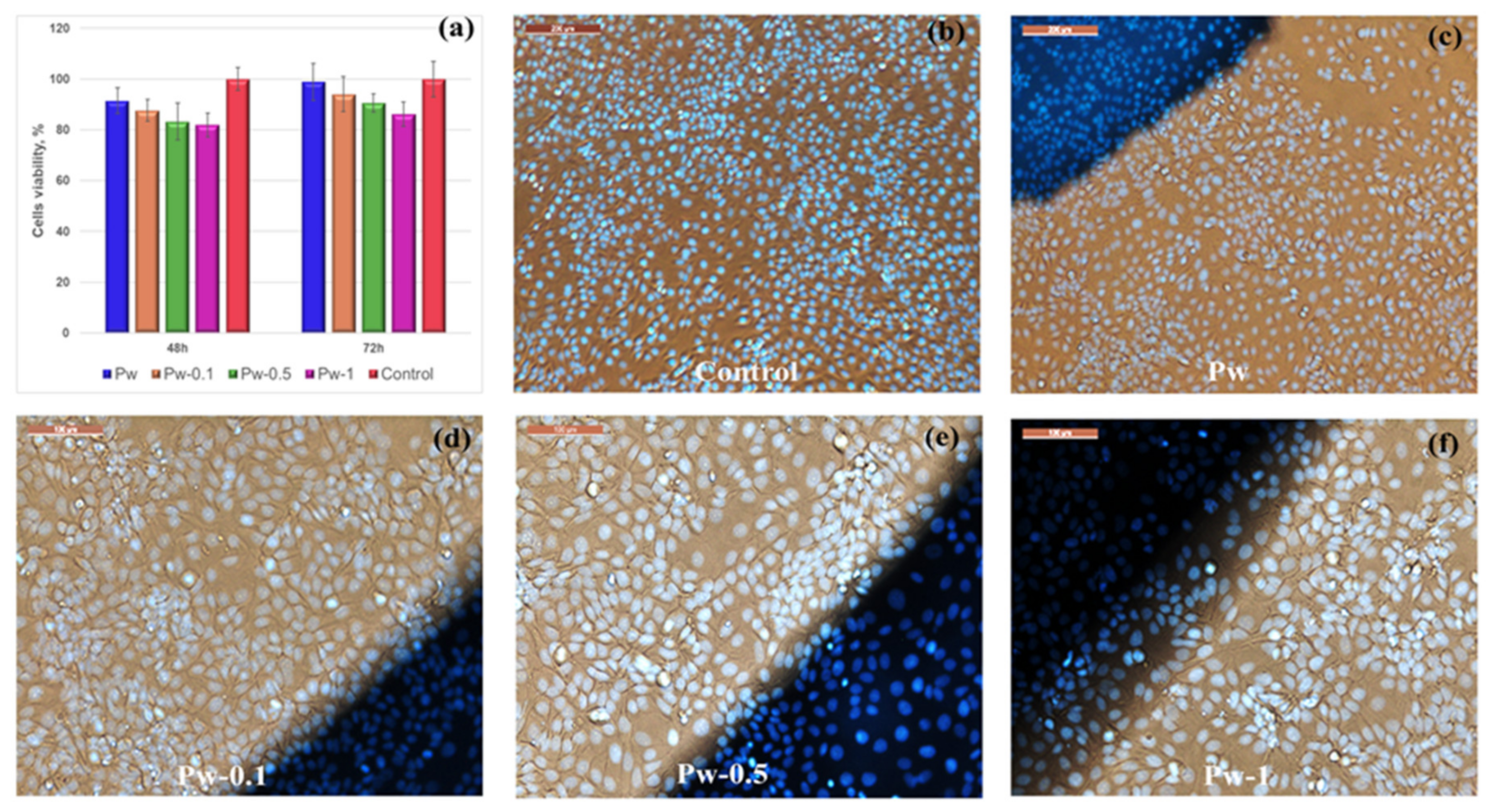
3.2.5. Cytotoxicity Evaluations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shiohara, A.; Prieto-Simon, B.; Voelcker, N.H. Porous polymeric membranes: Fabrication techniques and biomedical applications. J. Mater. Chem. B 2021, 9, 2129–2154. [Google Scholar] [CrossRef] [PubMed]
- Dmitrenko, M.E.; Penkova, A.V.; Atta, R.R.; Zolotarev, A.A.; Plisko, T.V.; Mazur, A.S.; Solovyev, N.D.; Ermakov, S.S. The development and study of novel membrane materials based on polyphenylene isophthalamide—Pluronic F127 composite. Mater. Des. 2019, 165, 107596. [Google Scholar] [CrossRef]
- Adamczak, M.; Kamińska, G.; Bohdziewicz, J. Preparation of polymer membranes by in situ interfacial polymerization. Int. J. Polym. Sci. 2019, 2019, 6217924. [Google Scholar] [CrossRef]
- Mori, Y. Functional Polymeric Membrane in Agriculture. In Functional Polymers in Food Science: From Technology to Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 2, pp. 33–45. [Google Scholar] [CrossRef]
- Puoci, F.; Iemma, F.; Spizzirri, U.G.; Cirillo, G.; Curcio, M.; Picci, N. Polymer in Agriculture: A Review. Am. J. Agric. Biol. Sci. 2008, 3, 299–314. [Google Scholar] [CrossRef]
- Gouanvé, F. Advances in Bio-Based Materials for Food Packaging Applications. Membranes 2022, 12, 735. [Google Scholar] [CrossRef]
- Turan, D. Water Vapor Transport Properties of Polyurethane Films for Packaging of Respiring Foods. Food Eng. Rev. 2021, 13, 54–65. [Google Scholar] [CrossRef]
- Radjabian, M.; Abetz, V. Advanced porous polymer membranes from self-assembling block copolymers. Prog. Polym. Sci. 2020, 102, 101219. [Google Scholar] [CrossRef]
- Jose, A.J.; Kappen, J.; Alagar, M. Polymeric membranes: Classification, preparation, structure physiochemical, and transport mechanisms. In Fundamental Biomaterials: Polymers; Elsevier: Cambridge, UK, 2018; pp. 21–35. ISBN 9780081021958. [Google Scholar]
- Khulbe, K.C.; Matsuura, T.; Feng, C. The Art of Making Polymeric Membranes. In Handbook of Polymers for Pharmaceutical Technologies; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 2, pp. 33–66. ISBN 9781119041412. [Google Scholar]
- Balakrishnan, P.; Geethamma, V.G.; Sreekala, M.S.; Thomas, S. Polymeric biomaterials: State-of-the-art and new challenges. In Fundamental Biomaterials: Polymers; Elsevier: Cambridge, UK, 2018; pp. 1–20. ISBN 9780081021958. [Google Scholar]
- Das, A.; Mahanwar, P. A brief discussion on advances in polyurethane applications. Adv. Ind. Eng. Polym. Res. 2020, 3, 93–101. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Barbalata Mandru, M.; Drobota, M.; Aflori, M.; Butnaru, M.; Spiridon, M.; Doroftei, F.; Aradoaei, M.; Ciobanu, R.C.; Vlad, S. Composite Materials Based on Iron Oxide Nanoparticles and Polyurethane for Improving the Quality of MRI. Polymers 2021, 13, 4316. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Bercea, M.; Vlad, S.; Barbalata Mandru, M.; Drobota, M.; Aflori, M.; Ciobanu, R.C. Preparation and characterization of electrospun magnetic poly(ether urethane) nanocomposite mats: Relationships between the viscosity of the polymer solutions and the electrospinning ability. Polymer 2022, 256, 125186. [Google Scholar] [CrossRef]
