
Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics

 , ,
, , 
Abstract
:1. Introduction
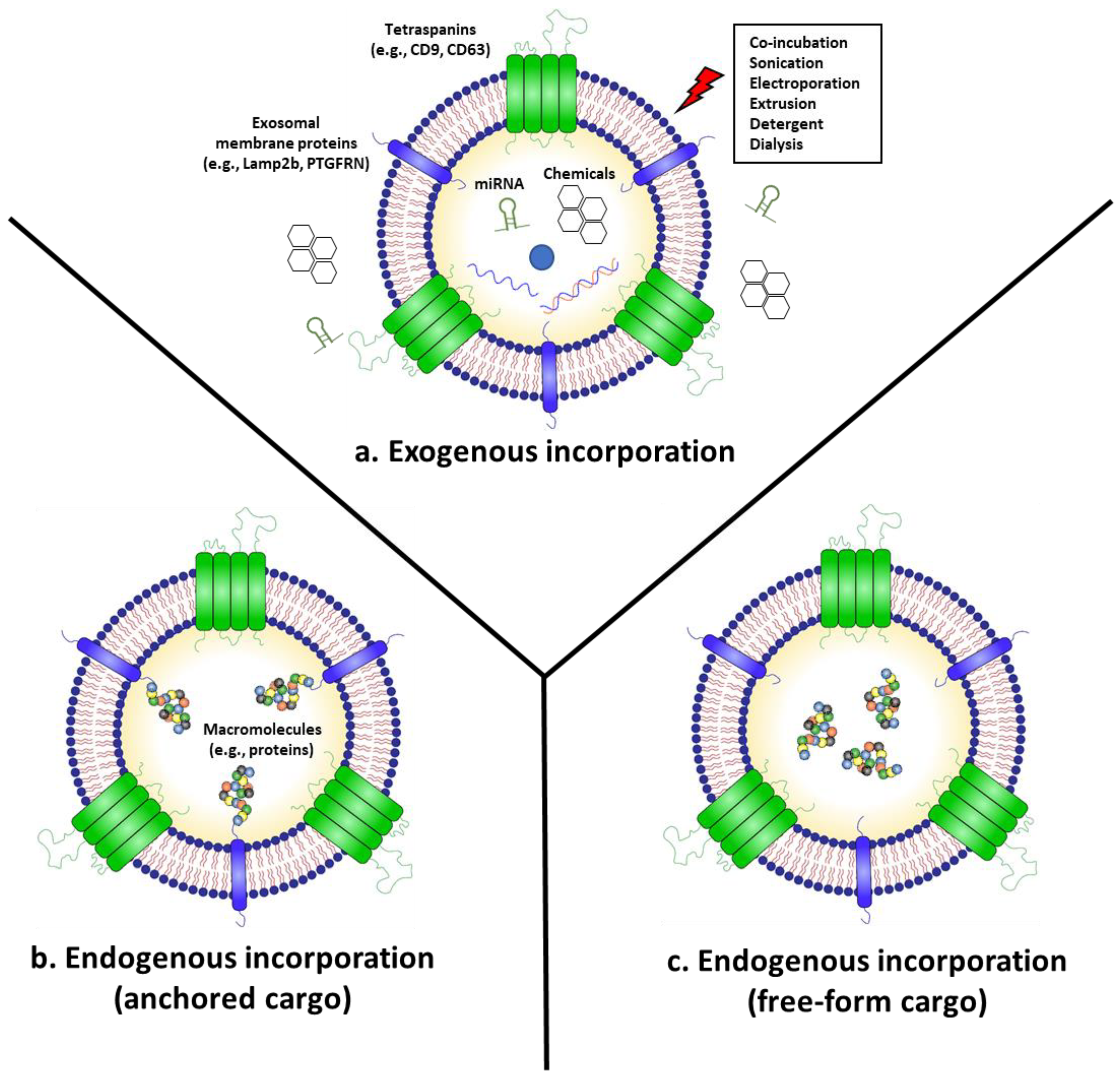
2. “Armed” Exosomes as Cancer Therapeutics
2.1. Anti-Cancer Exosomes Loaded with RNAs (miRNA, siRNA, and mRNA)
2.2. Chemotherapeutics-Armed Exosomes
2.3. Exosomes “Armed” with Anti-Cancer Proteins
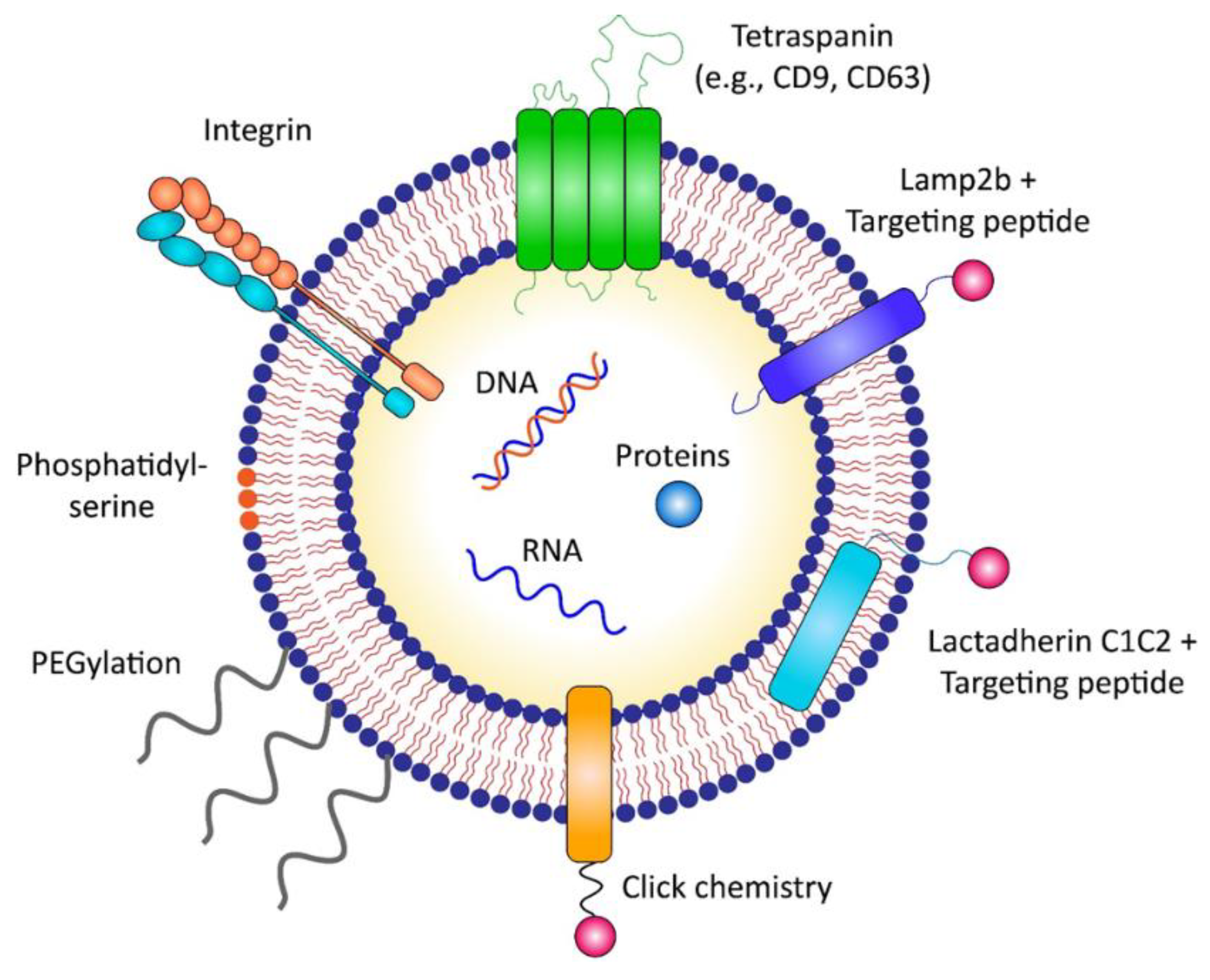
3. Strategies for Targeted Delivery of Exosomes to Cancer Cells
3.1. Passive Targeting
3.2. Active Targeting
3.2.1. Direct Surface Engineering of Exosomes
3.2.2. Indirect Engineering of Exosomes by Modifying Exosome-Producing Cells

{kind=link}
{kind=link}
{kind=link}
| Category | Method | Targeting Moiety | Target Cancer | Ref. |
|---|---|---|---|---|
| Direct engineering of exosomes | Click chemistry | Neuropillin-1-targeting RGE peptide (RGERPPR) | Glioma | [85] |
| PEGylation | Aminoethyl anisamide-PEG (AA-PEG) | Sigma receptor-overexpressing lung cancer | [88] | |
| Mixing with micelles | DMPE-PEG conjugated with anti-EGFR nanobody | EGFR-overexpressing tumor cells in vitro | [89] | |
| Indirect engineering of exosomes | Conjugation with C1C2 domain | Anti-Her2 scFv | HER2-expressing breast cancer | [93] |
| GPI anchorage | Anti-EGFR nanobody | EGFR-expressing breast cancer | [94] | |
| Conjugation with Lamp2b | αv integrin-targeting iRGD peptide | Breast cancer cell line | [44] | |
| NSCLC-homing peptide Tlyp-1 | Lung cancer cell line | [96] | ||
| HER2 targeting DARPins | HER2-expressing breast cancer | [97] | ||
| Conjugation with CD63 | Apo-A1 | Hepatocellular carcinoma | [98] | |
| Conjugation with CD47 | U87-targeting CDX peptide, GL261-targeting CREKA peptide | U87 glioblastoma cell, GL261 glioma cell | [34] |
4. Scalable Manufacturing of GMP-Grade Therapeutic Exosomes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Nam, G.H.; Choi, Y.; Kim, G.B.; Kim, S.; Kim, S.A.; Kim, I.S. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Adv. Mater. 2020, 32, e2002440. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as drug carriers for cancer therapy and challenges regarding exosome uptake. Biomed Pharm. 2020, 128, 110237. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Peng, H.; Ji, W.; Zhao, R.; Yang, J.; Lu, Z.; Li, Y.; Zhang, X. Exosome: A significant nano-scale drug delivery carrier. J. Mater. Chem. B 2020, 8, 7591–7608. [Google Scholar] [CrossRef]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef] [Green Version]
