
The Optimization of Dispersion and Application Techniques for Nanocarbon-Doped Mixed Matrix Gas Separation Membranes


 ,
, 
Abstract
:1. Introduction
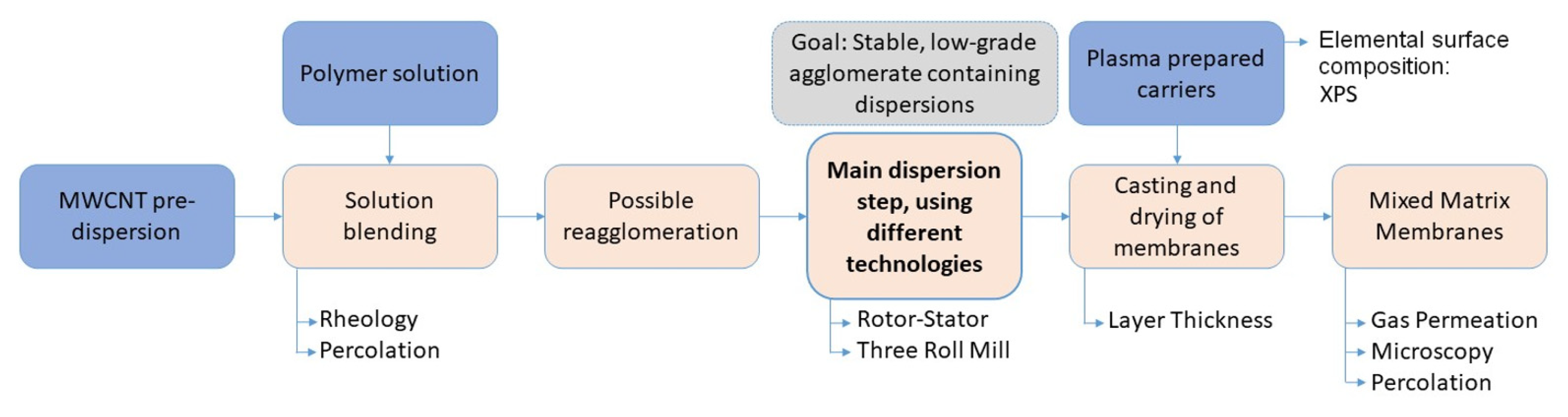
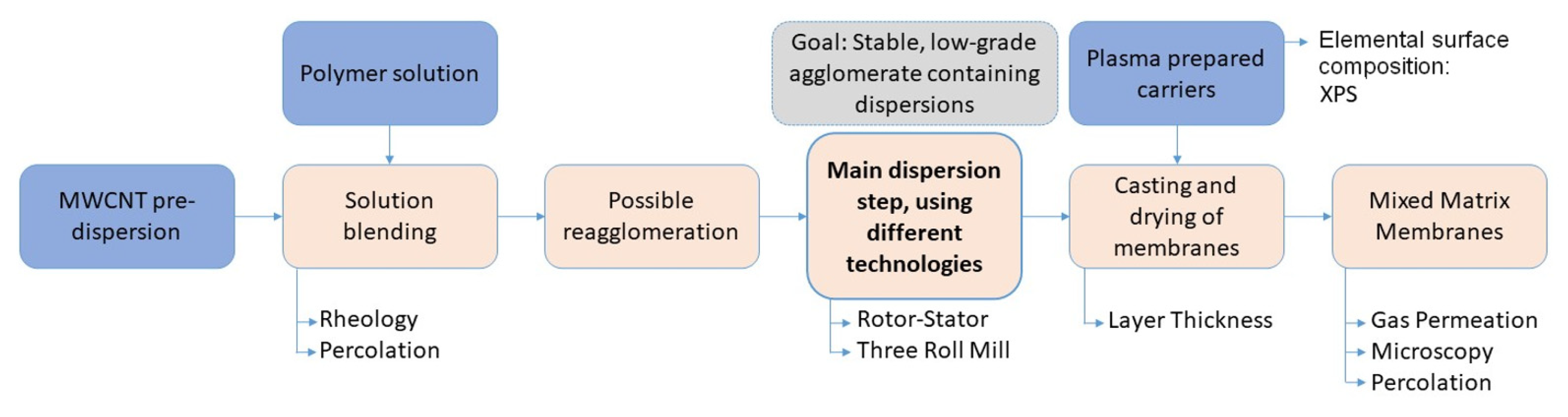
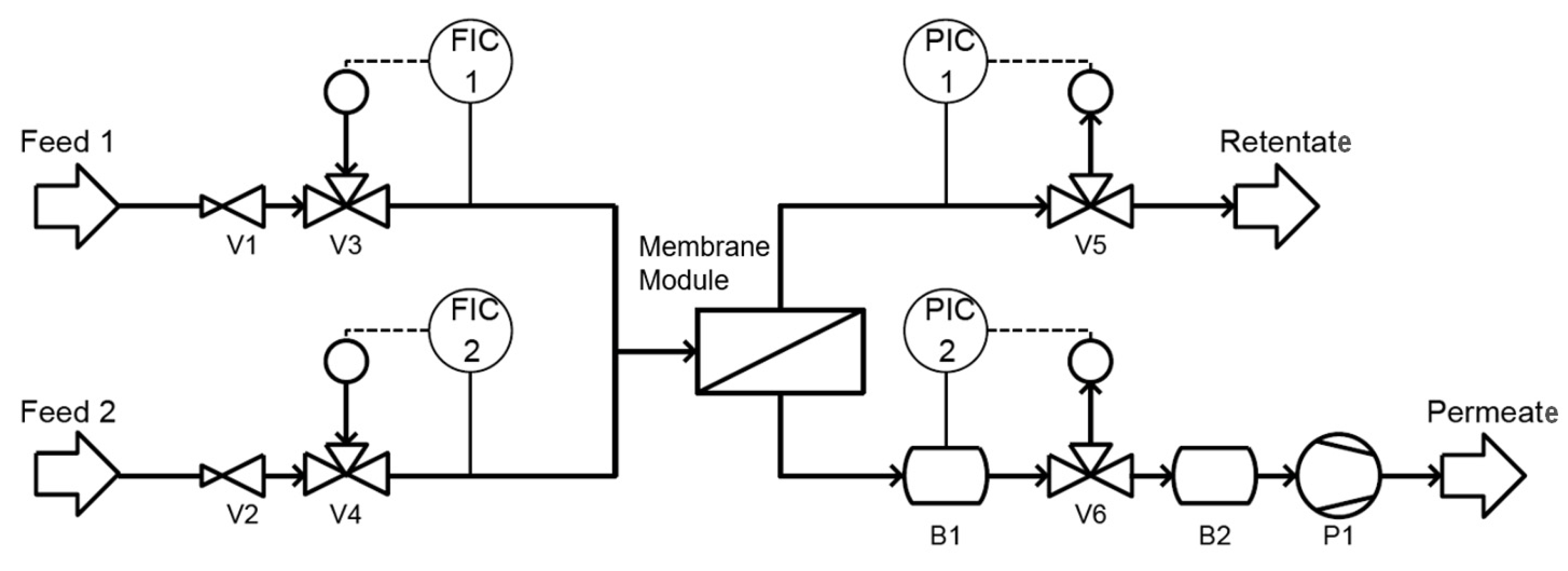
2. Materials and Methods
2.1. Solution Blending
2.2. Main Dispersion Step
2.3. Substrate (Carrier) Preparation
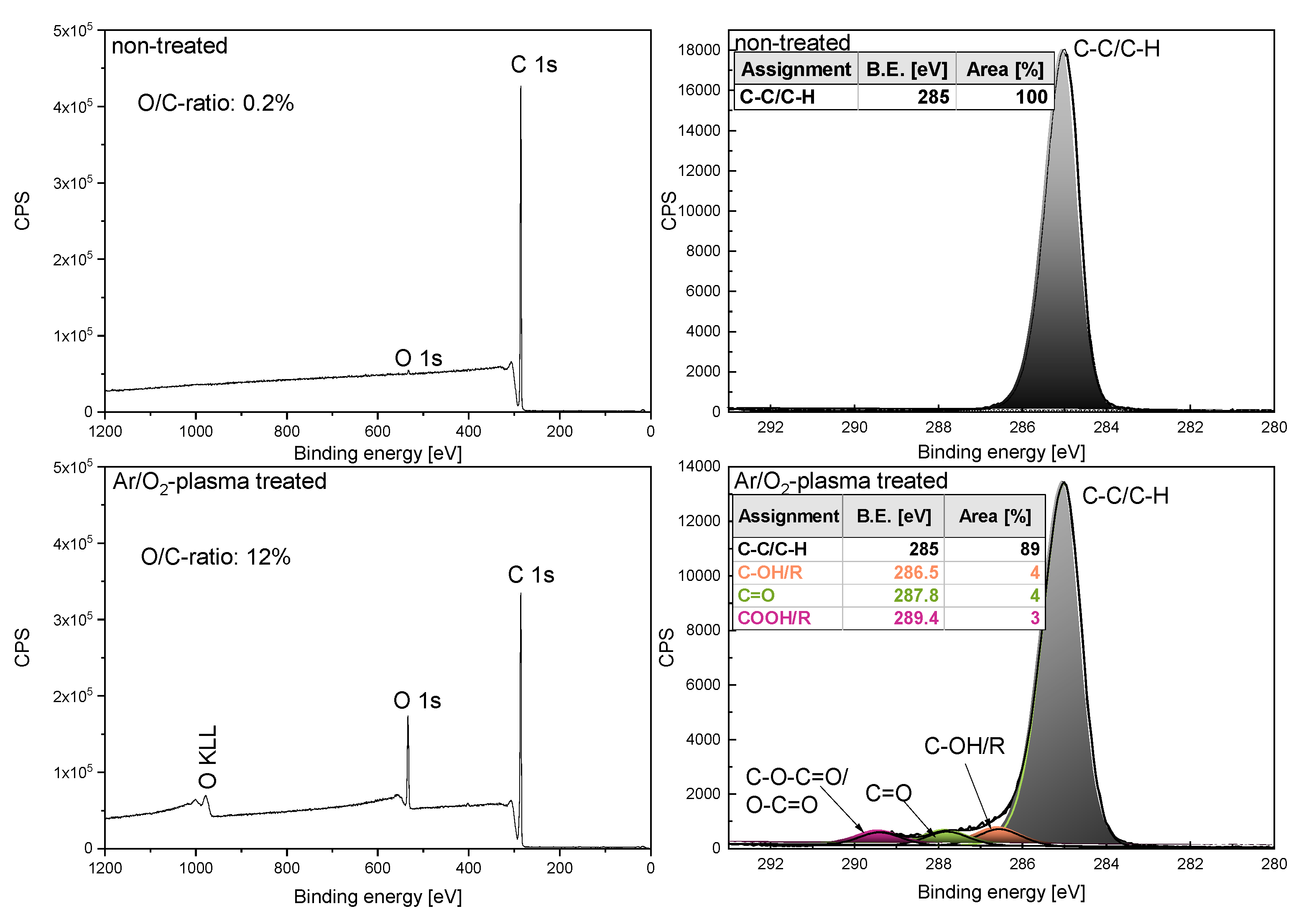
2.4. Plasma Treatment
2.5. X-ray Photoelectron Spectroscopy (XPS)
2.6. Membrane Preparation
2.7. The Analysis of Membrane Properties/Permeation Performance
2.8. Percolation Study
3. Results and Discussion
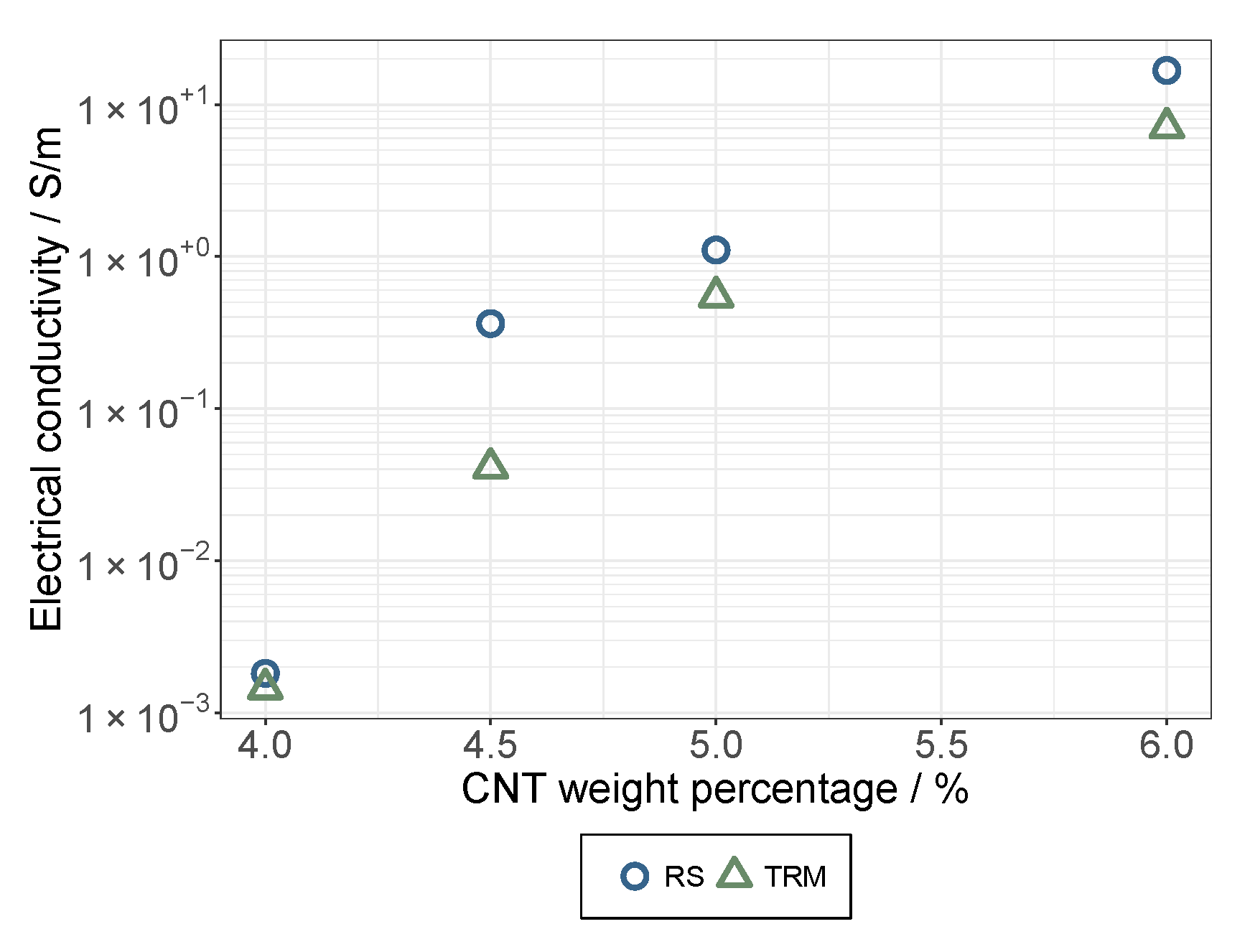
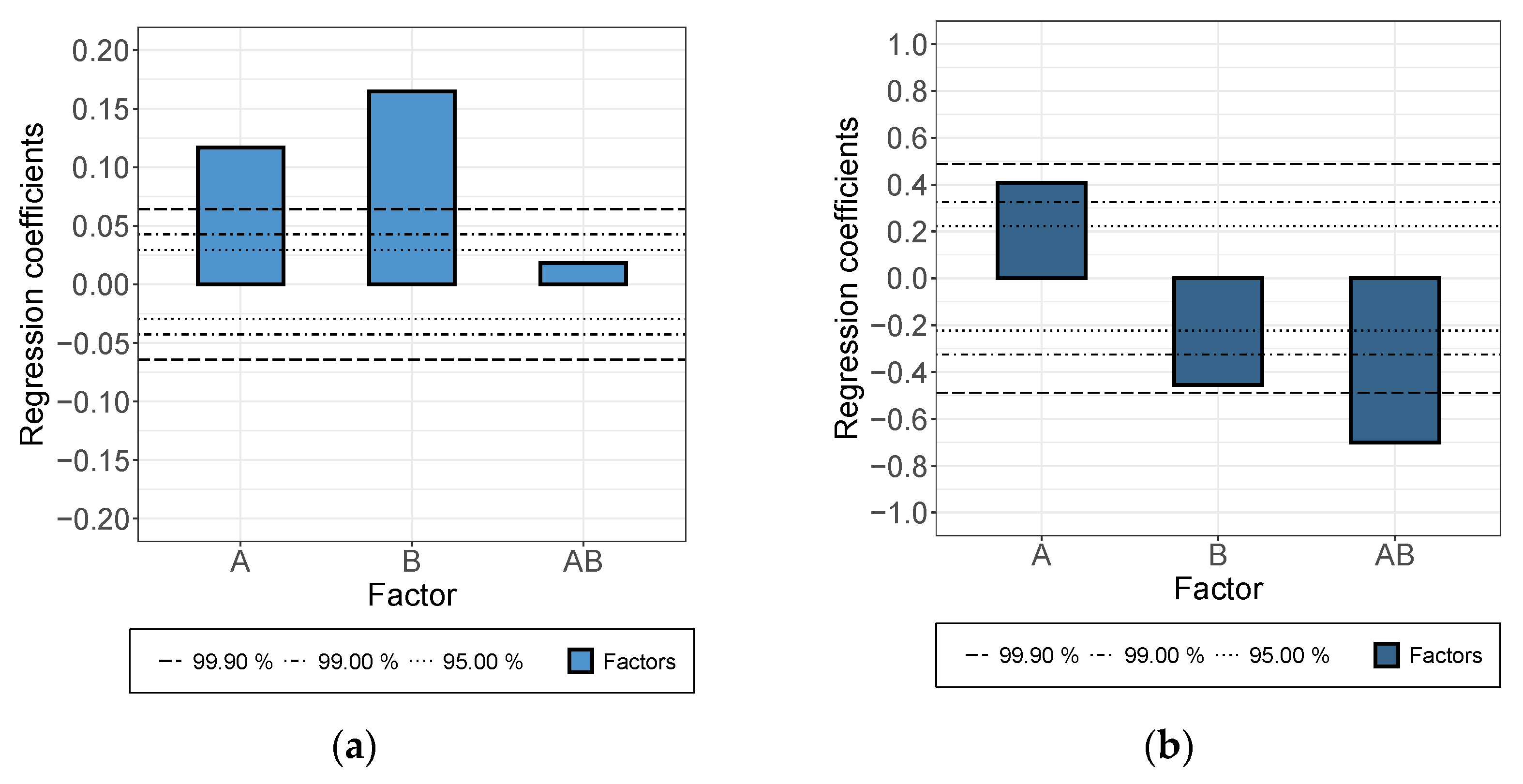
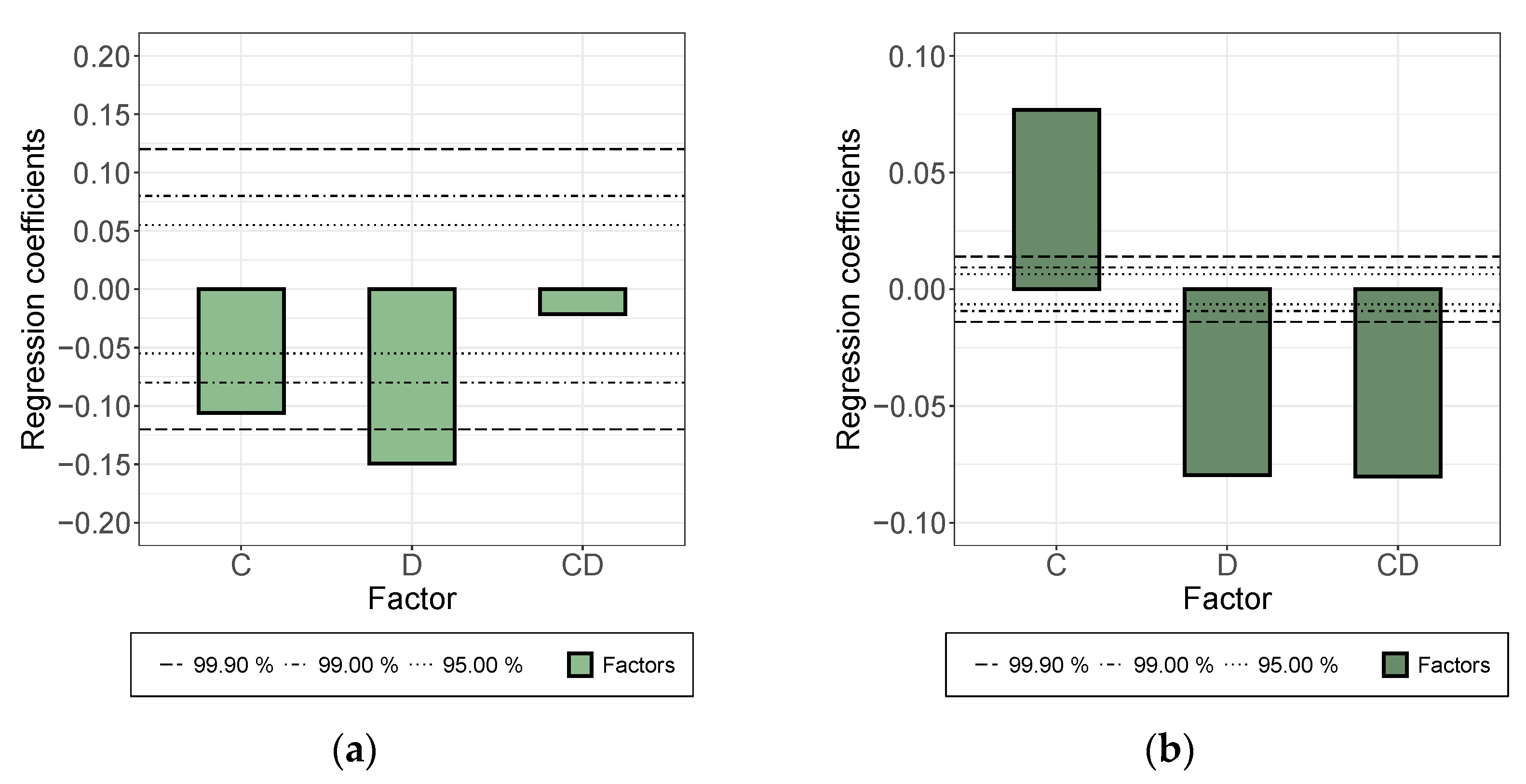
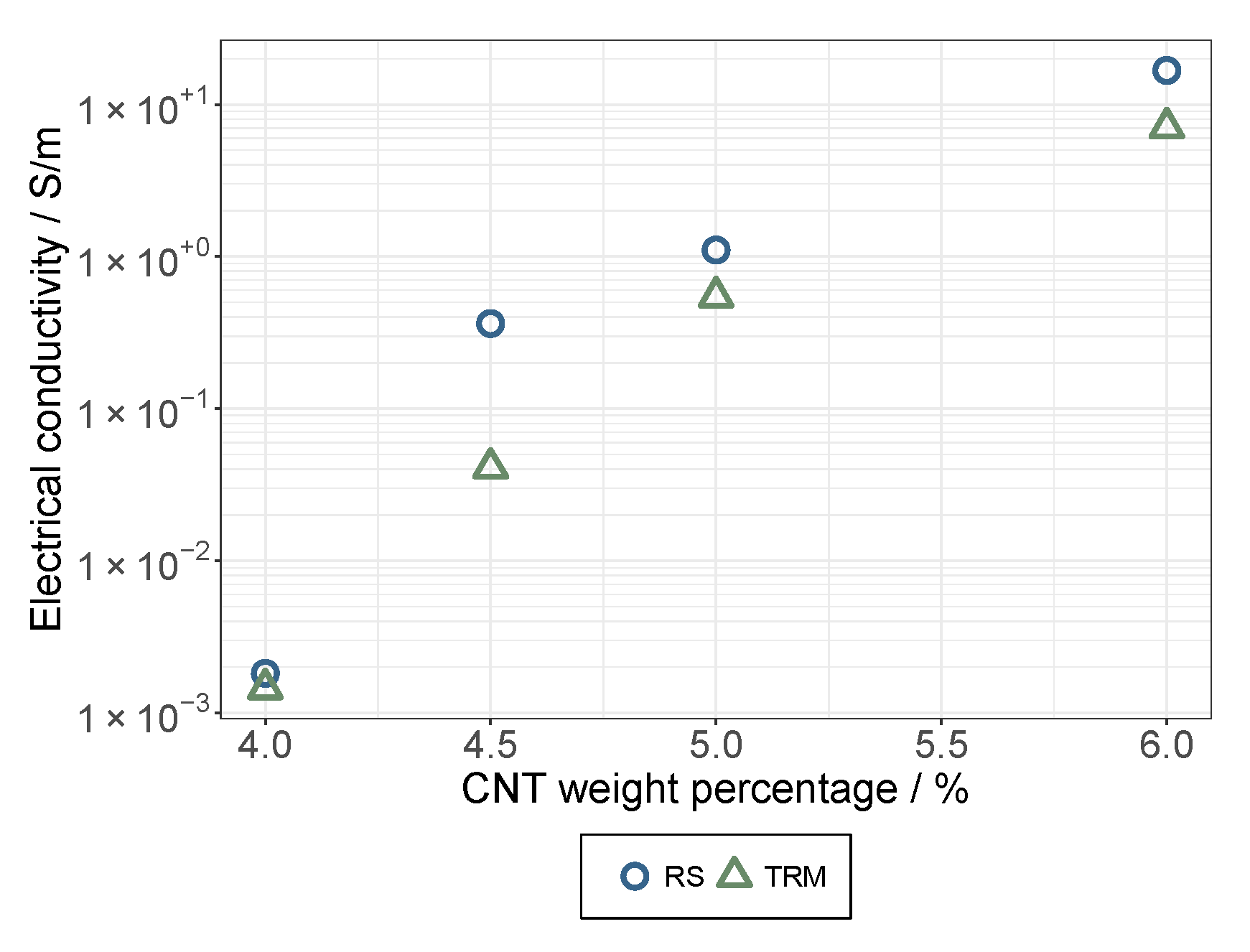
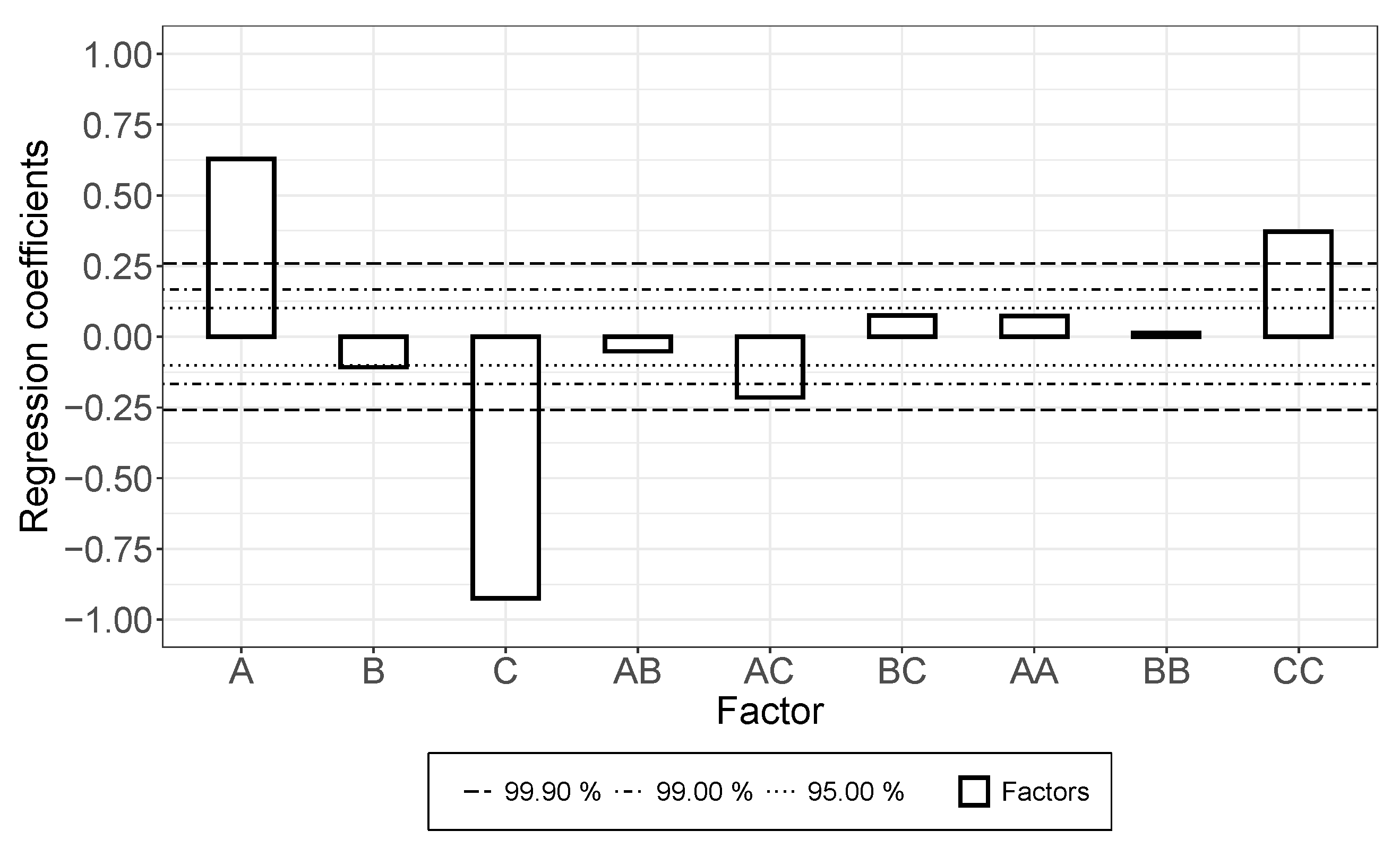
3.1. The Dispersion/Percolation Study
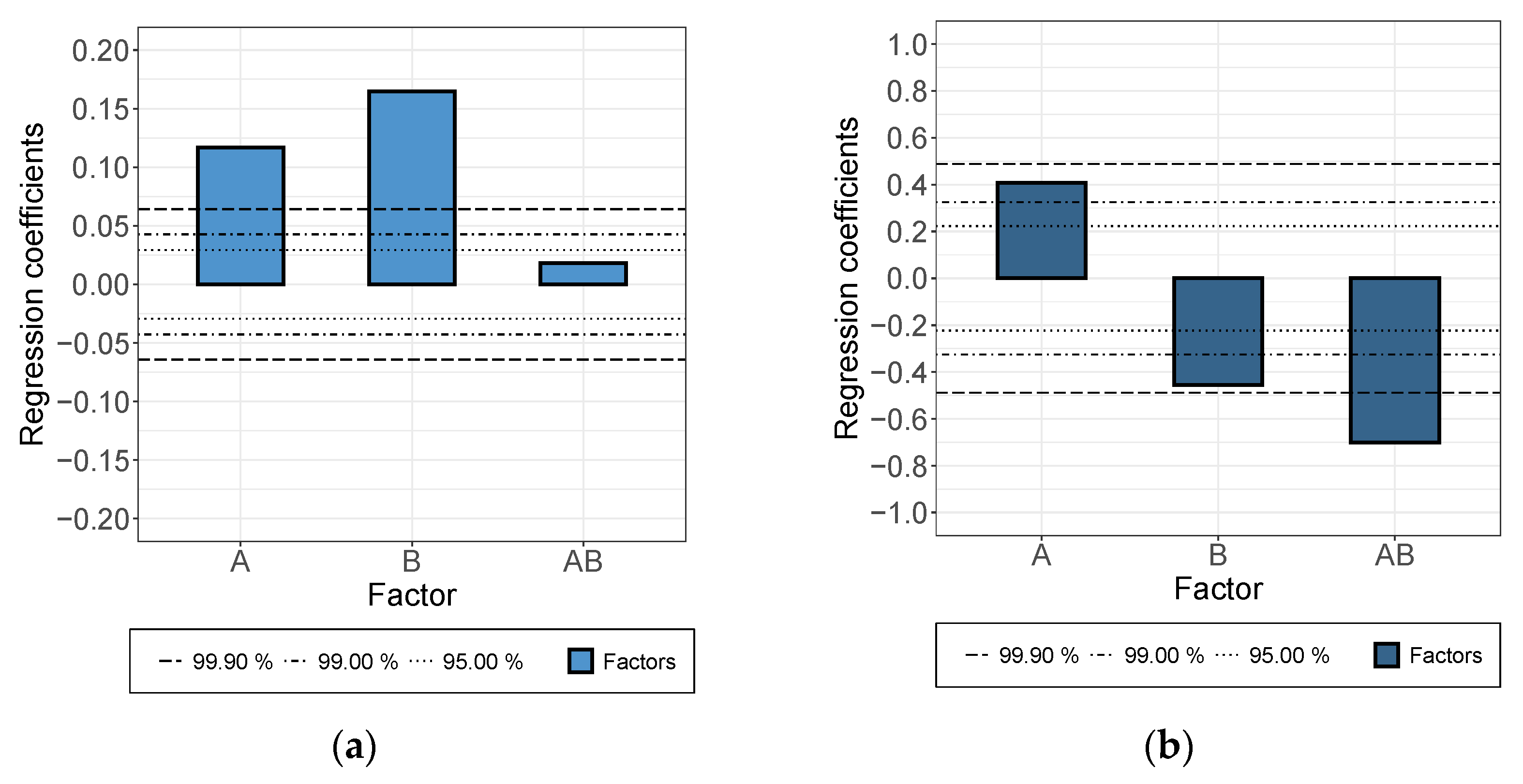
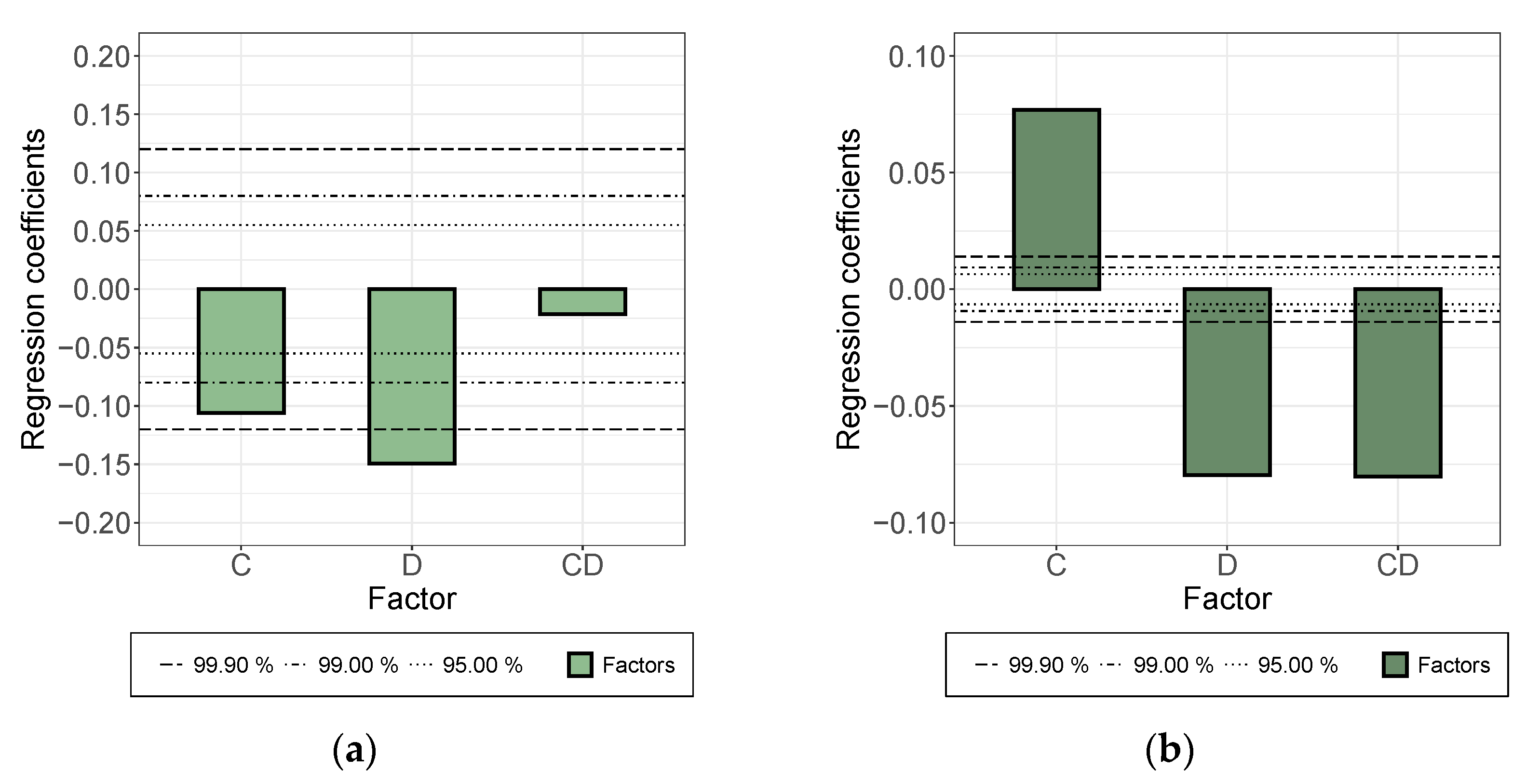
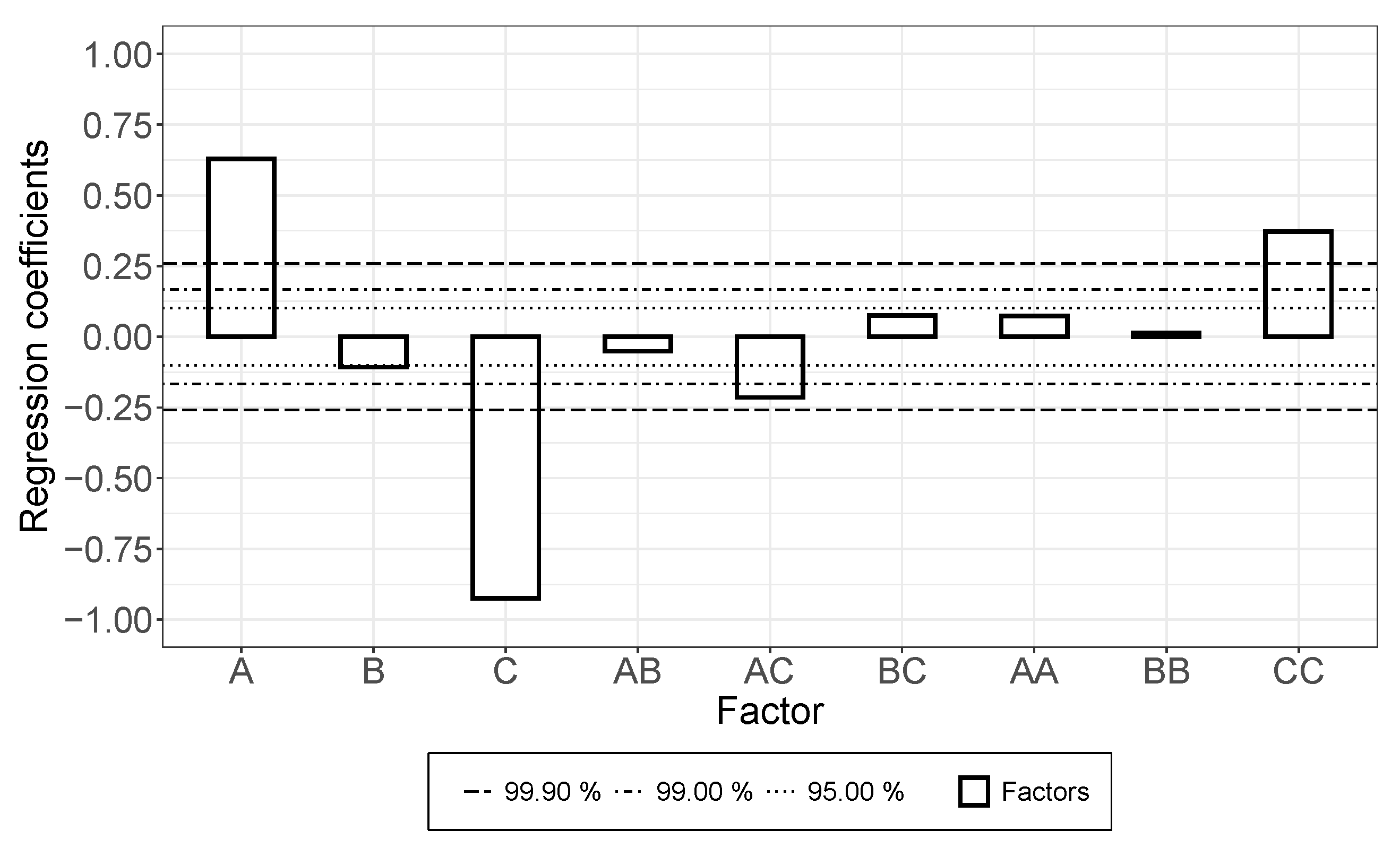
