
Computational Approaches to Alkaline Anion-Exchange Membranes for Fuel Cell Applications



Abstract
:1. Introduction
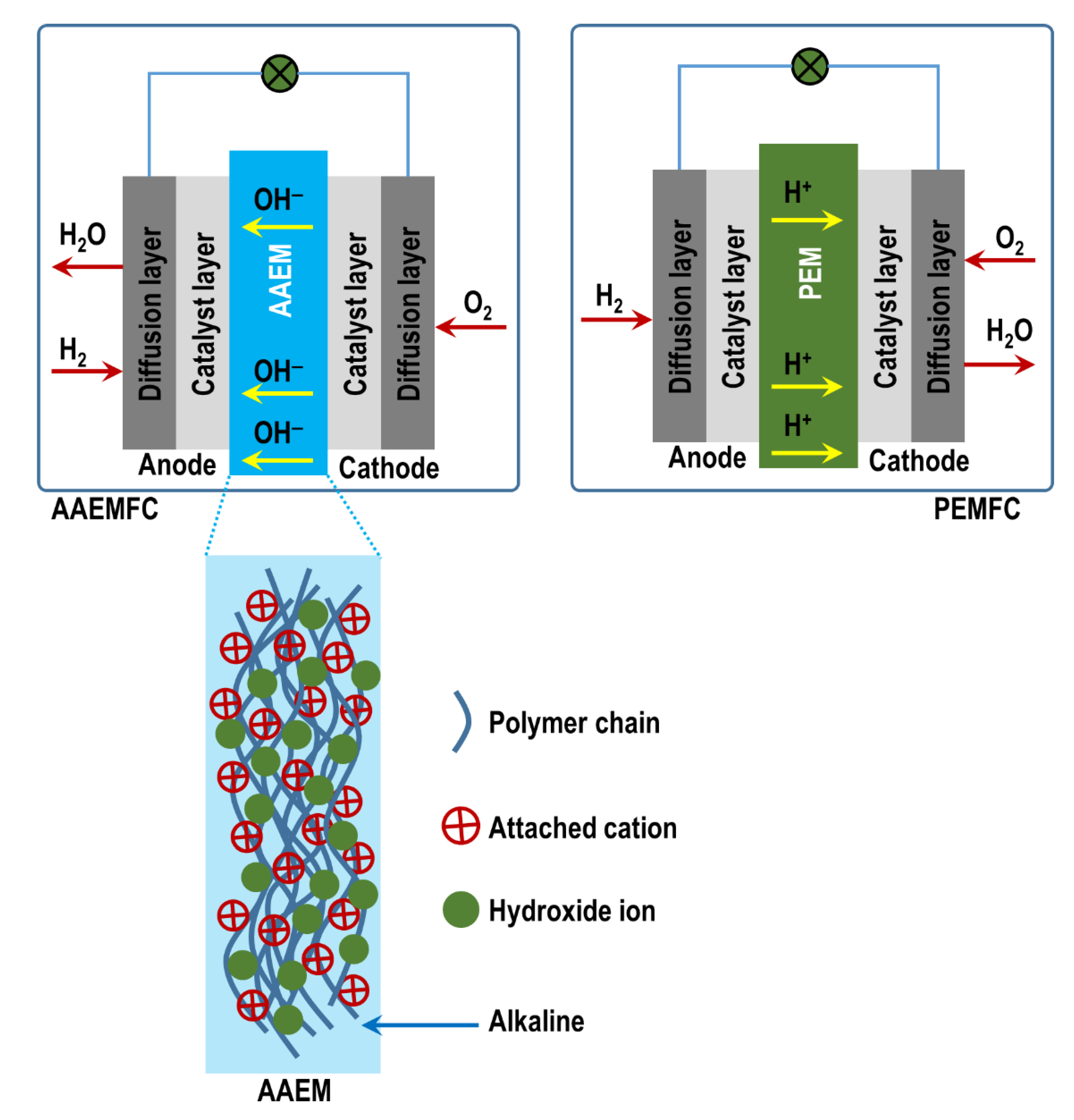
2. Alkaline Anion-Exchange Membrane Fuel Cells (AAEMFCs)
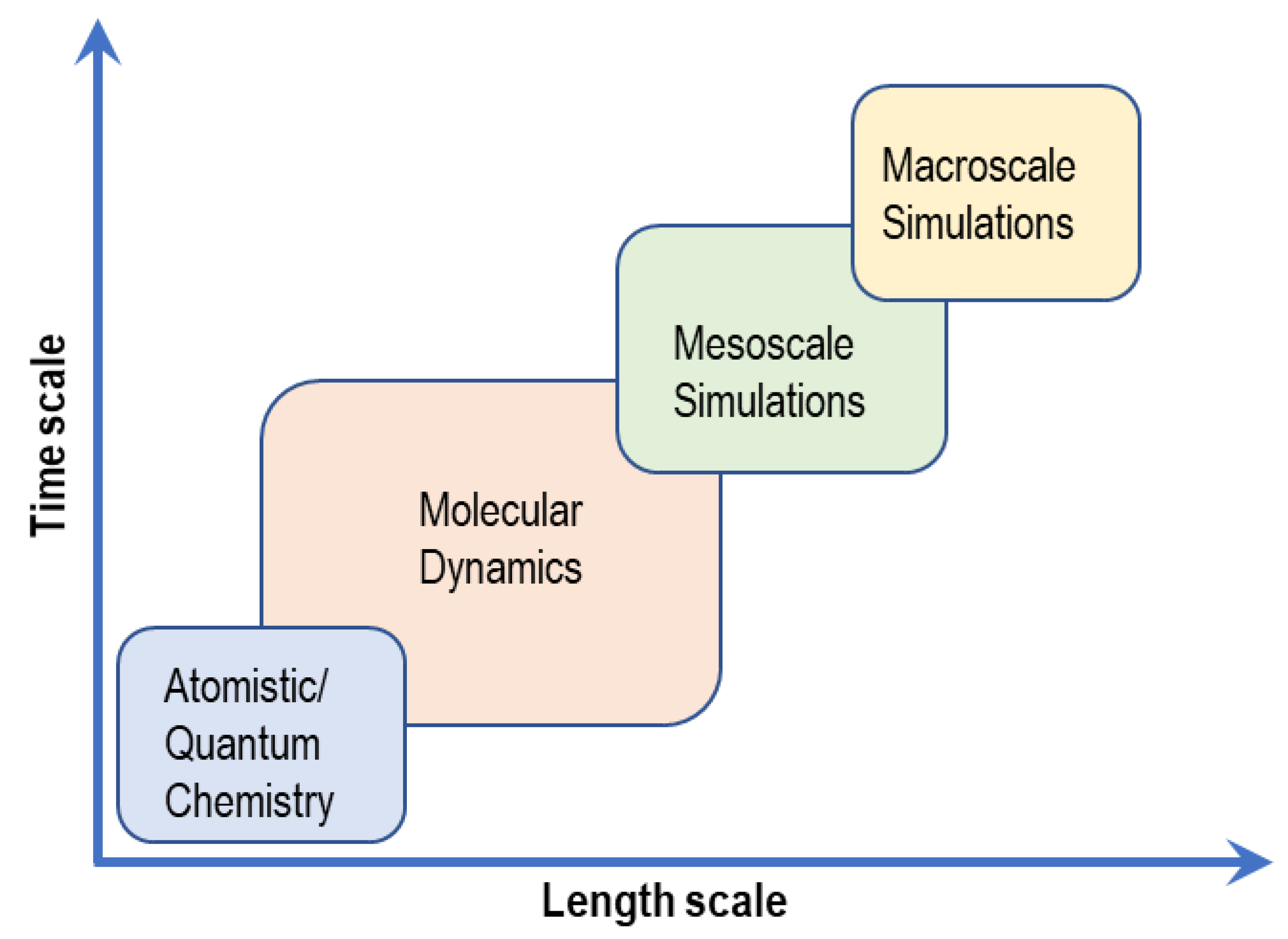
3. Computational Studies of AAEMs
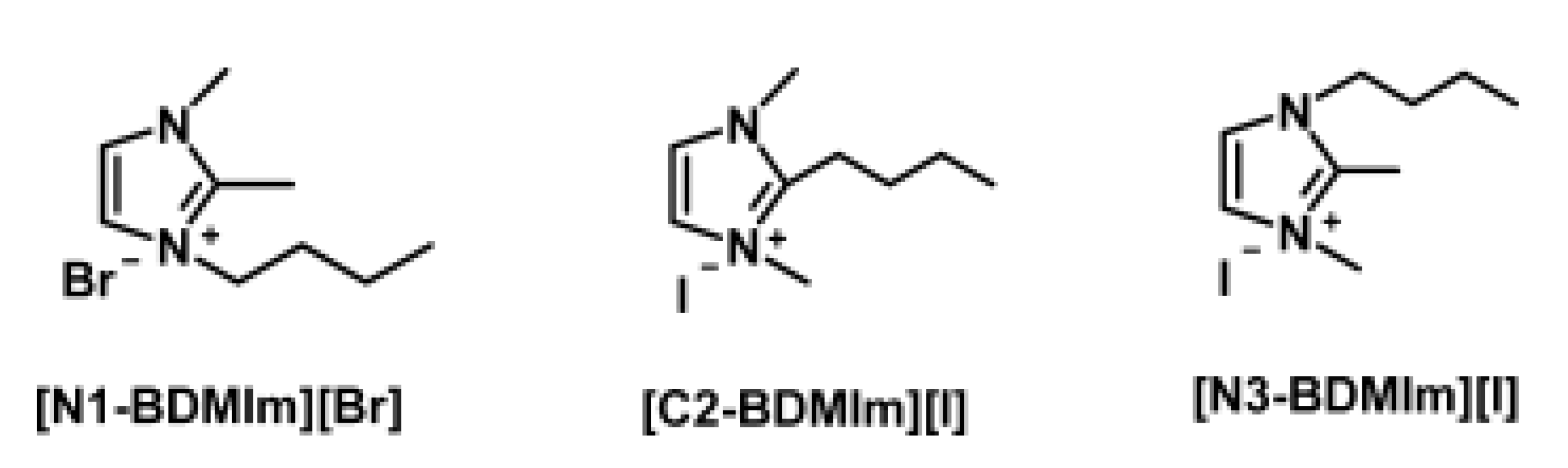
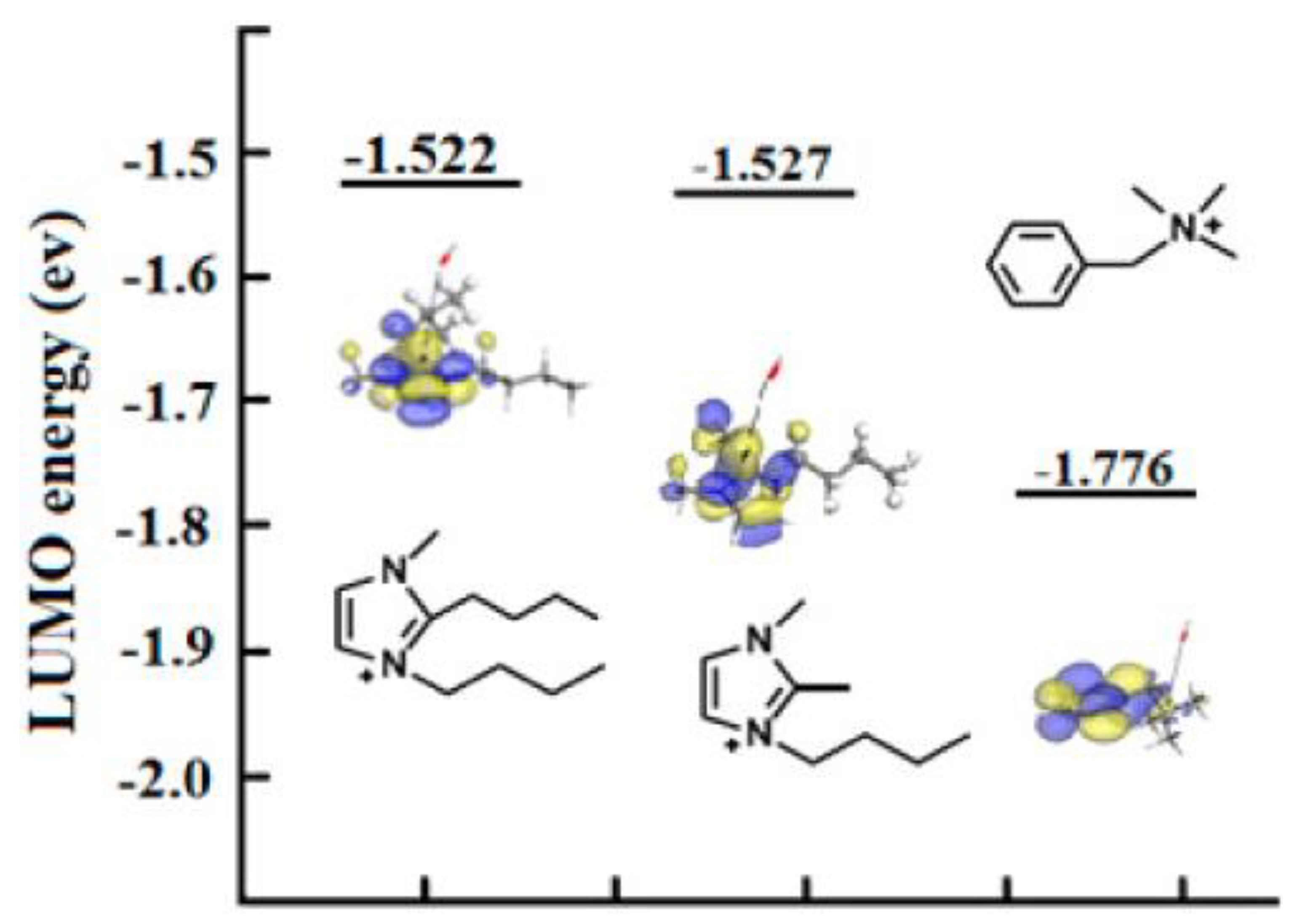
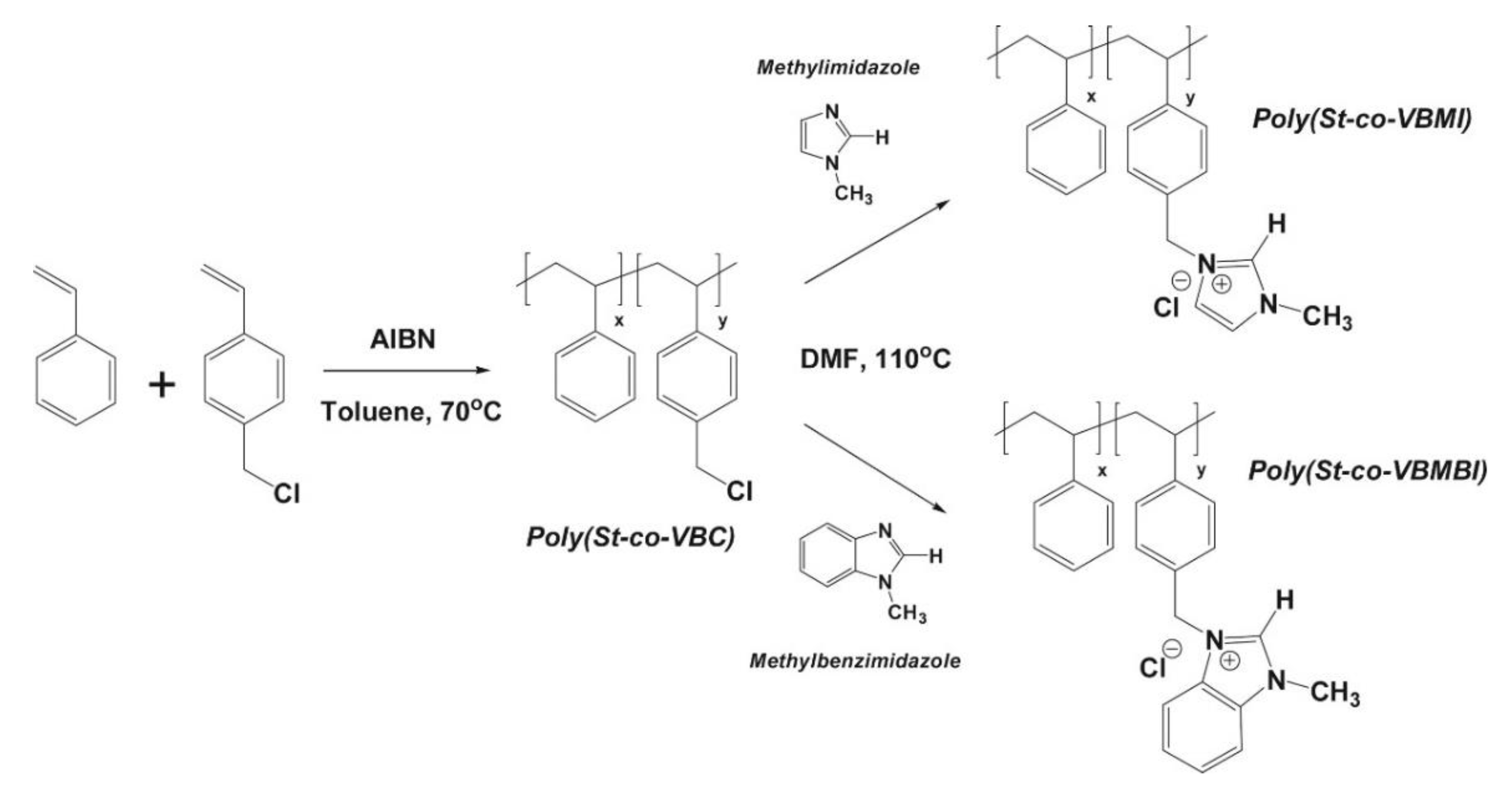
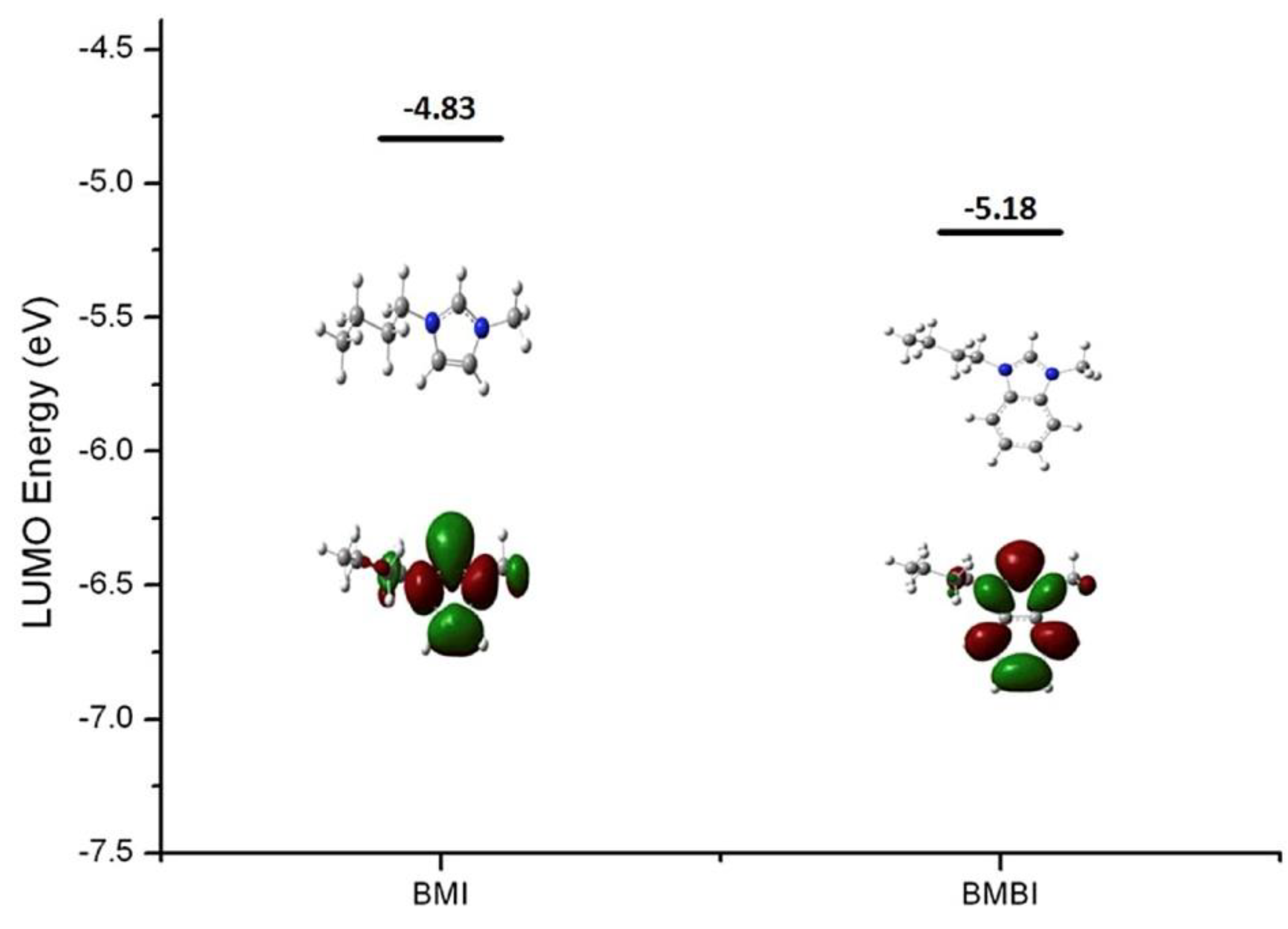
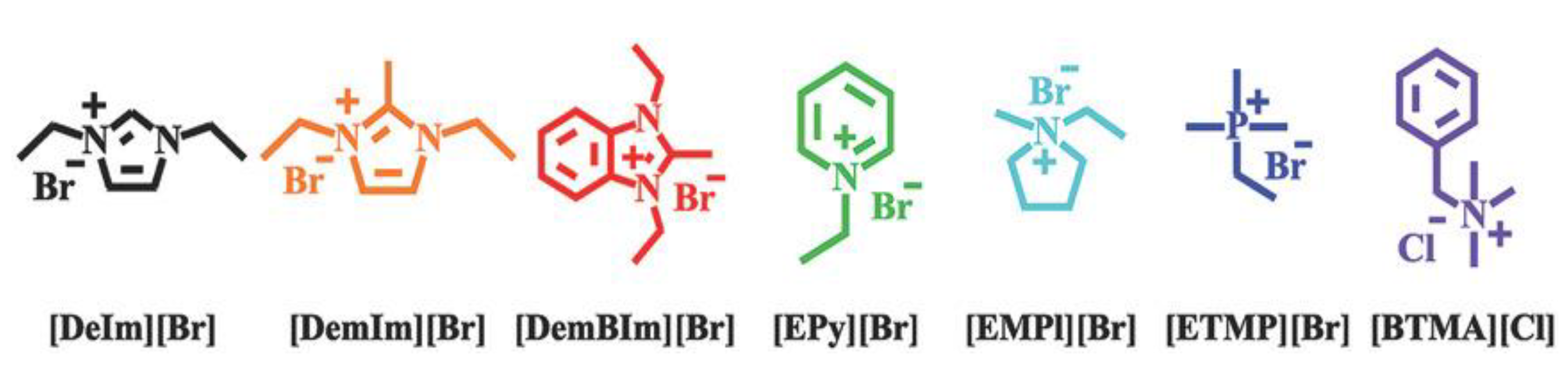
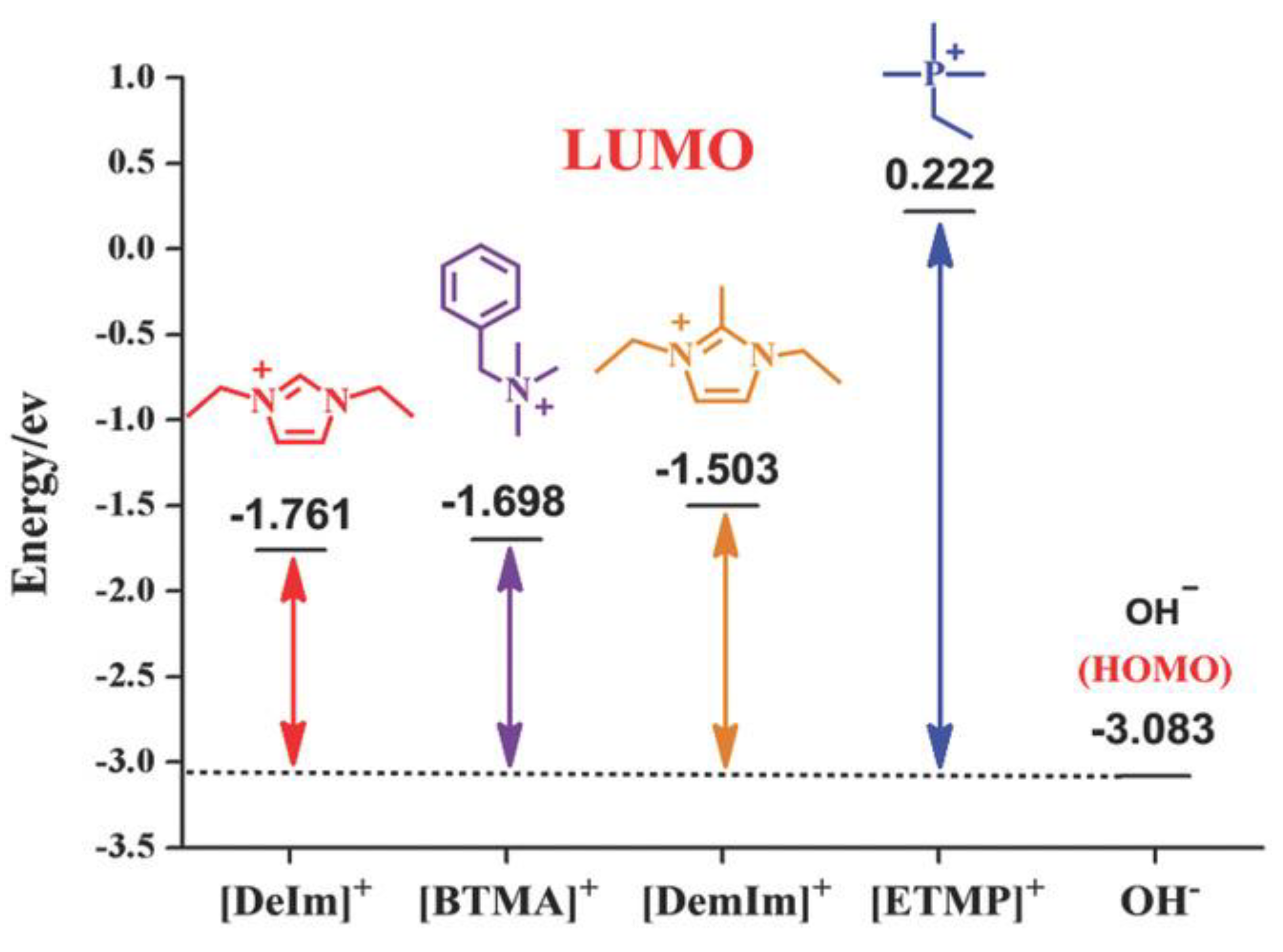
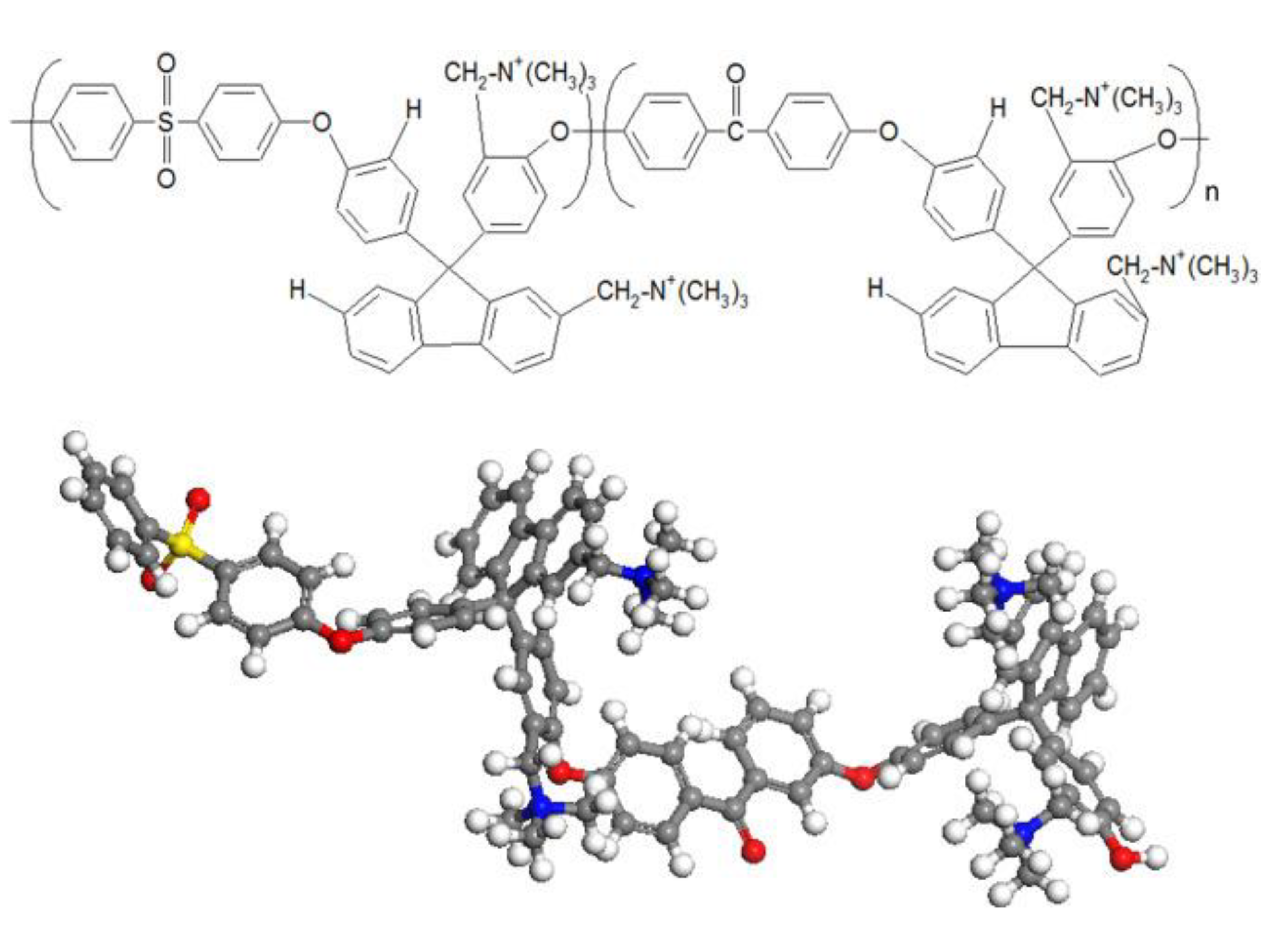
3.1. Atomistic/Quantum Chemistry Studies of AAEMs
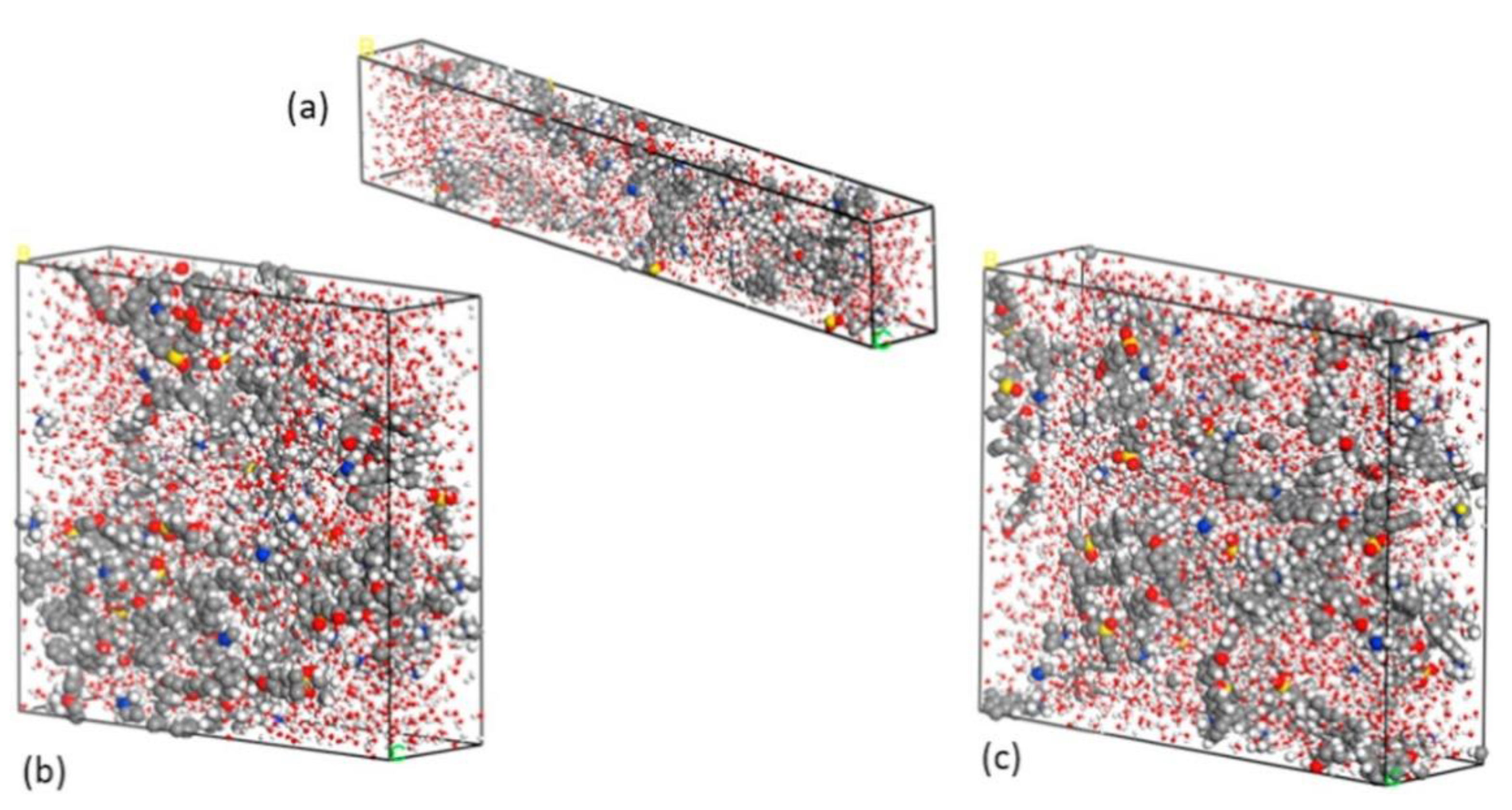
3.2. Molecular Dynamic Studies of AAEMs
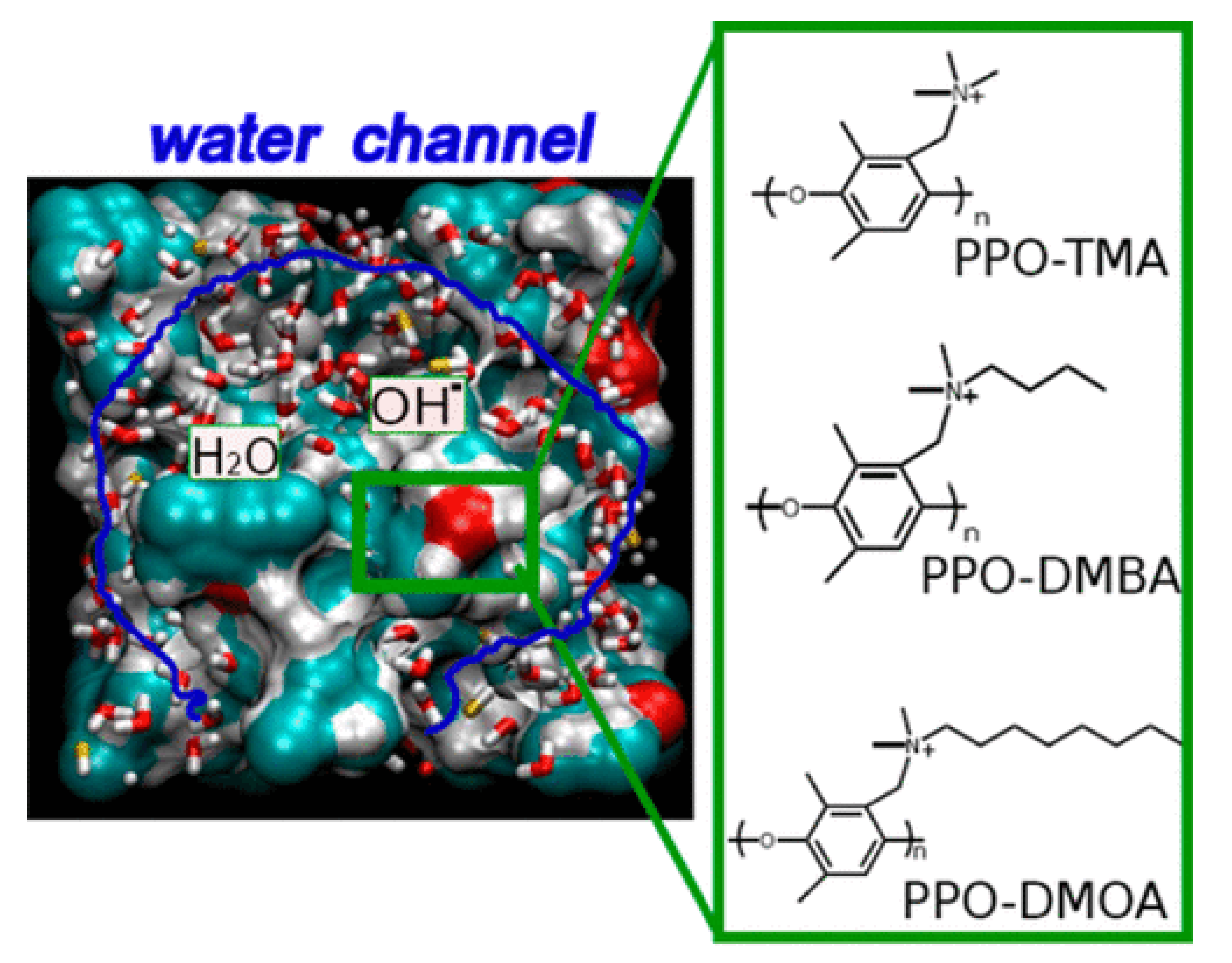
3.3. Mesoscale Studies of AAEMs
3.4. Application of Machine Learning in Modeling Properties of AAEMs for AAEMFCs
3.5. Consistency of Modeling Length Scales with Experiments
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Z.; Lin, B.; Yan, F. Anion-Exchange Membranes for Alkaline Fuel-Cell Applications: The Effects of Cations. Chem. Sus. Chem. 2018, 11, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Hickner, M.A.; Herring, A.M.; Coughlin, E.B. Anion exchange membranes: Current status and moving forward. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1727–1735. [Google Scholar] [CrossRef]
- Vincent, I.; Kruger, A.; Bessarabov, D. Development of efficient membrane electrode assembly for low cost hydrogen production by anion exchange membrane electrolysis. Int. J. Hydrogen Energy 2017, 42, 10752–10761. [Google Scholar] [CrossRef]
- Gao, X.; He, L.; Yu, H.; Xie, F.; Yang, Y.; Shao, Z. The non-precious metal ORR catalysts for the anion exchange membrane fuel cells application: A numerical simulation and experimental study. Int. J. Hydrogen Energy 2020, 45, 23353–23367. [Google Scholar] [CrossRef]
- Zhang, F.; Li, T.; Chen, W.; Yan, X.; Wu, X.; Jiang, X.; Zhang, Y.; Wang, X.; He, G. High-Performance Anion Exchange Membranes with Para-Type Cations on Electron-Withdrawing C=O Links Free Backbone. Macromolecules 2020, 53, 10988–10997. [Google Scholar] [CrossRef]