- Jafari, A.; Hassanajili, S.; Karimi, M.B.; Emami, A.; Ghaffari, F.; Azarpira, N. Effect of organic/inorganic nanoparticles on performance of polyurethane nanocomposites for potential wound dressing applications. J. Mech. Behav. Biomed. Mater. 2018, 88, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Antolín-Cerón, V.-H.; González-López, F.-J.; Astudillo-Sánchez, P.D.; Barrera-Rivera, K.-A.; Martínez-Richa, A. High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation. Polymers 2022, 14, 831. [Google Scholar] [CrossRef] [PubMed]
- Diez-Garcia, I.; Eceiza, A.; Tercjak, A. Improvement of Mechanical Properties and Self-Healing Efficiency by Ex-Situ Incorporation of TiO2 Nanoparticles to a Waterborne Poly(urethane-urea). Polymers 2019, 11, 1209. [Google Scholar] [CrossRef] [PubMed]
- Stroea, L.; Chibac-Scutaru, A.-L.; Melinte, V. Aliphatic Polyurethane Elastomers Quaternized with Silane-Functionalized TiO2 Nanoparticles with UV-Shielding Features. Polymers 2021, 13, 1318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Pan, M.; Wen, J.; Yuan, J.; Zhu, L.; Yu, H. Robust, Transparent, and Superhydrophobic Coating Fabricated with Waterborne Polyurethane and Inorganic Nanoparticle Composites. Ind. Eng. Chem. Res. 2019, 58, 8050–8060. [Google Scholar] [CrossRef]
- Zhong, Z.; Luo, S.; Yang, K.; Wu, X.; Ren, T. High-performance anionic waterborne polyurethane/Ag nanocomposites with excellent antibacterial property via in situ synthesis of Ag nanoparticles. RSC Adv. 2017, 7, 42296–42304. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Mandru, M.; Ciobanu, C.; Aflori, M. Opportumities and challenges in polymer-noble metal nanocomposites. In Intelligent Polymers for Nanomedicine and Biotechnologies, 1st ed.; Aflori, M., Ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 1–22. ISBN 9781315268903. [Google Scholar]
- Aflori, M.; Vlad, S.; Gradinaru, L.M.; Gradinaru, R.V.; Ciobanu, C. Recent developments in ZnO-polyurethane nanomaterials. In Intelligent Polymers for Nanomedicine and Biotechnologies, 1st ed.; Aflori, M., Ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 45–63. ISBN 9781351977449. [Google Scholar]
- Chen, X.; Yuan, J.; Huang, J.; Ren, K.; Liu, Y.; Lu, S.; Li, H. Large-scale fabrication of superhydrophobic polyurethane/nano-Al2O3 coatings by suspension flame spraying for anti-corrosion applications. Appl. Surf. Sci. 2014, 311, 864–869. [Google Scholar] [CrossRef]
- Tietze, R.; Zaloga, J.; Unterweger, H.; Lyer, S.; Friedrich, R.P.; Janko, C.; Pöttler, M.; Dürr, S.; Alexiou, C. Magnetic nanoparticle-based drug delivery for cancer therapy. Biochem. Biophys. Res. Commun. 2015, 468, 463–470. [Google Scholar] [CrossRef]