- Michael, A.; Bajracharya, S.D.; Yuen, P.S.; Zhou, H.; Star, R.A.; Illei, G.G.; Alevizos, I. Exosomes from human saliva as a source of microRNA biomarkers. Oral Dis. 2010, 16, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filen, J.J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Vojtech, L.; Woo, S.; Hughes, S.; Levy, C.; Ballweber, L.; Sauteraud, R.P.; Strobl, J.; Westerberg, K.; Gottardo, R.; Tewari, M.; et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014, 42, 7290–7304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Choi, Y.; Yim, H.Y.; Mirzaaghasi, A.; Yoo, J.K.; Choi, C. Biodistribution of Exosomes and Engineering Strategies for Targeted Delivery of Therapeutic Exosomes. Tissue Eng. Regen. Med. 2021, 18, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhang, W.; Zhang, H.; Zhang, F.; Chen, L.; Ma, L.; Larcher, L.M.; Chen, S.; Liu, N.; Zhao, Q.; et al. Progress, opportunity, and perspective on exosome isolation-efforts for efficient exosome-based theranostics. Theranostics 2020, 10, 3684–3707. [Google Scholar] [CrossRef]
- Nistico, N.; Maisano, D.; Iaccino, E.; Vecchio, E.; Fiume, G.; Rotundo, S.; Quinto, I.; Mimmi, S. Role of Chronic Lymphocytic Leukemia (CLL)-Derived Exosomes in Tumor Progression and Survival. Pharmaceuticals 2020, 13, 244. [Google Scholar] [CrossRef]
- Song, Y.; Kim, Y.; Ha, S.; Sheller-Miller, S.; Yoo, J.; Choi, C.; Park, C.H. The emerging role of exosomes as novel therapeutics: Biology, technologies, clinical applications, and the next. Am. J. Reprod. Immunol. 2021, 85, e13329. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, 6478. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Zhou, B.; Xu, K.; Zheng, X.; Chen, T.; Wang, J.; Song, Y.; Shao, Y.; Zheng, S. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct. Target. Ther. 2020, 5, 144. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. Exosomes as a Multicomponent Biomarker Platform in Cancer. Trends Cancer 2020, 6, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.P.; Khan, S.; Gilligan, K.E.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J. Extracell. Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Kase, Y.; Uzawa, K.; Wagai, S.; Yoshimura, S.; Yamamoto, J.I.; Toeda, Y.; Okubo, M.; Eizuka, K.; Ando, T.; Nobuchi, T.; et al. Engineered exosomes delivering specific tumor-suppressive RNAi attenuate oral cancer progression. Sci. Rep. 2021, 11, 5897. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Xu, H.; Liao, C.; Liang, S.; Ye, B.C. A Novel Peptide-Equipped Exosomes Platform for Delivery of Antisense Oligonucleotides. ACS Appl. Mater. Interfaces 2021, 13, 10760–10767. [Google Scholar] [CrossRef]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control. Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Mehryab, F.; Rabbani, S.; Shahhosseini, S.; Shekari, F.; Fatahi, Y.; Baharvand, H.; Haeri, A. Exosomes as a next-generation drug delivery system: An update on drug loading approaches, characterization, and clinical application challenges. Acta Biomater. 2020, 113, 42–62. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Gomari, H.; Forouzandeh Moghadam, M.; Soleimani, M.; Ghavami, M.; Khodashenas, S. Targeted delivery of doxorubicin to HER2 positive tumor models. Int. J. Nanomed. 2019, 14, 5679–5690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Che, S.P.Y.; LeBleu, V.S.; Kalluri, R. Effective delivery of STING agonist using exosomes suppresses tumor growth and enhances antitumor immunity. J. Biol. Chem. 2021, 296, 100523. [Google Scholar] [CrossRef]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Song, Y.; Park, C.H.; Choi, C. Platform technologies and human cell lines for the production of therapeutic exosomes. Extracell. Vesicles Circ. Nucleic Acids 2021, 2, 3–17. [Google Scholar] [CrossRef]
- Koh, E.; Lee, E.J.; Nam, G.H.; Hong, Y.; Cho, E.; Yang, Y.; Kim, I.S. Exosome-SIRPalpha, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 2017, 121, 121–129. [Google Scholar] [CrossRef]
- Dooley, K.; McConnell, R.E.; Xu, K.; Lewis, N.D.; Haupt, S.; Youniss, M.R.; Martin, S.; Sia, C.L.; McCoy, C.; Moniz, R.J.; et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol. Ther. 2021, 29, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Sia, C.L.; Kirwin, K.; Haupt, S.; Mahimkar, G.; Zi, T.; Xu, K.; Dooley, K.; Jang, S.C.; Choi, B.; et al. Exosome Surface Display of IL12 Results in Tumor-Retained Pharmacology with Superior Potency and Limited Systemic Exposure Compared with Recombinant IL12. Mol. Cancer Ther. 2021, 20, 523–534. [Google Scholar] [CrossRef]
- Yim, N.; Ryu, S.W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef]
- Shalitin, D.; Yang, H.; Mockler, T.C.; Maymon, M.; Guo, H.; Whitelam, G.C.; Lin, C. Regulation of Arabidopsis cryptochrome 2 by blue-light-dependent phosphorylation. Nature 2002, 417, 763–767. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef] [Green Version]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer-using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Koo, M.Y.; Park, J.; Lim, J.M.; Joo, S.Y.; Shin, S.P.; Shim, H.B.; Chung, J.; Kang, D.; Woo, H.A.; Rhee, S.G. Selective inhibition of the function of tyrosine-phosphorylated STAT3 with a phosphorylation site-specific intrabody. Proc. Natl. Acad. Sci. USA 2014, 111, 6269–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Fang, J.; Maeda, H. Development of next-generation macromolecular drugs based on the EPR effect: Challenges and pitfalls. Expert Opin. Drug Deliv. 2015, 12, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kibria, G.; Ramos, E.K.; Wan, Y.; Gius, D.R.; Liu, H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Mol. Pharm. 2018, 15, 3625–3633. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Bogart, L.K.; Pourroy, G.; Murphy, C.J.; Puntes, V.; Pellegrino, T.; Rosenblum, D.; Peer, D.; Levy, R. Nanoparticles for imaging, sensing, and therapeutic intervention. ACS Nano 2014, 8, 3107–3122. [Google Scholar] [CrossRef]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Hu, S.Q.; Huang, K.; Su, T.; Li, Z.H.; Vandergriff, A.; Cores, J.; Dinh, P.U.; Allen, T.; Shen, D.L.; et al. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474–3487. [Google Scholar] [CrossRef]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lazaro-Ibanez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.O.; Jo, H.; Yu, J.H.; Gambhir, S.S.; Pratx, G. Development and MPI tracking of novel hypoxia-targeted theranostic exosomes. Biomaterials 2018, 177, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Berenguer, J.; Lagerweij, T.; Zhao, X.W.; Dusoswa, S.; van der Stoop, P.; Westerman, B.; de Gooijer, M.C.; Zoetemelk, M.; Zomer, A.; Crommentuijn, M.H.W.; et al. Glycosylated extracellular vesicles released by glioblastoma cells are decorated by CCL18 allowing for cellular uptake via chemokine receptor CCR8. J. Extracell. Vesicles 2018, 7, 1446660. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Heon, M.; Zhao, Z.; He, M. Microfluidic engineering of exosomes: Editing cellular messages for precision therapeutics. Lab Chip 2018, 18, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Rayamajhi, S.; Aryal, S. Surface functionalization strategies of extracellular vesicles. J. Mater. Chem. B 2020, 8, 4552–4569. [Google Scholar] [CrossRef]