- Tao, Z.; Wang, C.; Zhao, X.; Li, J.; Guiver, M.D.; Tao, Z.; Wang, C.; Li, J.; Zhao, X.; Guiver, M.D. Progress in High-Performance Anion Exchange Membranes Based on the Design of Stable Cations for Alkaline Fuel Cells. Adv. Mater. Technol. 2021, 6, 2001220. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, Y.C.; Wu, Z.P.; Chen, G.; Yang, F.; Zhu, S.; Siddharth, K.; Kong, Z.; Lu, A.; Li, J.C.; et al. Recent Advances in Electrocatalysts for Proton Exchange Membrane Fuel Cells and Alkaline Membrane Fuel Cells. Adv. Mater. 2021, 33, 2006292. [Google Scholar] [CrossRef]
- Hren, M.; Božič, M.; Fakin, D.; Kleinschek, K.S.; Gorgieva, S. Alkaline membrane fuel cells: Anion exchange membranes and fuels. Sustain. Energy Fuels 2021, 5, 604–637. [Google Scholar] [CrossRef]
- Sarapuu, A.; Kibena-Põldsepp, E.; Borghei, M.; Tammeveski, K. Electrocatalysis of oxygen reduction on heteroatom-doped nanocarbons and transition metal-nitrogen-carbon catalysts for alkaline membrane fuel cells. J. Mater. Chem. A 2018, 6, 776–804. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, Y.S. Quaternized aryl ether-free polyaromatics for alkaline membrane fuel cells: Synthesis, properties, and performance-a topical review. J. Mater. Chem. A 2018, 6, 15456–15477. [Google Scholar] [CrossRef]
- Yang, Y.; Peltier, C.R.; Zeng, R.; Schimmenti, R.; Li, Q.; Huang, X.; Yan, Z.; Potsi, G.; Selhorst, R.; Lu, X.; et al. Electrocatalysis in Alkaline Media and Alkaline Membrane-Based Energy Technologies. Chem. Rev. 2022, 122, 6117–6321. [Google Scholar] [CrossRef] [PubMed]
- Karibayev, M.; Kalybekkyzy, S.; Wang, Y.; Mentbayeva, A. Molecular Modeling in Anion Exchange Membrane Research: A Brief Review of Recent Applications. Molecules 2022, 27, 3574. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Pan, J.; Guo, J.; Yan, F. The Alkaline Stability of Anion Exchange Membrane for Fuel Cell Applications: The Effects of Alkaline Media. Adv. Sci. 2018, 5, 1800065. [Google Scholar] [CrossRef] [PubMed]