- Sobczak, M.; Kędra, K. Biomedical Polyurethanes for Anti-Cancer Drug Delivery Systems: A Brief, Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 8181. [Google Scholar] [CrossRef]
- Lammers, T.; Mertens, M.E.; Schuster, P.; Rahimi, K.; Shi, Y.; Schulz, V.; Kuehne, A.J.C.; Jockenhoevel, S.; Kiessling, F. Fluorinated Polyurethane Scaffolds for 19F Magnetic Resonance Imaging. Chem. Mater. 2017, 29, 2669–2671. [Google Scholar] [CrossRef]
- Oraby, H.; Tantawy, H.R.; Correa-Duarte, M.A.; Darwish, M.; Elsaidy, A.; Naeem, I.; Senna, M.H. Tuning Electro-Magnetic Interference Shielding Efficiency of Customized Polyurethane Composite Foams Taking Advantage of rGO/Fe3O4 Hybrid Nanocomposites. Nanomaterials 2022, 12, 2805. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.G.; Pereira, I.M.; Silva, T.I.; da Silva, M.R.; Rocha, R.A.; Silva, M.C. Magnetic foams from polyurethane and magnetite applied as attenuators of electromagnetic radiation in X band. J. Appl. Polym. Sci. 2021, 138, 49629. [Google Scholar] [CrossRef]
- Kirchberg, S.; Abdin, Y.; Ziegmann, G. Influence of particle shape and size on the wetting behavior of soft magnetic micropowders. Powder Technol. 2011, 207, 311–317. [Google Scholar] [CrossRef]
- Yoonessi, M.; Peck, J.A.; Bail, J.L.; Rogers, R.B.; Lerch, B.A.; Meador, M.A. Transparent large-strain thermoplastic polyurethane magnetoactive nanocomposites. ACS Appl. Mater. Interfaces 2011, 3, 2686–2693. [Google Scholar] [CrossRef]
- Vargas, P.C.; Merlini, C.; da Silva Ramôa, S.D.A.; Arenhart, R.; de Oliveira Barra, G.M.; Soares, B.G. Conductive composites based on polyurethane and nanostructured conductive filler of montmorillonite/polypyrrole for electromagnetic shielding applications. Mater. Res. 2018, 21, e20180014. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Vlad, S.; Barbalata-Mandru, M.; Drobota, M.; Spiridon, M.; Aflori, M.; Butnaru, M.; Ionel, A.; Ciobanu, R.C. Polyurethane Composites Based on Iron Oxide Nanoparticles as Precursor in MRI. In Proceedings of the 2021 International Conference on e-Health and Bioengineering (EHB), Iasi, Romania, 18–19 November 2021; IEEE: New York, NY, USA, 2021; pp. 1–4. [Google Scholar]
- Mansourpanah, Y.; Habili, E.M. Investigation and characterization of TiO2-TFC nanocomposite membranes; membrane preparation and UV studies. J. Membr. Sci. Res. 2015, 1, 26–33. [Google Scholar]
- Kudr, J.; Haddad, Y.; Richtera, L.; Heger, Z.; Cernak, M.; Adam, V.; Zitka, O. Magnetic nanoparticles: From design and synthesis to real world applications. Nanomaterials 2017, 7, 243. [Google Scholar] [CrossRef]
- Kausar, A. Polymeric materials filled with hematite nanoparticle: Current state and prospective application. Polym. Technol. Mater. 2020, 59, 323–338. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Z.; Yu, T.; Jiang, C.; Kim, W.S. Recent progress on magnetic iron oxide nanoparticles: Synthesis, surface functional strategies and biomedical applications. Sci. Technol. Adv. Mater. 2015, 16, 23501. [Google Scholar] [CrossRef]