- While a longer treatment time in the rotor-stator increased the conductivity in the dispersion, it decreased in the applied layer. The comminution of the agglomerates (positive) was evidently overcompensated here by a shortening of the MWCNTs and this became effective during the rearrangement during drying. This was also indicated by the significant interaction between parameters A and B.
- An increase in roller speed in the TRM worsened the conductivity in the suspension, but increased it significantly in the applied layer. The reason for this opposite trend has not yet been clarified. The starting point could be the structural viscosity effects of the suspension in which the increased roller speed reduced the viscosity due to strong shear, so that there was less agglomerate shear. As they dry, the MWCNTs can rearrange themselves and, due to the smaller volume, can result in a higher probability of the connection between MWCNTs.
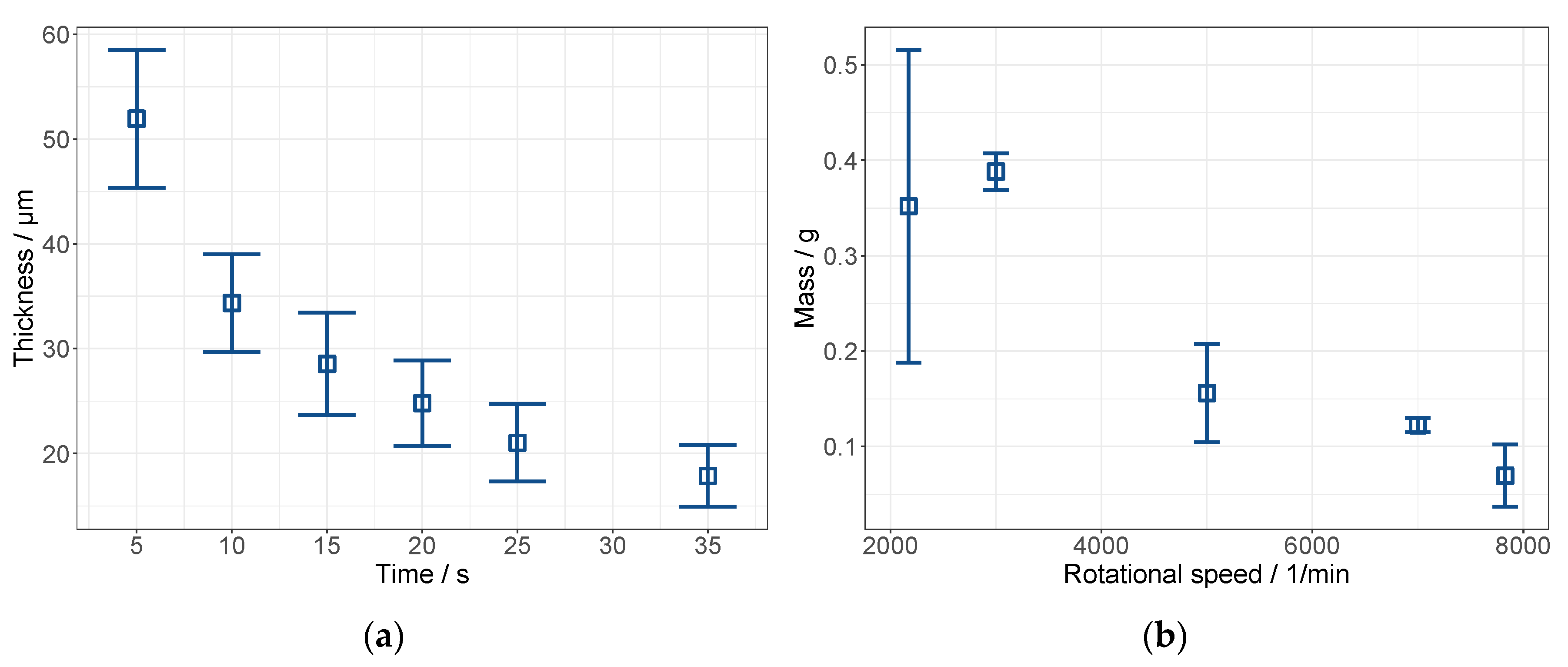
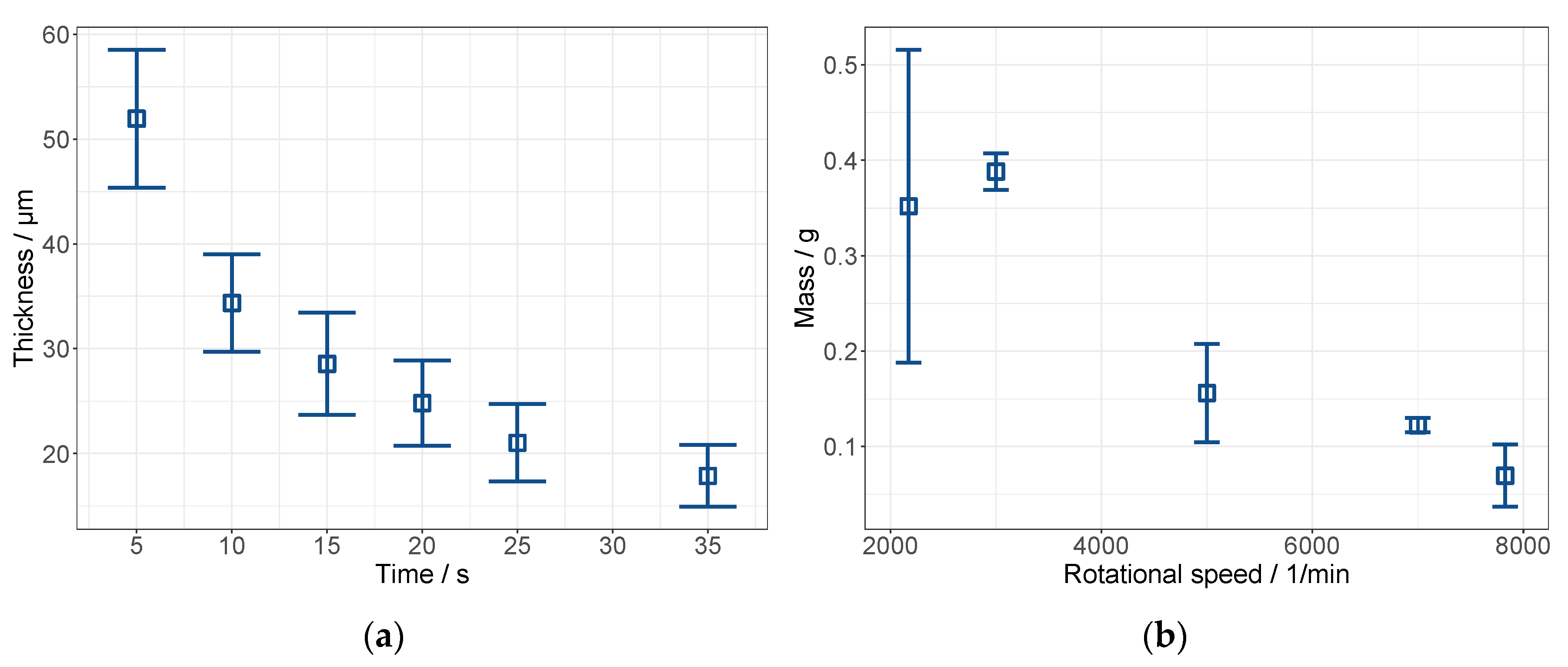
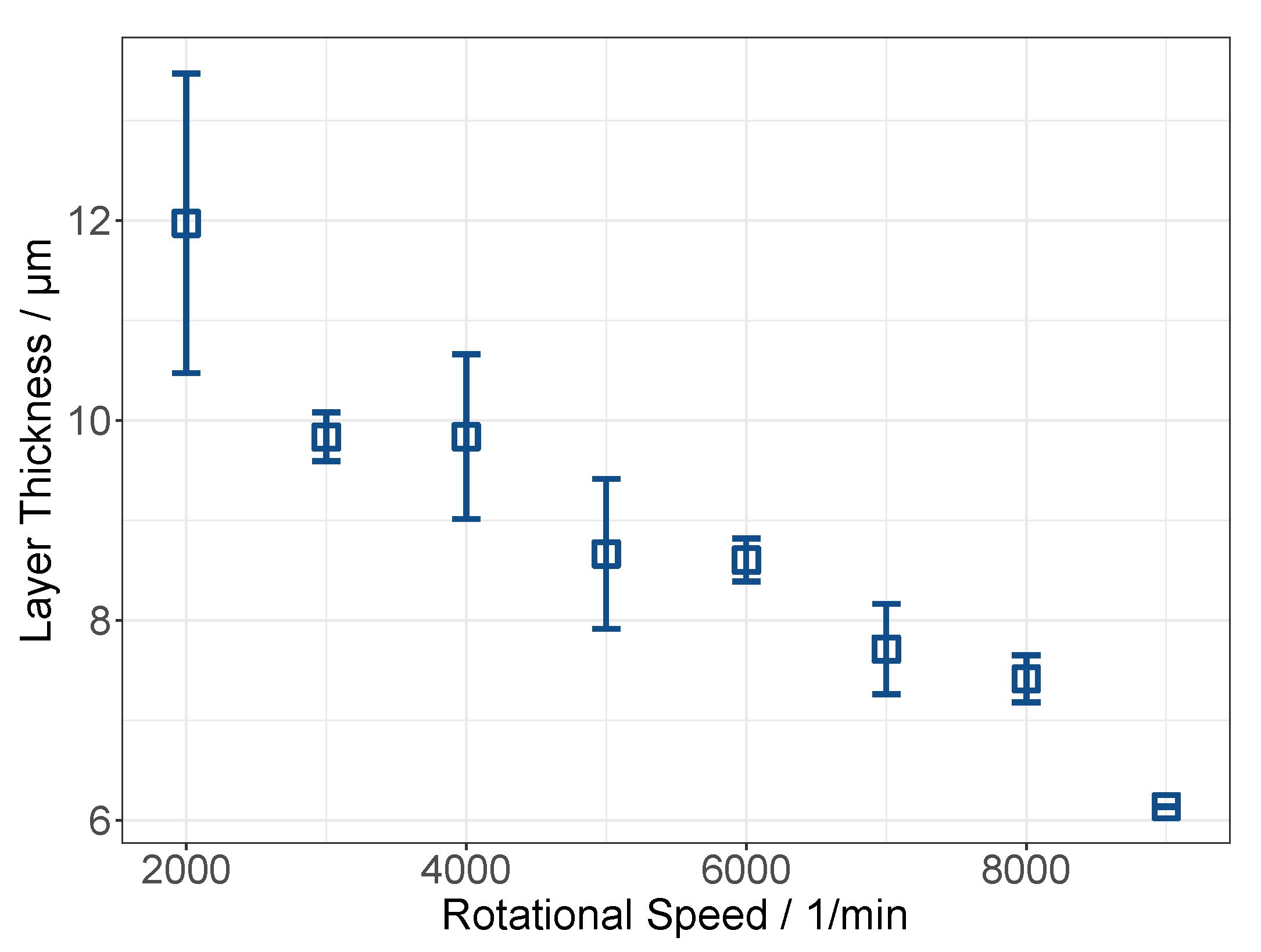