- Mamlouk, M.; Manolova, M. Chapter 6: Alkaline Anionic Exchange Membrane Water Electrolysers. In Electrochemical Methods for Hydrogen Production; Energy and Environment Series; Royal Society of Chemistry: London, UK, 2019; pp. 180–252. [Google Scholar]
- Henkensmeier, D.; Najibah, M.; Harms, C.; Žitka, J.; Hnát, J.; Bouzek, K. Overview: State-of-the Art Commercial Membranes for Anion Exchange Membrane Water Electrolysis. J. Electrochem. Energy Convers. Storage 2021, 18, 024001. [Google Scholar] [CrossRef]
- Vincent, I.; Bessarabov, D. Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renew. Sustain. Energy Rev. 2018, 81, 1690–1704. [Google Scholar] [CrossRef]
- Du, N.; Roy, C.; Peach, R.; Turnbull, M.; Thiele, S.; Bock, C. Anion-Exchange Membrane Water Electrolyzers. Chem. Rev. 2022, 122, 11830–11895. [Google Scholar] [CrossRef] [PubMed]
- Hibbs 2022, M.R. Alkaline stability of poly(phenylene)-based anion exchange membranes with various cations. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1736–1742. [Google Scholar] [CrossRef]
- Sepehr, F.; Liu, H.; Luo, X.; Bae, C.; Tuckerman, M.E.; Hickner, M.A.; Paddison, S.J. Mesoscale Simulations of Anion Exchange Membranes Based on Quaternary Ammonium Tethered Triblock Copolymers. Macromolecules 2017, 50, 4397–4405. [Google Scholar] [CrossRef]
- Espiritu, R.; Tan, J.L.; Lim, L.H.; Arco, S. Density functional theory study on the degradation of fuel cell anion exchange membranes via removal of vinylbenzyl quaternary ammonium head group. J. Phys. Org. Chem. 2020, 33, e4049. [Google Scholar] [CrossRef]
- Vijayakumar, V.; Nam, S.Y. Recent advancements in applications of alkaline anion exchange membranes for polymer electrolyte fuel cells. J. Ind. Eng. Chem. 2019, 70, 70–86. [Google Scholar] [CrossRef]
- Mohanty, A.D.; Tignor, S.E.; Krause, J.A.; Choe, Y.K.; Bae, C. Systematic Alkaline Stability Study of Polymer Backbones for Anion Exchange Membrane Applications. Macromolecules 2016, 49, 3361–3372. [Google Scholar] [CrossRef]