- Ali, A.; Zafar, H.; Zia, M.; ul Haq, I.; Phull, A.R.; Ali, J.S.; Hussain, A. Synthesis, characterization, applications, and challenges of iron oxide nanoparticles. Nanotechnol. Sci. Appl. 2016, 9, 49–67. [Google Scholar] [CrossRef]
- Erbil, Y. Surface Chemistry of Solid and Liquid Interfaces; Erbil, Y., Ed.; Blackwell Publishing Ltd.: Oxford, UK, 2016; ISBN 1-4051-1968-3. [Google Scholar]
- ASTM E96/E96M-05; Standard Test Methods for Water Vapor Transmission of Materials. ASTM International: West Conshohocken, PA, USA, 1995; pp. 1–8.
- Chun, H.L.; Yuan, L.; Laurentia Setiawan, R.W. Fabrication of Polymeric and Composite membranes. In Membranes Fabrication; CRC Press: Boca Raton, FL, USA, 2015; ISBN 9780429161889. [Google Scholar]
- Uragami, T. Preparation Methods of Membranes. In Science and Technology of Separation Membranes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 59–85. ISBN 9781118932551. [Google Scholar] [CrossRef]
- Ciobanu, G.; Favier, L.; Harja, M. Asymmetric Cellulose Acetate Membranes Used in Separation Applications. J. Appl. Life Sci. Environ. 2021, 185, 70–76. [Google Scholar] [CrossRef]
- Ismail, A.F.; Yean, L.P. Review on the development of defect-free and ultrathin-skinned asymmetric membranes for gas separation through manipulation of phase inversion and rheological factors. J. Appl. Polym. Sci. 2003, 88, 442–451. [Google Scholar] [CrossRef]
- Tan, X.M.; Rodrigue, D. A review on porous polymeric membrane preparation. Part II: Production techniques with polyethylene, polydimethylsiloxane, polypropylene, polyimide, and polytetrafluoroethylene. Polymers 2019, 11, 1310. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Abetz, V. Nonequilibrium Processes in Polymer Membrane Formation: Theory and Experiment. Chem. Rev. 2021, 121, 14189–14231. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Song, H.M. Bottom-up self-assembly of nanofibers in the surfactant mixture of CTAB and Pluronics. AIP Adv. 2021, 11, 125031. [Google Scholar] [CrossRef]
- Krupka, T.M.; Exner, A.A. Structural parameters governing activity of Pluronic triblock copolymers in hyperthermia cancer therapy. Int. J. Hyperth. 2011, 27, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Gülmüs, S.A.; Yilmaz, L. Effect of temperature and membrane preparation parameters on gas permeation properties of polymethacrylates. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 3025–3033. [Google Scholar] [CrossRef]
- Oprea, S.; Ciobanu, C. Effect of the Temperature of Polyurethane Wet-Casting Membrane Formation on the Physico-Mechanical Properties. High Perform. Polym. 2008, 20, 208–220. [Google Scholar] [CrossRef]
- Dong, X.; Lu, D.; Harris, T.A.L.; Escobar, I.C. Polymers and solvents used in membrane fabrication: A review focusing on sustainable membrane development. Membranes 2021, 11, 309. [Google Scholar] [CrossRef]