3.2. Pre Tests on Spin Coating: The Establishment of a Novel Application Method for MMMs
3.2.1. Cellulose Acetate System
3.2.2. Polyimide System
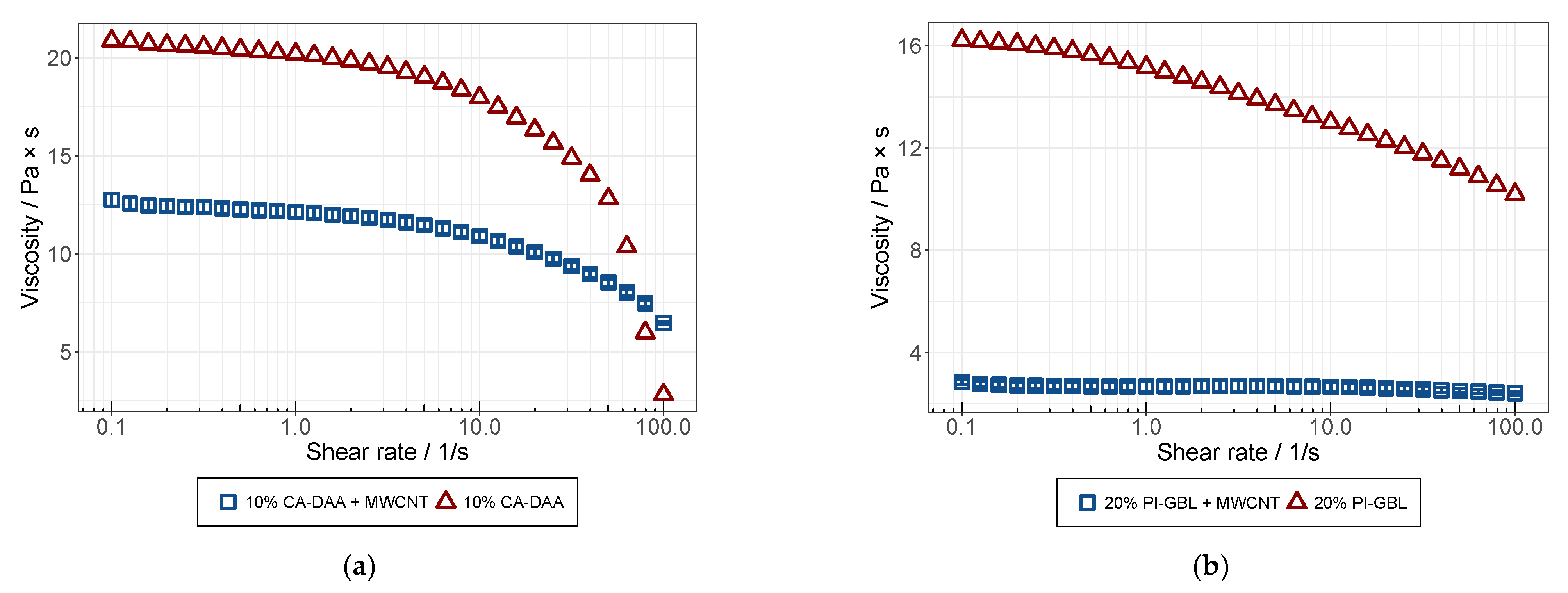
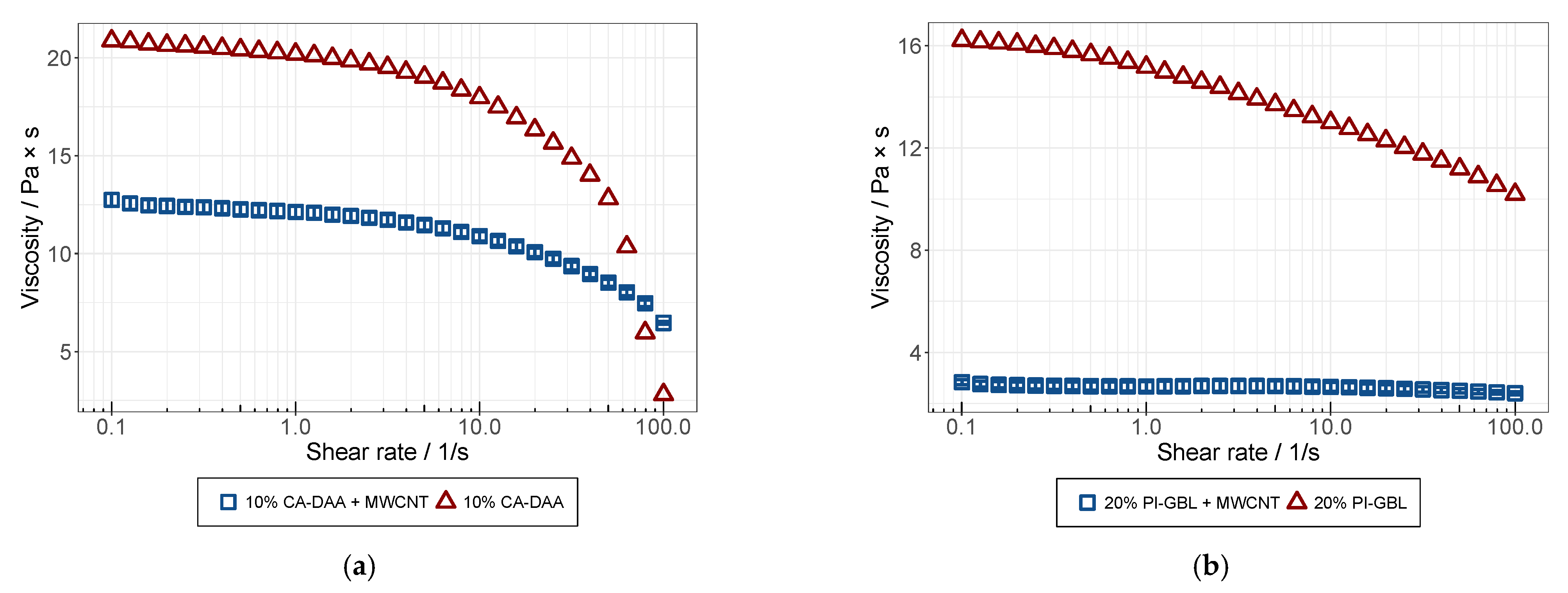
3.3. Rheological Analysis
3.4. Membrane Preparation
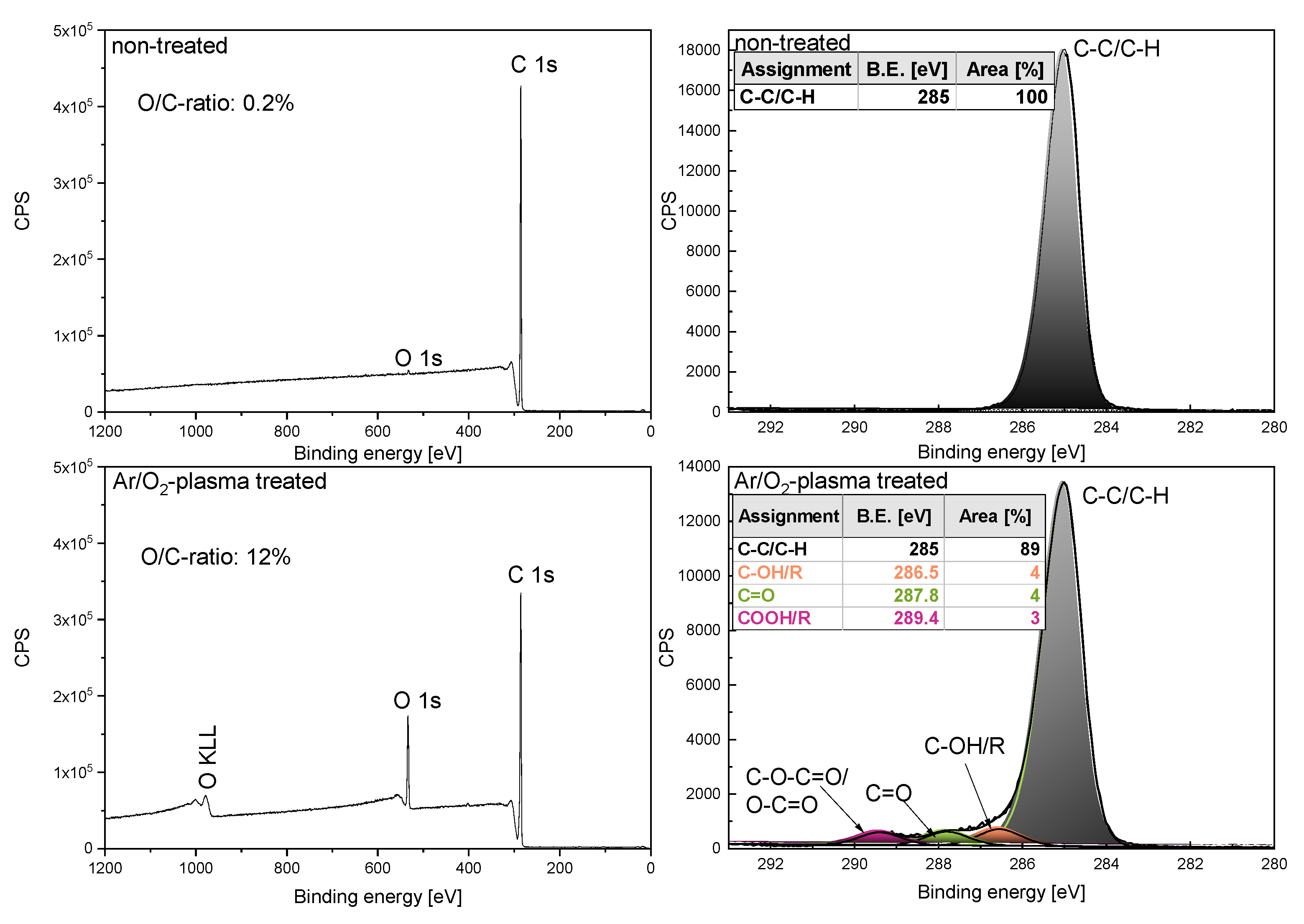
3.5. Plasma Treatment
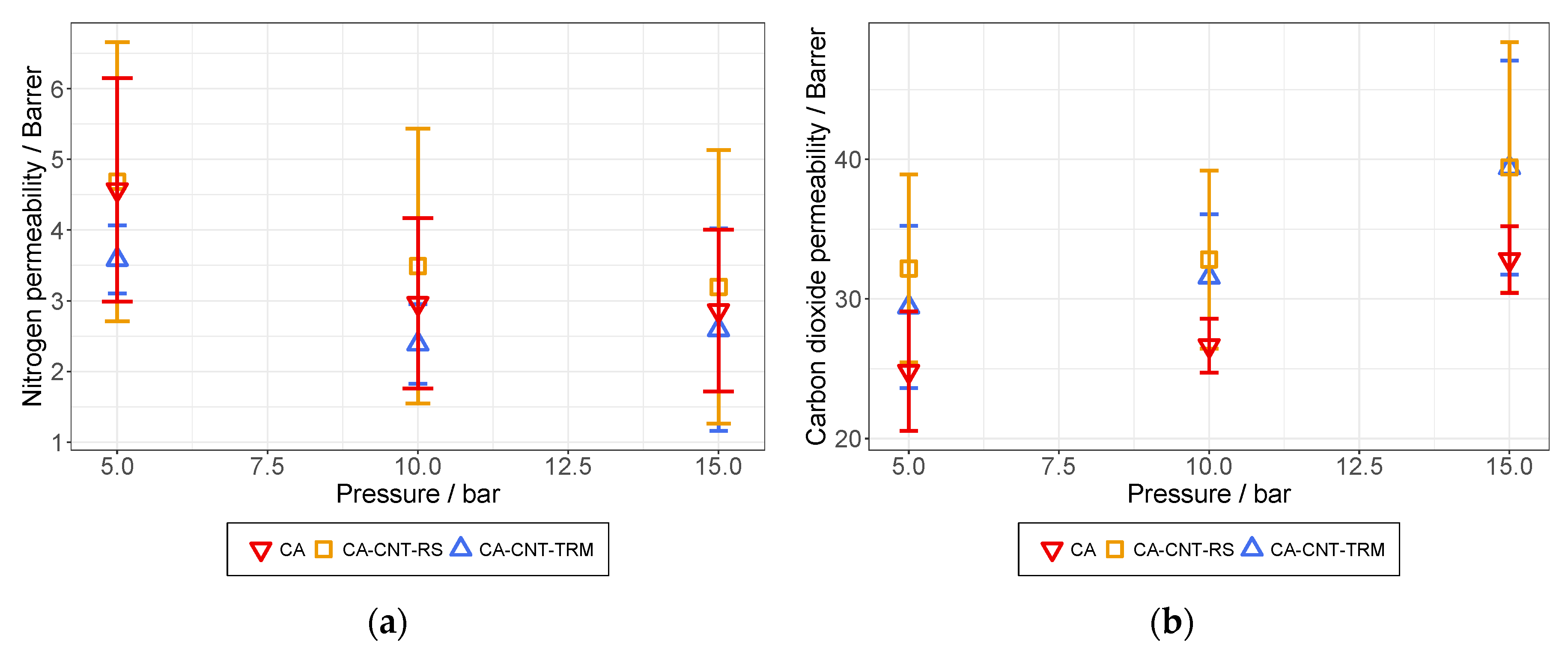
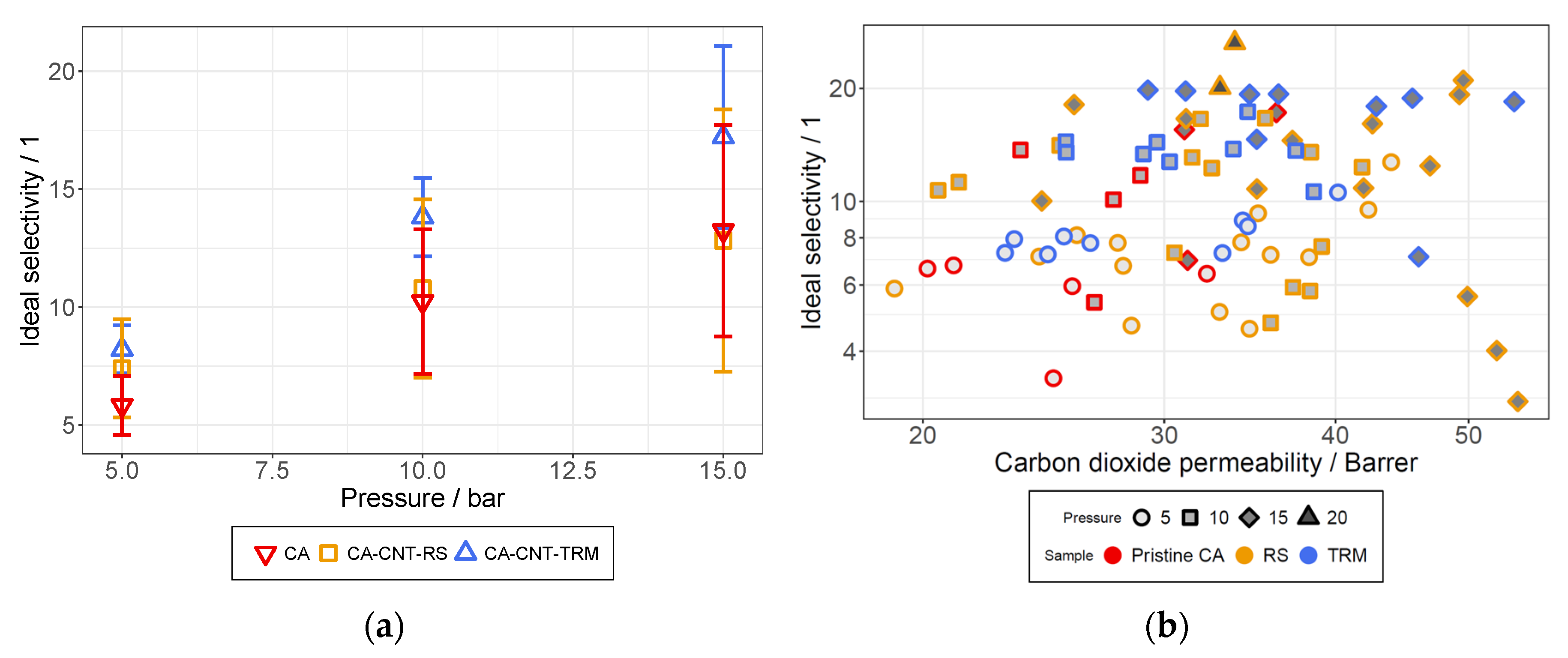
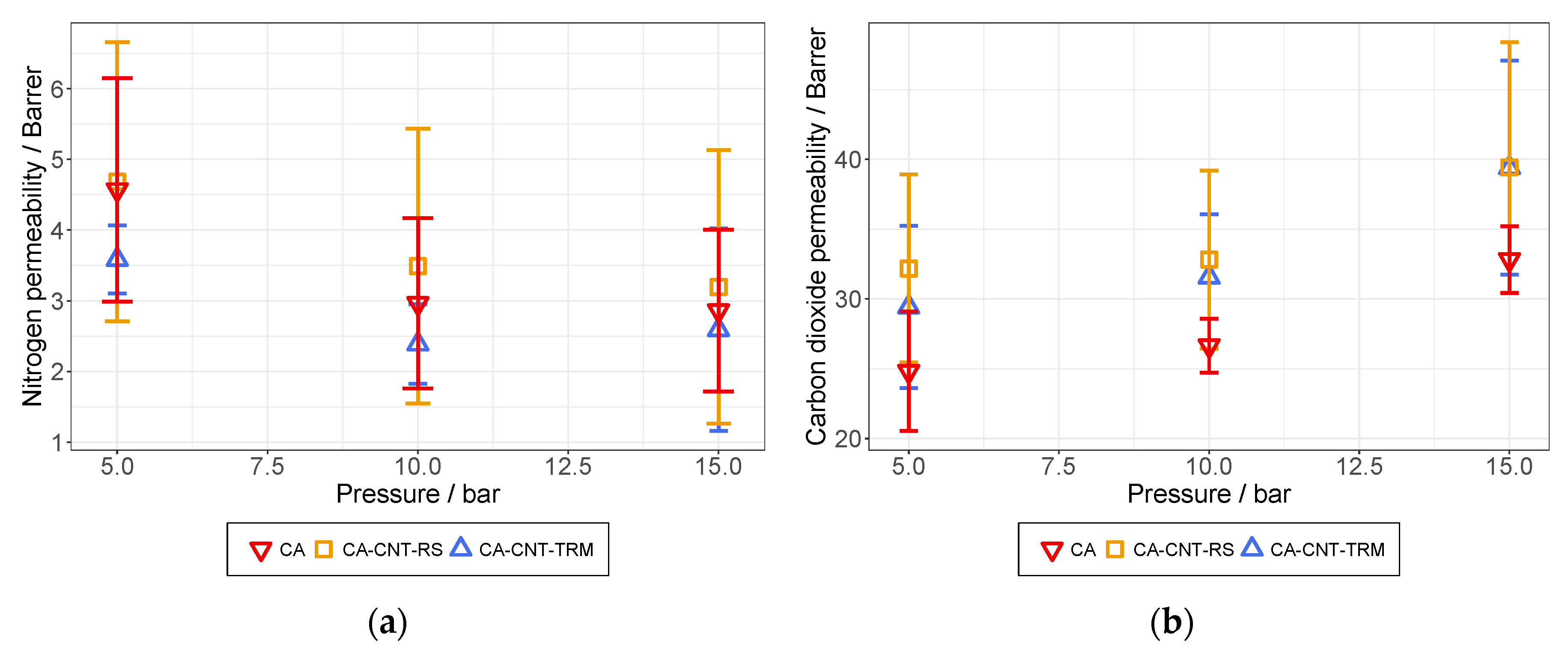
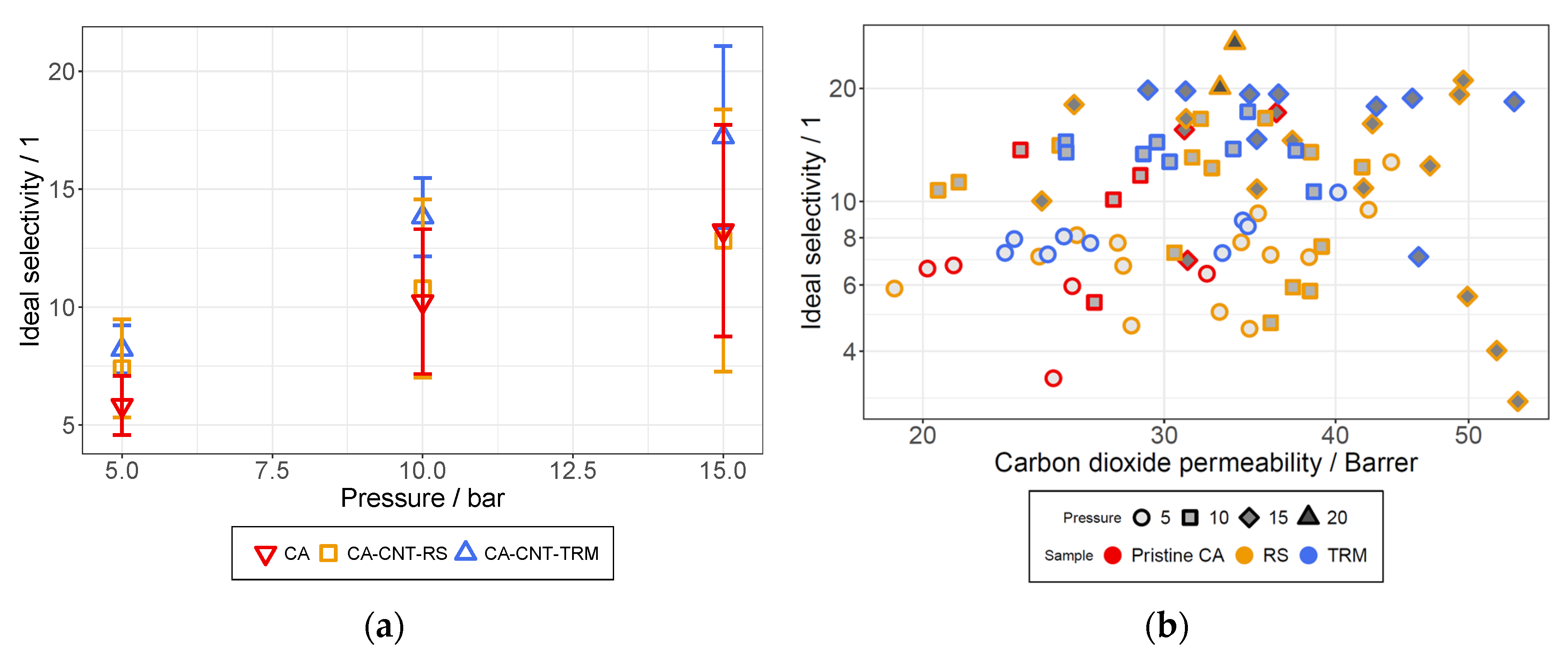
3.6. Permeation Analysis
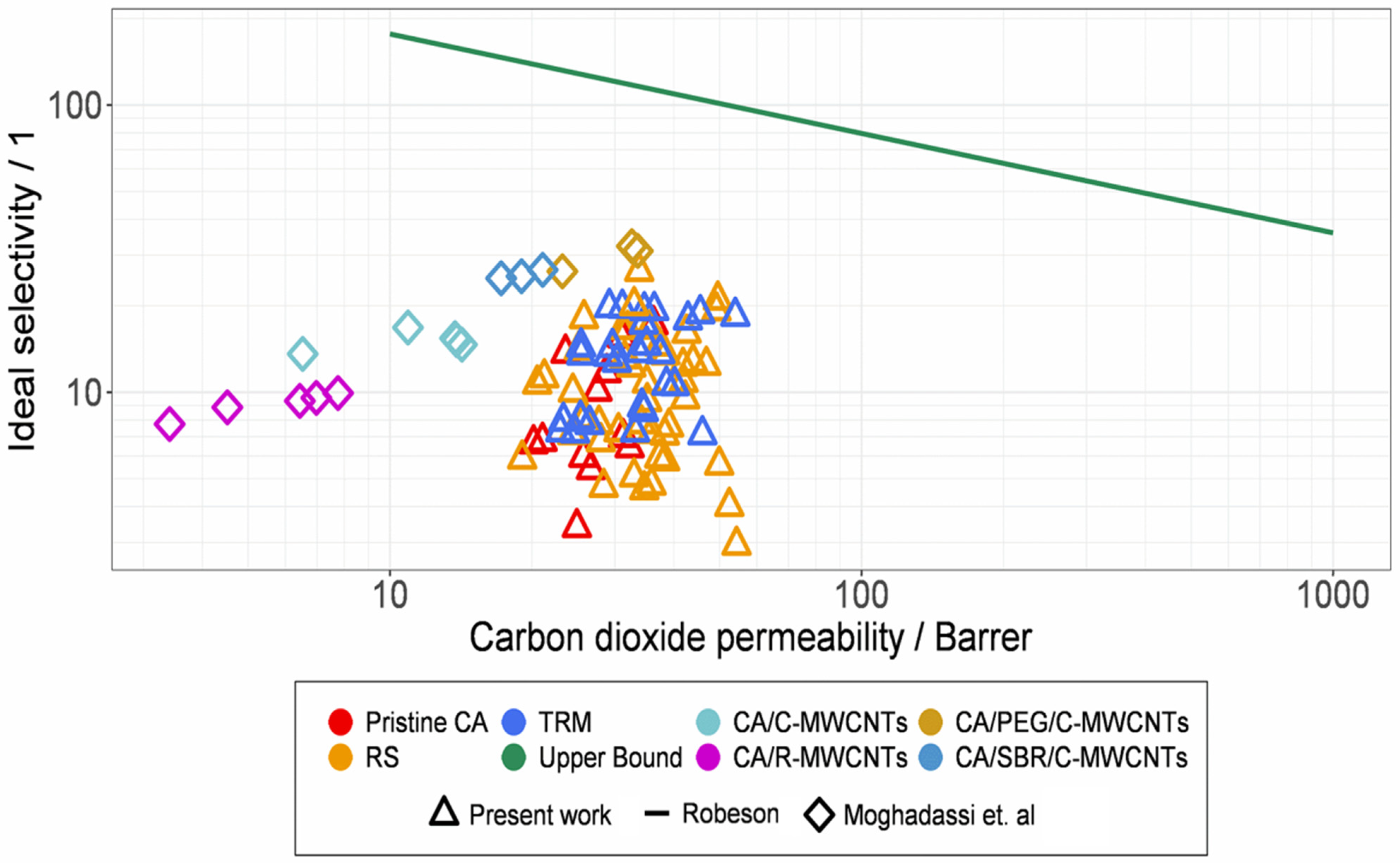
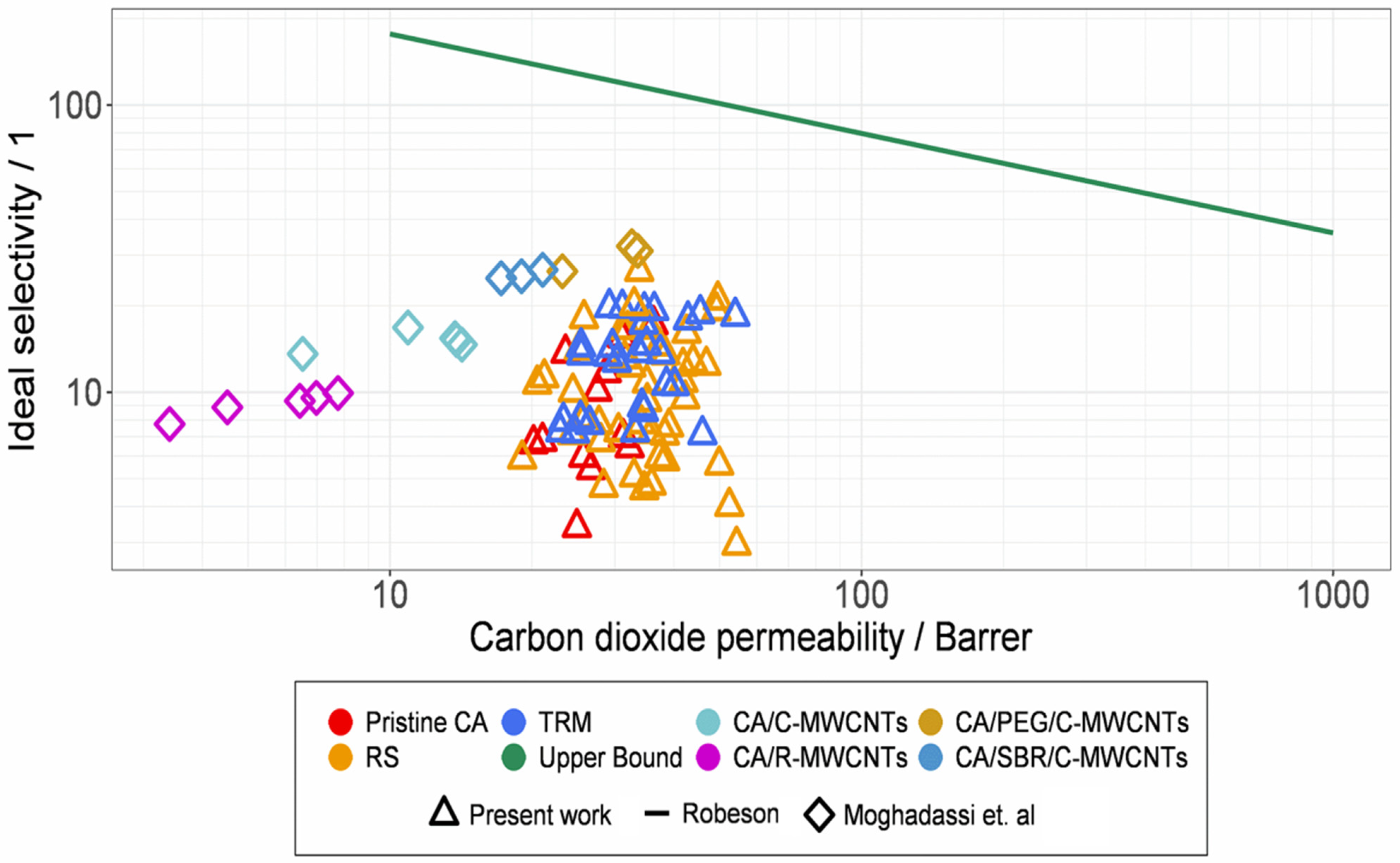
3.7. Comparisons and Classifications with Former Results
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, G.; Bhoria, N.; AlHallaq, S.; Abdala, A.; Mittal, V. Polymer membranes for acid gas removal from natural gas. Sep. Purif. Technol. 2016, 158, 333–356. [Google Scholar] [CrossRef]
- Nakao, S.-I.; Yogo, K.; Goto, K.; Kai, T.; Yamada, H. Advanced CO2 Capture Technologies; Springer International Publishing: Cham, Germany, 2019; ISBN 978-3-030-18857-3. [Google Scholar]