- Pan, J.; Sun, Z.; Zhu, H.; Cao, H.; Wang, B.; Zhao, J.; Yan, F. Synthesis and characterization of main-chain type polyimidazolium-based alkaline anion exchange membranes. J. Memb. Sci. 2020, 610, 118283. [Google Scholar] [CrossRef]
- Choe, Y.K.; Fujimoto, C.; Lee, K.S.; Dalton, L.T.; Ayers, K.; Henson, N.J.; Kim, Y.S. Alkaline stability of benzyl trimethyl ammonium functionalized polyaromatics: A computational and experimental study. Chem. Mater. 2014, 26, 5675–5682. [Google Scholar] [CrossRef]
- Gu, F.; Dong, H.; Li, Y.; Si, Z.; Yan, F. Highly stable N3-substituted imidazolium-based alkaline anion exchange membranes: Experimental studies and theoretical calculations. Macromolecules 2014, 47, 208–216. [Google Scholar] [CrossRef]
- Si, Z.; Qiu, L.; Dong, H.; Gu, F.; Li, Y.; Yan, F. Effects of substituents and substitution positions on alkaline stability of imidazolium cations and their corresponding anion-exchange membranes. ACS Appl. Mater. Interfaces 2014, 6, 4346–4355. [Google Scholar] [CrossRef]
- You, W.; Ganley, J.M.; Ernst, B.G.; Peltier, C.R.; Ko, H.Y.; DiStasio, R.A.; Knowles, R.R.; Coates, G.W. Expeditious synthesis of aromatic-free piperidinium-functionalized polyethylene as alkaline anion exchange membranes. Chem. Sci. 2021, 12, 3898–3910. [Google Scholar] [CrossRef]
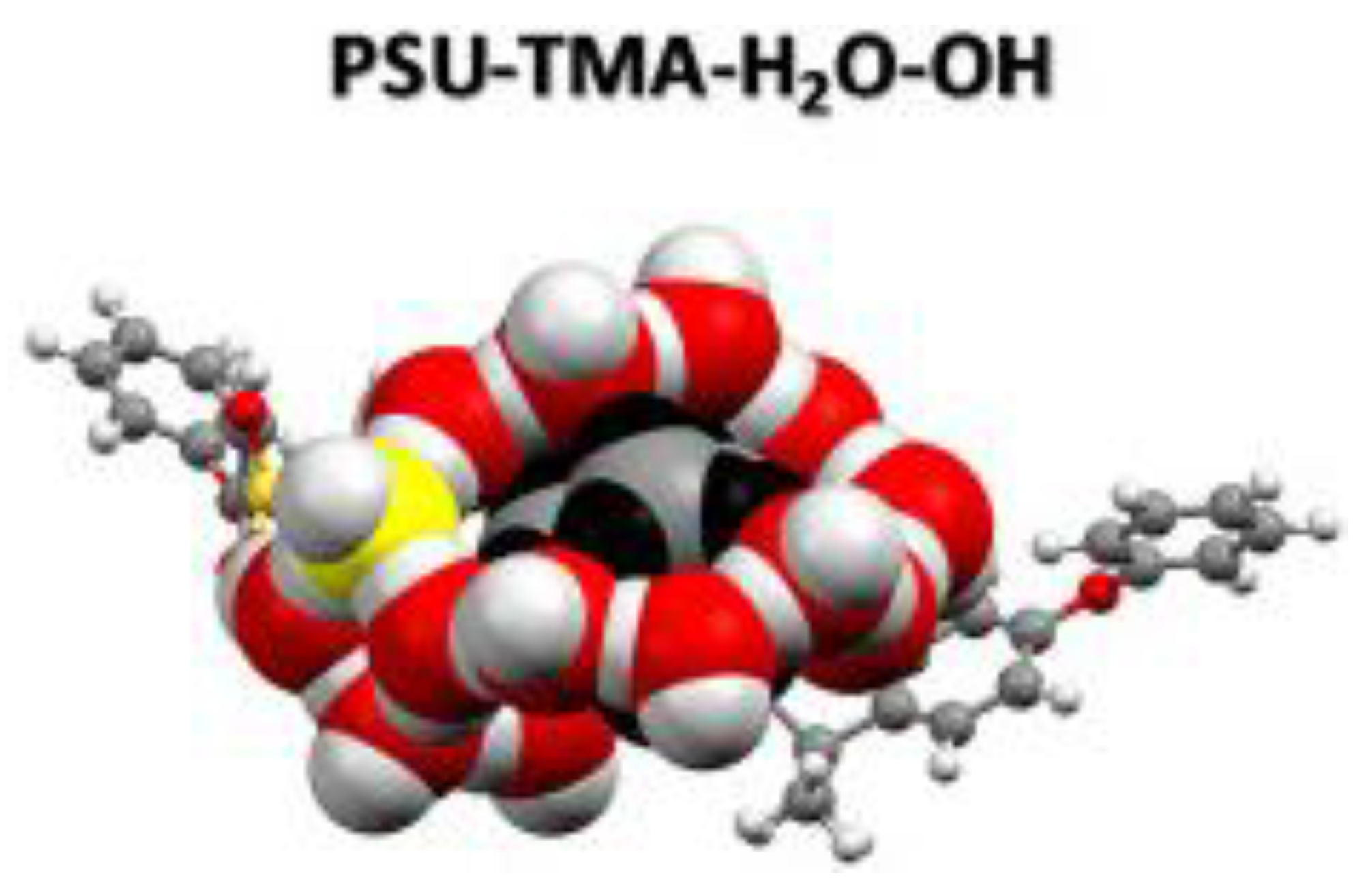
- Luque Di Salvo, J.; De Luca, G.; Cipollina, A.; Micale, G. Effect of ion exchange capacity and water uptake on hydroxide transport in PSU-TMA membranes: A DFT and molecular dynamics study. J. Memb. Sci. 2020, 599, 117837. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Zhang, J.; Lu, S.; Xiang, Y. Effect of side chain on the electrochemical performance of poly (ether ether ketone) based anion-exchange membrane: A molecular dynamics study. J. Memb. Sci. 2020, 605, 118105. [Google Scholar] [CrossRef]
- Park, C.H.; Kim, T.H.; Kim, D.J.; Nam, S.Y. Molecular dynamics simulation of the functional group effect in hydrocarbon anionic exchange membranes. Int. J. Hydrogen Energy 2017, 42, 20895–20903. [Google Scholar] [CrossRef]