- Mortaheb, H.R.; Baghban Salehi, M.; Rajabzadeh, M. Optimized hybrid PVDF/graphene membranes for enhancing performance of AGMD process in water desalination. J. Ind. Eng. Chem. 2021, 99, 407–421. [Google Scholar] [CrossRef]
- Ciobanu, G.; Ciobanu, O. Mixed-matrix membranes based on polyurethane containing nanohydroxyapatite and its potential applications. J. Appl. Polym. Sci. 2015, 132, 41813. [Google Scholar] [CrossRef]
- Garcia, J.U.; Iwama, T.; Chan, E.Y.; Tree, D.R.; Delaney, K.T.; Fredrickson, G.H. Mechanisms of Asymmetric Membrane Formation in Nonsolvent-Induced Phase Separation. ACS Macro Lett. 2020, 9, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.D.; Pleinemann, K.V.; Behling, R.D. Preparation and characterization of thin-film zeolite-PDMS composite membranes. J. Memb. Sci. 1992, 73, 119–128. [Google Scholar] [CrossRef]
- Agrawal, N.; Thakur, O.P.; Singh, A.K. Role of shape, size and concentration of filler particles on filler matrix interface: A mathematical analysis. Mater. Today Proc. 2021, 44, 890–894. [Google Scholar] [CrossRef]
- Osman, M.A.; Alamoush, R.A.; Kushnerev, E.; Seymour, K.G.; Shawcross, S.; Yates, J.M. In-Vitro Phenotypic Response of Human Osteoblasts to Different Degrees of Titanium Surface Roughness. Dent. J. 2022, 10, 140. [Google Scholar] [CrossRef]
- Boyan, B.D.; Hummert, T.W.; Dean, D.D.; Schwartz, Z. Role of material surfaces in regulating bone and cartilage cell response. Biomaterials 1996, 17, 137–146. [Google Scholar] [CrossRef]
- Chappard, D.; Degasne, I.; Huré, G.; Legrand, E.; Audran, M.; Baslé, M.F. Image analysis measurements of roughness by texture and fractal analysis correlate with contact profilometry. Biomaterials 2003, 24, 1399–1407. [Google Scholar] [CrossRef]
- dos Santos, L.M.; Ligabue, R.; Dumas, A.; Le Roux, C.; Micoud, P.; Meunier, J.; Martin, F.; Einloft, S. New magnetic nanocomposites: Polyurethane/ Fe3O4-synthetic talc. Eur. Polym. J. 2015, 69, 38–49. [Google Scholar] [CrossRef]
- Gradinaru, L.M.; Barbalata-Mandru, M.; Drobota, M.; Aflori, M.; Spiridon, M.; Gradisteanu Pircalabioru, G.; Bleotu, C.; Butnaru, M.; Vlad, S. Preparation and Evaluation of Nanofibrous Hydroxypropyl Cellulose and β-Cyclodextrin Polyurethane Composite Mats. Nanomaterials 2020, 10, 754. [Google Scholar] [CrossRef]
- Chen, L.; Yan, C.; Zheng, Z. Functional polymer surfaces for controlling cell behaviors. Mater. Today 2018, 21, 38–59. [Google Scholar] [CrossRef]
- Schmidt, H.; Marcinkowska, D.; Cieślak, M. Testing water vapour permeability through porous membranes. Fibres Text. East. Eur. 2005, 13, 66–68. [Google Scholar]
- Baldo, L.G.M.; Lenzi, M.K.; Eiras, D. Water vapor permeation and morphology of polysulfone membranes prepared by phase inversion. Polímeros 2020, 30, 1–8. [Google Scholar] [CrossRef]