- Rezakazemi, M.; Ebadi Amooghin, A.; Montazer-Rahmati, M.M.; Ismail, A.F.; Matsuura, T. State-of-the-art membrane based CO2 separation using mixed matrix membranes (MMMs): An overview on current status and future directions. Prog. Polym. Sci. 2014, 39, 817–861. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Guan, J.; Yang, L.; Ren, Y.; Nasir, N.; Wu, H.; Chen, Z.; Jiang, Z. 110th Anniversary: Mixed Matrix Membranes with Fillers of Intrinsic Nanopores for Gas Separation. Ind. Eng. Chem. Res. 2019, 58, 7706–7724. [Google Scholar] [CrossRef]
- Chung, T.-S.; Jiang, L.Y.; Li, Y.; Kulprathipanja, S. Mixed matrix membranes (MMMs) comprising organic polymers with dispersed inorganic fillers for gas separation. Prog. Polym. Sci. 2007, 32, 483–507. [Google Scholar] [CrossRef]
- Robeson, L.M. The upper bound revisited. J. Membr. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- Vu, D.Q.; Koros, W.J.; Miller, S.J. Mixed matrix membranes using carbon molecular sieves. J. Membr. Sci. 2003, 211, 311–334. [Google Scholar] [CrossRef]
- Moore, T.T.; Mahajan, R.; Vu, D.Q.; Koros, W.J. Hybrid membrane materials comprising organic polymers with rigid dispersed phases. AIChE J. 2004, 50, 311–321. [Google Scholar] [CrossRef]
- Asif, K.; Lock, S.S.M.; Taqvi, S.A.A.; Jusoh, N.; Yiin, C.L.; Chin, B.L.F.; Loy, A.C.M. A Molecular Simulation Study of Silica/Polysulfone Mixed Matrix Membrane for Mixed Gas Separation. Polymers 2021, 13, 2199. [Google Scholar] [CrossRef]
- Wong, K.K.; Jawad, Z.A.; Chin, B.L.F. A polyethylene glycol (PEG)—Polyethersulfone (PES)/multi-walled carbon nanotubes (MWCNTs) polymer blend mixed matrix membrane for CO2/N2 separation. J. Polym. Res. 2021, 28, 7276. [Google Scholar] [CrossRef]
- Juber, F.A.H.; Jawad, Z.A.; Teoh, G.H.; Ahmad, A.L.; Chin, B.L.F. Development of novel blend poly (Ethylene Glycol)/Poly(Ethersulfone) polymeric membrane using N-Methyl-2-Pyrollidone and dimethylformamide solvents for facilitating CO2/N2 gas separation. Mater. Today 2021, 46, 1963–1970. [Google Scholar] [CrossRef]