- Dubey, V.; Maiti, A.; Daschakraborty, S. Predicting the solvation structure and vehicular diffusion of hydroxide ion in an anion exchange membrane using nonreactive molecular dynamics simulation. Chem. Phys. Lett. 2020, 755, 137802. [Google Scholar] [CrossRef]
- Dekel, D.R.; Willdorf, S.; Ash, U.; Amar, M.; Pusara, S.; Dhara, S.; Srebnik, S.; Diesendruck, C.E. The critical relation between chemical stability of cations and water in anion exchange membrane fuel cells environment. J. Power Sources 2018, 375, 351–360. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Wang, F.; Wang, Z.; Zhu, H. Poly(aryl piperidinium) anion exchange membranes with cationic extender sidechain for fuel cells. J. Memb. Sci. 2022, 653, 120448. [Google Scholar] [CrossRef]
- Chen, W.; Wu, X.; Li, T.; Yan, X.; Zhang, Y.; Wang, X.; Zhang, F.; Zhang, S.; He, G. Structural contribution of cationic groups to water sorption in anion exchange membranes: A combined DFT and MD simulation study. Chem. Eng. Sci. 2021, 244, 116791. [Google Scholar] [CrossRef]
- Chen, W.; Fu, Z.; Wu, X.; Li, T.; Yan, X.; Wang, X.; Cui, F.; Zhang, S.; He, G. Micro-phase separation promoted by electrostatic field in electrospinning of alkaline polymer electrolytes: DFT and MD simulations. Chem. Eng. Sci. 2022, 248, 117171. [Google Scholar] [CrossRef]
- Tanaka, M.; Koike, M.; Miyatake, K.; Watanabe, M. Anion conductive aromatic ionomers containing fluorenyl groups. Macromolecules 2010, 43, 2657–2659. [Google Scholar] [CrossRef]
- Takaba, H.; Hisabe, T.; Shimizu, T.; Alam, M.K. Molecular modeling of OH− transport in poly(arylene ether sulfone ketone)s containing quaternized ammonio-substituted fluorenyl groups as anion exchange membranes. J. Memb. Sci. 2017, 522, 237–244. [Google Scholar] [CrossRef]
- Grew, K.N.; Chiu, W.K.S. A Dusty Fluid Model for Predicting Hydroxyl Anion Conductivity in Alkaline Anion Exchange Membranes. J. Electrochem. Soc. 2010, 157, B327. [Google Scholar] [CrossRef]
- Wang, C.; Mo, B.; He, Z.; Xie, X.; Zhao, C.X.; Zhang, L.; Shao, Q.; Guo, X.; Wujcik, E.K.; Guo, Z. Hydroxide ions transportation in polynorbornene anion exchange membrane. Polymer 2018, 138, 363–368. [Google Scholar] [CrossRef]