- Lainioti, G.; Bounos, G.; Voyiatzis, G.; Kallitsis, J. Enhanced Water Vapor Transmission through Porous Membranes Based on Melt Blending of Polystyrene Sulfonate with Polyethylene Copolymers and Their CNT Nanocomposites. Polymers 2016, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhou, Z.; Chen, G.X.; Li, Q. Regulating the dielectric property of percolative composites via a core–shell-structured ionic liquid/carbon nanotube hybrid. J. Mater. Sci. 2019, 54, 7096–7109. [Google Scholar] [CrossRef]
- Debnath, T.; Saha, P.; Patra, N.; Das, S.; Sutradhar, S. Hydrothermal process assists undoped and Cr-doped semiconducting ZnO nanorods: Frontier of dielectric property. J. Appl. Phys. 2018, 123, 194101. [Google Scholar] [CrossRef]
- Cetiner, S.; Sirin, S.; Olariu, M.; Sarac, A.S. Frequency and Temperature Dependence of Dielectric Behaviors for Conductive Acrylic Composites. Adv. Polym. Technol. 2016, 35, 1–10. [Google Scholar] [CrossRef]
- Cao, D.; Li, H.; Pan, L.; Li, J.; Wang, X.; Jing, P.; Cheng, X.; Wang, W.; Wang, J.; Liu, Q. High saturation magnetization of γ 3-Fe2O3 nanoparticles by a facile one-step synthesis approach. Sci. Rep. 2016, 6, 32360. [Google Scholar] [CrossRef]
- Di Palma, L.; Bavasso, I.; Sarasini, F.; Tirillò, J.; Puglia, D.; Dominici, F.; Torre, L.; Galluzzi, A.; Polichetti, M.; Ramazanov, M.A.; et al. Effect of nano-magnetite particle content on mechanical, thermal and magnetic properties of polypropylene composites. Polym. Compos. 2018, 39, E1742–E1750. [Google Scholar] [CrossRef]
- Shahrousvand, M.; Hoseinian, M.S.; Ghollasi, M.; Karbalaeimahdi, A.; Salimi, A.; Tabar, F.A. Flexible magnetic polyurethane/Fe2O3 nanoparticles as organic-inorganic nanocomposites for biomedical applications: Properties and cell behavior. Mater. Sci. Eng. C 2017, 74, 556–567. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | G/t (g/h) | R2 | A (cm2) | WVP (g/m2h) |
|---|---|---|---|---|
| Pw | 0.0497 | 0.9981 | 10 | 49.7 ± 0.9 |
| Pw-0.1 | 0.0526 | 0.9980 | 52.6 ± 1.2 | |
| Pw-0.5 | 0.0613 | 0.9979 | 61.3 ± 1.3 | |
| Pw-1 | 0.0665 | 0.9973 | 66.5 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gradinaru, L.M.; Vlad, S.; Ciobanu, R.C. The Development and Study of Some Composite Membranes Based on Polyurethanes and Iron Oxide Nanoparticles. Membranes 2022, 12, 1127. https://doi.org/10.3390/membranes12111127
Gradinaru LM, Vlad S, Ciobanu RC. The Development and Study of Some Composite Membranes Based on Polyurethanes and Iron Oxide Nanoparticles. Membranes. 2022; 12(11):1127. https://doi.org/10.3390/membranes12111127
Chicago/Turabian StyleGradinaru, Luiza Madalina, Stelian Vlad, and Romeo Cristian Ciobanu. 2022. "The Development and Study of Some Composite Membranes Based on Polyurethanes and Iron Oxide Nanoparticles" Membranes 12, no. 11: 1127. https://doi.org/10.3390/membranes12111127
APA StyleGradinaru, L. M., Vlad, S., & Ciobanu, R. C. (2022). The Development and Study of Some Composite Membranes Based on Polyurethanes and Iron Oxide Nanoparticles. Membranes, 12(11), 1127. https://doi.org/10.3390/membranes12111127