- Ahmad, A.L.; Jawad, Z.A.; Low, S.C.; Zein, S.H.S. A cellulose acetate/multi-walled carbon nanotube mixed matrix membrane for CO2/N2 separation. J. Membr. Sci. 2014, 451, 55–66. [Google Scholar] [CrossRef]
- Esser, T.; Wolf, T.; Schubert, T.; Benra, J.; Forero, S.; Maistros, G.; Barbe, S.; Theodorakopoulos, G.V.; Karousos, D.S.; Sapalidis, A.A.; et al. CO2/CH4 and He/N2 Separation Properties and Water Permeability Valuation of Mixed Matrix MWCNTs-Based Cellulose Acetate Flat Sheet Membranes: A Study of the Optimization of the Filler Material Dispersion Method. Nanomaterials 2021, 11, 280. [Google Scholar] [CrossRef]
- Yonkoski, R.K.; Soane, D.S. Model for spin coating in microelectronic applications. J. Appl. Phys. 1992, 72, 725–740. [Google Scholar] [CrossRef] [Green Version]
- Meyerhofer, D. Characteristics of resist films produced by spinning. J. Appl. Phys. 1978, 49, 3993–3997. [Google Scholar] [CrossRef] [Green Version]
- Emslie, A.G.; Bonner, F.T.; Peck, L.G. Flow of a Viscous Liquid on a Rotating Disk. J. Appl. Phys. 1958, 29, 858–862. [Google Scholar] [CrossRef]
- Schubert, D.W.; Dunkel, T. Spin coating from a molecular point of view: Its concentration regimes, influence of molar mass and distribution. Mater. Res. Innov. 2003, 7, 314–321. [Google Scholar] [CrossRef]
- Clausi, M.; Santonicola, M.G.; Laurenzi, S. Fabrication of carbon-based nanocomposite films by spin-coating process: An experimental and modeling study of the film thickness. Compos. Part A 2016, 88, 86–97. [Google Scholar] [CrossRef]
- Lee, U.G.; Kim, W.-B.; Han, D.H.; Chung, H.S. A Modified Equation for Thickness of the Film Fabricated by Spin Coating. Symmetry 2019, 11, 1183. [Google Scholar] [CrossRef] [Green Version]
- Burmann, P.; Zornoza, B.; Téllez, C.; Coronas, J. Mixed matrix membranes comprising MOFs and porous silicate fillers prepared via spin coating for gas separation. Chem. Eng. Sci. 2014, 107, 66–75. [Google Scholar] [CrossRef]
- Handbook of Industrial Mixing: Science and Practice; Wiley-Interscience: Hoboken, NJ, USA, 2010; ISBN 0-471-26919-0.