- Zhang, W.; Van Duin, A.C.T. ReaxFF Reactive Molecular Dynamics Simulation of Functionalized Poly(phenylene oxide) Anion Exchange Membrane. J. Phys. Chem. C 2015, 119, 27727–27736. [Google Scholar] [CrossRef]
- Pan, J.; Zhu, H.; Cao, H.; Wang, B.; Zhao, J.; Sun, Z.; Yan, F. Flexible cationic side chains for enhancing the hydroxide ion conductivity of olefinic-type copolymer-based anion exchange membranes: An experimental and theoretical study. J. Memb. Sci. 2021, 620, 118794. [Google Scholar] [CrossRef]
- Wang, J.; Han, Y.; Xu, Z.; Yang, X.; Ramakrishna, S.; Liu, Y. Dissipative Particle Dynamics Simulation: A Review on Investigating Mesoscale Properties of Polymer Systems. Macromol. Mater. Eng. 2021, 306, 2000724. [Google Scholar] [CrossRef]
- Zhu, Z.; Luo, X.; Paddison, S.J. DPD simulations of anion exchange membranes functionalized with various cationic groups and associated anions. Solid State Ionics 2019, 340, 115011. [Google Scholar] [CrossRef]
- Lee, M.T. Designing Anion Exchange Membranes with Enhanced Hydroxide Ion Conductivity by Mesoscale Simulations. J. Phys. Chem. C 2020, 124, 4470–4482. [Google Scholar] [CrossRef]
- Lee, M.T. Exploring Side-Chain Designs for Enhanced Ion Conductivity of Anion-Exchange Membranes by Mesoscale Simulations. J. Phys. Chem. C 2019, 123, 10802–10815. [Google Scholar] [CrossRef]
- Marino, M.G.; Kreuer, K.D. Alkaline Stability of Quaternary Ammonium Cations for Alkaline Fuel Cell Membranes and Ionic Liquids. ChemSusChem 2015, 8, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Edson, J.B.; Macomber, C.S.; Pivovar, B.S.; Boncella, J.M. Hydroxide based decomposition pathways of alkyltrimethylammonium cations. J. Memb. Sci. 2012, 399–400, 49–59. [Google Scholar] [CrossRef]
- Luo, X.; Liu, H.; Bae, C.; Tuckerman, M.E.; Hickner, M.A.; Paddison, S.J. Mesoscale Simulations of Quaternary Ammonium-Tethered Triblock Copolymers: Effects of the Degree of Functionalization and Styrene Content. J. Phys. Chem. C 2020, 124, 16315–16323. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, B.; Liu, Z.; Zhang, G.; Xie, B.; Tongsh, C.; Xi, F.; Chen, W.; Jiao, K. Numerical investigation of design and operating parameter effects on permeability-differentiated alkaline fuel cell with metal foam flow field. Appl. Therm. Eng. 2022, 207, 118183. [Google Scholar] [CrossRef]
- Satjaritanun, P.; O’Brien, M.; Kulkarni, D.; Shimpalee, S.; Capuano, C.; Ayers, K.E.; Danilovic, N.; Parkinson, D.Y.; Zenyuk, I.V. Observation of Preferential Pathways for Oxygen Removal through Porous Transport Layers of Polymer Electrolyte Water Electrolyzers. iScience 2022, 23, 101783. [Google Scholar] [CrossRef]
- Jäger, M.O.J.; Ranawat, Y.S.; Canova, F.F.; Morooka, E.V.; Foster, A.S. Efficient Machine-Learning-Aided Screening of Hydrogen Adsorption on Bimetallic Nanoclusters. ACS Comb. Sci. 2020, 22, 768–781. [Google Scholar] [CrossRef]
- Wu, L.; Guo, T.; Li, T. Machine learning-accelerated prediction of overpotential of oxygen evolution reaction of single-atom catalysts. iScience 2021, 24, 102398. [Google Scholar] [CrossRef] [PubMed]
- Ran, N.; Sun, B.; Qiu, W.; Song, E.; Chen, T.; Liu, J. Identifying Metallic Transition-Metal Dichalcogenides for Hydrogen Evolution through Multilevel High-Throughput Calculations and Machine Learning. J. Phys. Chem. Lett. 2021, 12, 2102–2111. [Google Scholar] [CrossRef]
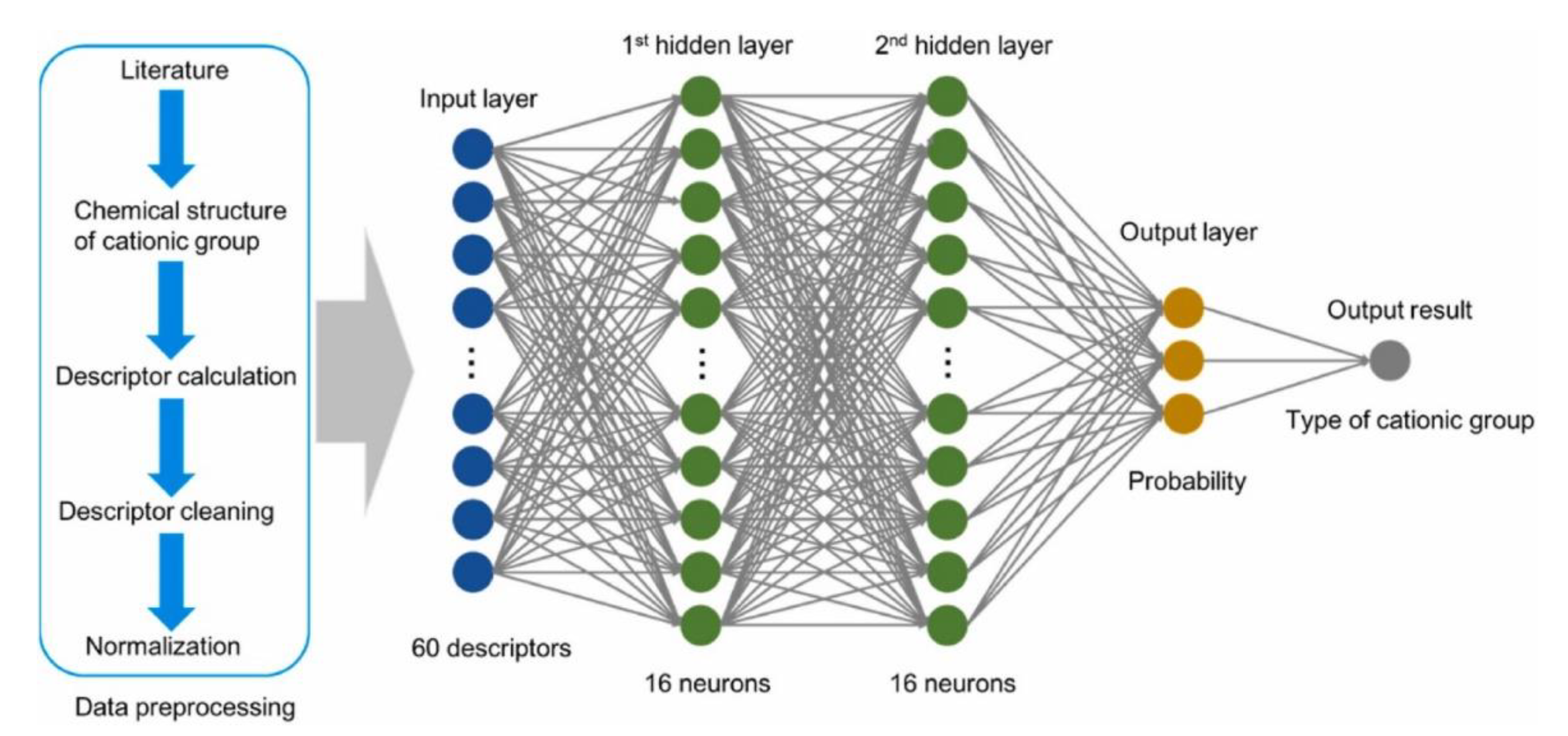
- Zhai, F.-H.; Zhan, Q.-Q.; Yang, Y.-F.; Ye, N.-Y.; Wan, R.-Y.; Wang, J.; Chen, S.; He, R.-H. A deep learning protocol for analyzing and predicting ionic conductivity of anion exchange membranes. J. Memb. Sci. 2022, 642, 119983. [Google Scholar] [CrossRef]
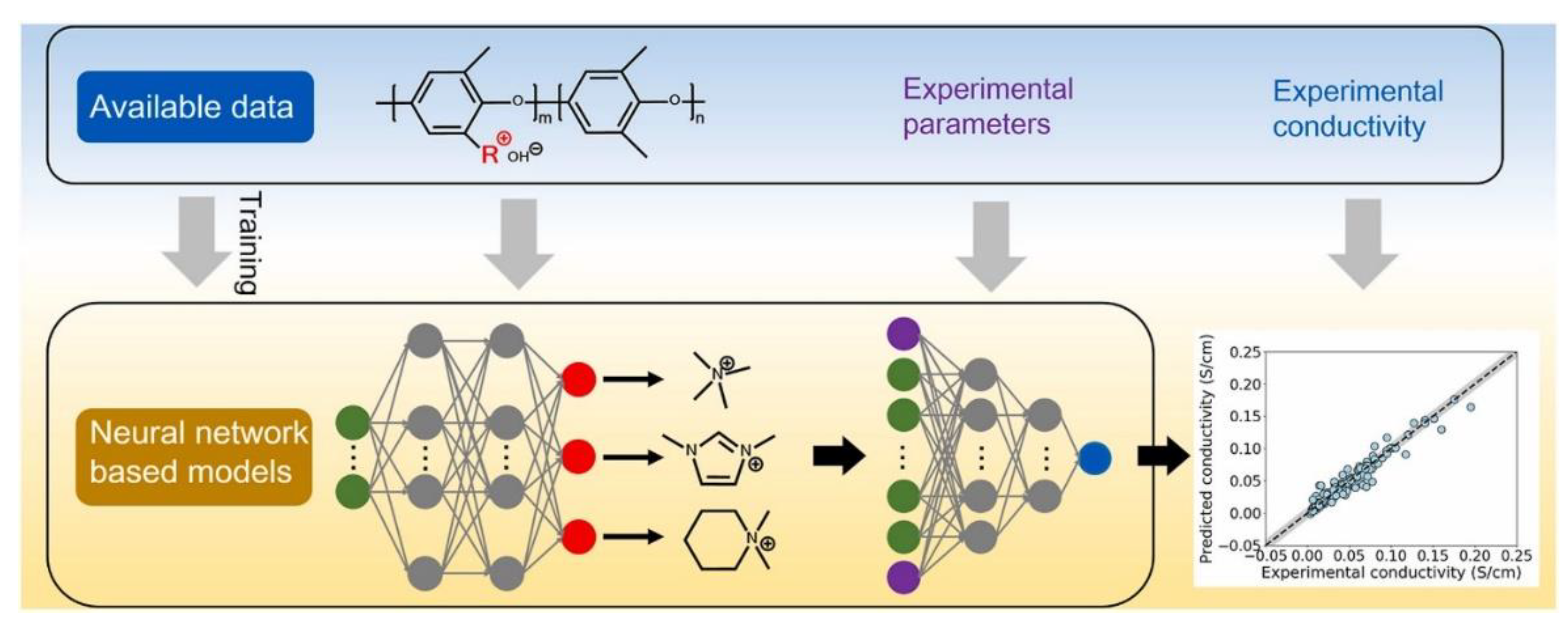
- Zou, X.; Pan, J.; Sun, Z.; Wang, B.; Jin, Z.; Xu, G.; Yan, F. Machine learning analysis and prediction models of alkaline anion exchange membranes for fuel cells. Energy Environ. Sci. 2021, 14, 3965–3975. [Google Scholar] [CrossRef]
- Maurya, S.; Dumont, J.H.; Villarrubia, C.N.; Matanovic, I.; Li, D.; Kim, Y.S.; Noh, S.; Han, J.; Bae, C.; Miller, H.A.; et al. Surface Adsorption Affects the Performance of Alkaline Anion-Exchange Membrane Fuel Cells. ACS Catal. 2018, 8, 9429–9439. [Google Scholar] [CrossRef]