- Håkansson, A. Rotor-Stator Mixers: From Batch to Continuous Mode of Operation—A Review. Processes 2018, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Thostenson, E.T.; Chou, T.-W. Processing-structure-multi-functional property relationship in carbon nanotube/epoxy composites. Carbon 2006, 44, 3022–3029. [Google Scholar] [CrossRef]
- Gojny, F.H.; Wichmann, M.H.G.; Köpke, U.; Fiedler, B.; Schulte, K. Carbon nanotube-reinforced epoxy-composites: Enhanced stiffness and fracture toughness at low nanotube content. Compos. Sci. Technol. 2004, 64, 2363–2371. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Bilotti, E.; Peijs, T. Optimization of Three-Roll Mill Parameters for In-Situ Exfoliation of Graphene. MRS Adv. 2016, 1, 1389–1394. [Google Scholar] [CrossRef]
- Schröder, K.; Meyer-Plath, A.; Keller, D.; Besch, W.; Babucke, G.; Ohl, A. Plasma-Induced Surface Functionalization of Polymeric Biomaterials in Ammonia Plasma. Contrib. Plasma. Phys. 2001, 41, 562–572. [Google Scholar] [CrossRef]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database; Wiley: Chichester, UK, 1992; ISBN 0-471-93592-1. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Lomax, M. Permeation of gases and vapours through polymer films and thin sheet—Part I. Polym. Test. 1980, 1, 105–147. [Google Scholar] [CrossRef]
- Naftaly, M.; Das, S.; Gallop, J.; Pan, K.; Alkhalil, F.; Kariyapperuma, D.; Constant, S.; Ramsdale, C.; Hao, L. Sheet Resistance Measurements of Conductive Thin Films: A Comparison of Techniques. Electronics 2021, 10, 960. [Google Scholar] [CrossRef]
- Miccoli, I.; Edler, F.; Pfnür, H.; Tegenkamp, C. The 100th anniversary of the four-point probe technique: The role of probe geometries in isotropic and anisotropic systems. J. Phys. Condens. Matter 2015, 27, 223201. [Google Scholar] [CrossRef]
- Buschhorn, S.T.; Wichmann, M.H.G.; Sumfleth, J.; Schulte, K.; Pegel, S.; Kasaliwal, G.R.; Villmow, T.; Krause, B.; Göldel, A.; Pötschke, P. Charakterisierung der Dispersionsgüte von Carbon Nanotubes in Polymer-Nanokompositen. Chem. Ing. Tech. 2011, 83, 767–781. [Google Scholar] [CrossRef]
- Earp, B.; Simpson, J.; Phillips, J.; Grbovic, D.; Vidmar, S.; McCarthy, J.; Luhrs, C.C. Electrically Conductive CNT Composites at Loadings below Theoretical Percolation Values. Nanomaterials 2019, 9, 491. [Google Scholar] [CrossRef] [Green Version]
- Lenth, R.V. Response-Surface Methods in R, Using rsm. J. Stat. Soft. 2009, 32, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kleppmann, W. Versuchsplanung: Produkte und Prozesse Optimieren, 10, Überarbeitete Auflage; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2020; ISBN 3446463976. [Google Scholar]
- Schröder, T. Rheologie der Kunststoffe; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2018; ISBN 978-3-446-45405-7. [Google Scholar]
- Dunstan, D.E. The viscosity-radius relationship for concentrated polymer solutions. Sci. Rep. 2019, 9, 543. [Google Scholar] [CrossRef]
- Ohl, A.; Schröder, K. Plasma assisted surface modification of biointerfaces. In Low Temperature Plasma Physics: Fundamental Aspects and Applications; Hippler, R., Kersten, H., Schmidt, M., Schoenbach, K.H., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 803–819. [Google Scholar]
- Baeva, M.; Stankov, M.; Trautvetter, T.; Methling, R.; Hempel, F.; Loffhagen, D.; Foest, R. The effect of oxygen admixture on the properties of microwave generated plasma in Ar–O2: A modelling study. J. Phys. D 2021, 54, 355205. [Google Scholar] [CrossRef]
- Hempel, F.; Steffen, H.; Busse, B.; Finke, B.; Barbara, J.; Quade, A.; Rebl, H.; Bergemann, C.; Weltmann, K.-D.; Schroder, K. On the Application of Gas Discharge Plasmas for the Immobilization of Bioactive Molecules for Biomedical and Bioengineering Applications. In Biomedical Engineering—Frontiers and Challenges; Fazel, R., Ed.; InTechOpen: London, UK, 2011; ISBN 978-953-307-309-5. Available online: https://www.intechopen.com/books/508 (accessed on 30 November 2021).
- Mokbul, H.M.; Dirk, H. Substrate independent dyeing of synthetic textiles treated with low-pressure plasmas. Text. Dyeing. Croat. 2011, 173–194. [Google Scholar]
- Jokinen, V.; Suvanto, P.; Franssila, S. Oxygen and nitrogen plasma hydrophilization and hydrophobic recovery of polymers. Biomicrofluidics 2012, 6, 16501. [Google Scholar] [CrossRef] [Green Version]
- Ebadi Amooghin, A.; Omidkhah, M.; Kargari, A. Enhanced CO2 transport properties of membranes by embedding nano-porous zeolite particles into Matrimid®5218 matrix: Supplementary information. RSC Adv. 2015, 5, 8552–8565. [Google Scholar] [CrossRef]
- Moghadassi, A.R.; Rajabi, Z.; Hosseini, S.M.; Mohammadi, M. Fabrication and modification of cellulose acetate based mixed matrix membrane: Gas separation and physical properties. J. Ind. Eng. Chem. 2014, 20, 1050–1060. [Google Scholar] [CrossRef]
- Stern, S.A.; de Meringo, A.H. Solubility of carbon dioxide in cellulose acetate at elevated pressures. J. Polym. Sci. Polym. Phys. Ed. 1978, 16, 735–751. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer in Solution | Polymer in Dispersion | Solvent in Dispersion | CNT in Dispersion |
|---|---|---|---|
| PI 20% | 15.79% | 83.89% | 0.32% |
| CA 10% | 8.82% | 91.0% | 0.18% |
| Three Roll Mill | Rotor-Stator | ||
|---|---|---|---|
| Rotational Speed min−1 | Gap Width µm | Rotational Speed min−1 | Duration of Treatment Min |
| 100 | 5 | 2000 | 10 |
| 300 | 15 | 6000 | 30 |
| Norm. Value | Rotational Speed min−1 | Applied Volume mL |
|---|---|---|
| −α * | 2172 | 1 |
| −1 | 3000 | 1.5 |
| 0 | 5000 | 2 |
| +1 | 7000 | 2.5 |
| +α * | 7828 | 3 |
| Rotational Speed min−1 | Wet Layer Thickness µm | Dry Layer Thickness µm |
|---|---|---|
| 2172 | 46.9 ± 21.9 | 3.89 ± 2.14 |
| 3000 | 51.8 ± 2.55 | 4.69 ± 0.36 |
| 5000 | 20.8 ± 6.86 | 2.23 ± 0.97 |
| 7000 | 16.3 ± 0.98 | 1.54 ± 0.13 |
| 7828 | 9.27 ± 4.34 | 1.4 ± 0.7 |
| Dispersion Method | Rotor Stator | Three-Roll-Mill |
|---|---|---|
| Median agglomerate size | 1.38 ± 0.075 µm | 1.58 ± 0.51 µm |
| Number of agglomerates | 92.3 ± 19.6 × 10−3 mm−2 | 29 ± 10 × 10−3 mm−2 |
| Relative agglomerate area | 14.0 ± 3.8% | 6.17 ± 4.57% |
| Norm. Value | Rotational Speed min−1 | PI-Concentration wt.% | Coating Time s |
|---|---|---|---|
| −α * | 1734 | 12.37 | 21 |
| −1 | 3000 | 13 | 30 |
| 0 | 5000 | 14 | 45 |
| +1 | 7000 | 15 | 60 |
| +α * | 8266 | 15.63 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammerstein, R.; Schubert, T.; Braun, G.; Wolf, T.; Barbe, S.; Quade, A.; Foest, R.; Karousos, D.S.; Favvas, E.P. The Optimization of Dispersion and Application Techniques for Nanocarbon-Doped Mixed Matrix Gas Separation Membranes. Membranes 2022, 12, 87. https://doi.org/10.3390/membranes12010087
Hammerstein R, Schubert T, Braun G, Wolf T, Barbe S, Quade A, Foest R, Karousos DS, Favvas EP. The Optimization of Dispersion and Application Techniques for Nanocarbon-Doped Mixed Matrix Gas Separation Membranes. Membranes. 2022; 12(1):87. https://doi.org/10.3390/membranes12010087
Chicago/Turabian StyleHammerstein, Ruben, Tim Schubert, Gerd Braun, Tobias Wolf, Stéphan Barbe, Antje Quade, Rüdiger Foest, Dionysios S. Karousos, and Evangelos P. Favvas. 2022. "The Optimization of Dispersion and Application Techniques for Nanocarbon-Doped Mixed Matrix Gas Separation Membranes" Membranes 12, no. 1: 87. https://doi.org/10.3390/membranes12010087
APA StyleHammerstein, R., Schubert, T., Braun, G., Wolf, T., Barbe, S., Quade, A., Foest, R., Karousos, D. S., & Favvas, E. P. (2022). The Optimization of Dispersion and Application Techniques for Nanocarbon-Doped Mixed Matrix Gas Separation Membranes. Membranes, 12(1), 87. https://doi.org/10.3390/membranes12010087