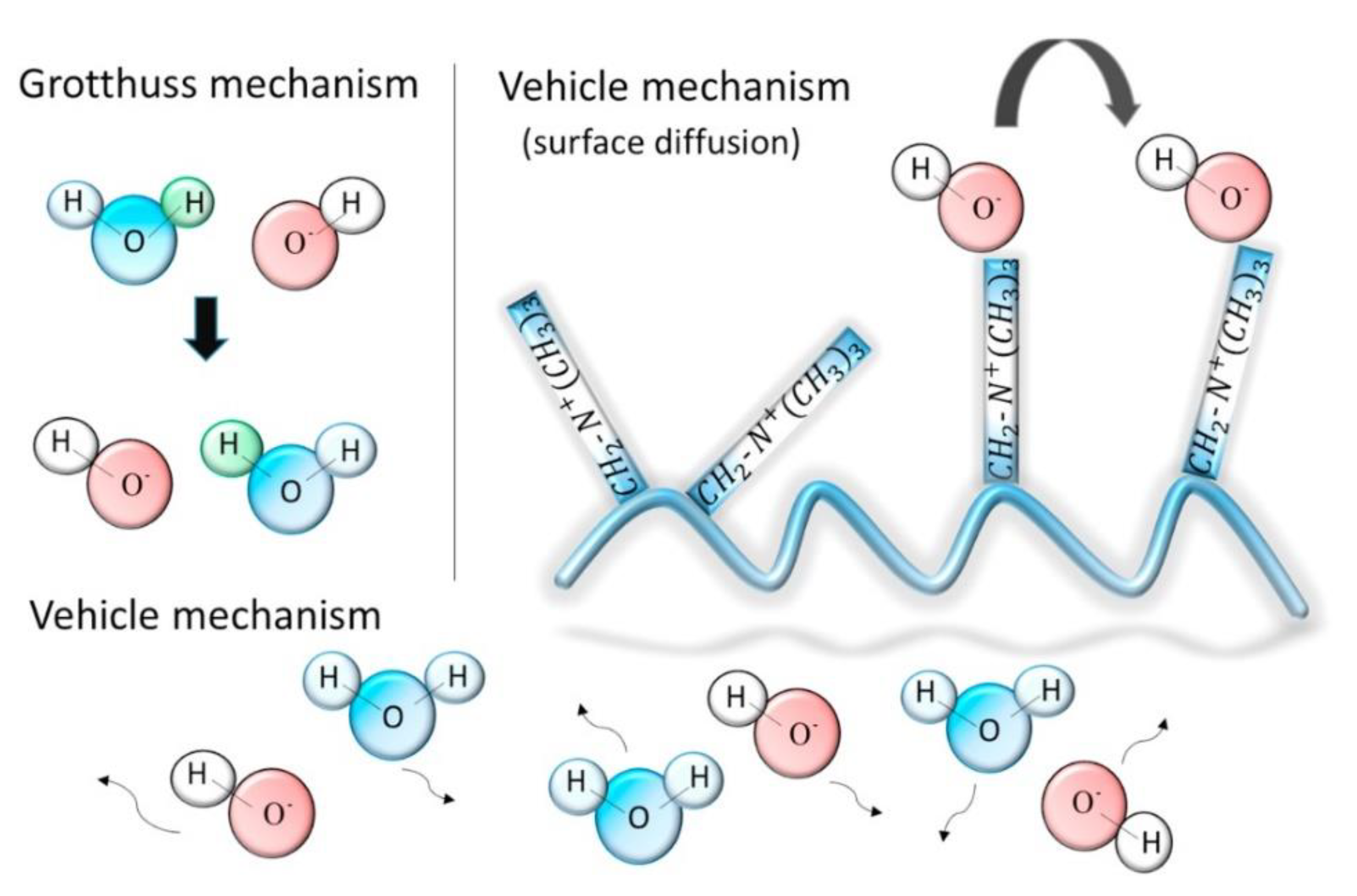
- Agmon, N. Mechanism of hydroxide mobility. Chem. Phys. Lett. 2000, 319, 247–252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scale | Tools | Reference | Phenomena |
|---|---|---|---|
| Density functional theory (DFT) | DFT | [13,22,23,24,26,27,28] | Chemical stability, alkaline stability, polymers interactions, OH– adsorption and diffusion, OH– transport, nucleophilic attack, HOMO and LUMO energy |
| Molecular dynamics (MD) | Coarse-grained MD, first-principles MD, ab initio MD, classical MD, force fields, ReaxFF (reactive MD) | [8,29,30,31,32,33,34,35,36,37,38,39,40,41,42] | OH– mobility and transport mechanism, cation head groups, polymer backbones, ionic conductivity, hydration (water uptake), ion exchange capacity (IEC) |
| Mesoscale simulations | Coarse-grained MD, dissipative particle dynamics | [19,43,44,45,46,47,48,49] | Alkaline stability, water uptake, IEC, hydrated morphology and microstructure, ionic conductivity |
| Machine learning | [50,51,52,53,54,55,56] | Alkaline stability, polymer configurations, catalysts, OH– transport |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouma, C.N.M.; Obodo, K.O.; Bessarabov, D. Computational Approaches to Alkaline Anion-Exchange Membranes for Fuel Cell Applications. Membranes 2022, 12, 1051. https://doi.org/10.3390/membranes12111051
Ouma CNM, Obodo KO, Bessarabov D. Computational Approaches to Alkaline Anion-Exchange Membranes for Fuel Cell Applications. Membranes. 2022; 12(11):1051. https://doi.org/10.3390/membranes12111051
Chicago/Turabian StyleOuma, Cecil Naphtaly Moro, Kingsley Onyebuchi Obodo, and Dmitri Bessarabov. 2022. "Computational Approaches to Alkaline Anion-Exchange Membranes for Fuel Cell Applications" Membranes 12, no. 11: 1051. https://doi.org/10.3390/membranes12111051
APA StyleOuma, C. N. M., Obodo, K. O., & Bessarabov, D. (2022). Computational Approaches to Alkaline Anion-Exchange Membranes for Fuel Cell Applications. Membranes, 12(11), 1051. https://doi.org/10.3390/membranes12111051