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.C.; Choi, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Therapeutics and as a Drug Delivery Platform. Stem Cells Transl. Med. 2019, 8, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface functionalization of exosomes using click chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasubramanian, L.; Kumar, P.; Wang, A. Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules 2019, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algar, W.R.; Prasuhn, D.E.; Stewart, M.H.; Jennings, T.L.; Blanco-Canosa, J.B.; Dawson, P.E.; Medintz, I.L. The controlled display of biomolecules on nanoparticles: A challenge suited to bioorthogonal chemistry. Bioconjug Chem. 2011, 22, 825–858. [Google Scholar] [CrossRef] [PubMed]
- Villata, S.; Canta, M.; Cauda, V. EVs and Bioengineering: From Cellular Products to Engineered Nanomachines. Int. J. Mol. Sci. 2020, 21, 6048. [Google Scholar] [CrossRef]
- Nwe, K.; Brechbiel, M.W. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radiopharm. 2009, 24, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef]
- Chen, L.; Miao, W.; Tang, X.; Zhang, H.; Wang, S.; Luo, F.; Yan, J. The expression and significance of neuropilin-1 (NRP-1) on glioma cell lines and glioma tissues. J. Biomed. Nanotechnol. 2013, 9, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Susa, F.; Limongi, T.; Dumontel, B.; Vighetto, V.; Cauda, V. Engineered Extracellular Vesicles as a Reliable Tool in Cancer Nanomedicine. Cancers 2019, 11, 1979. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.; Heijnen, H.F.G.; van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longatti, A.; Schindler, C.; Collinson, A.; Jenkinson, L.; Matthews, C.; Fitzpatrick, L.; Blundy, M.; Minter, R.; Vaughan, T.; Shaw, M.; et al. High affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles. Nanoscale 2018, 10, 14230–14244. [Google Scholar] [CrossRef] [Green Version]
- Kooijmans, S.A.; Aleza, C.G.; Roffler, S.R.; van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- de Gassart, A.; Geminard, C.; Fevrier, B.; Raposo, G.; Vidal, M. Lipid raft-associated protein sorting in exosomes. Blood 2003, 102, 4336–4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Duan, J.; Liu, R.; Du, Y.; Luo, Q.; Cui, Y.; Su, Z.; Xu, J.; Xie, Y.; Lu, W. Engineered targeting tLyp-1 exosomes as gene therapy vectors for efficient delivery of siRNA into lung cancer cells. Asian J. Pharm. Sci. 2020, 15, 461–471. [Google Scholar] [CrossRef]
- Gomari, H.; Forouzandeh Moghadam, M.; Soleimani, M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco Targets Ther. 2018, 11, 5753–5762. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Kan, S.; Zhu, Y.; Feng, S.; Feng, W.; Gao, S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomed. 2018, 13, 585–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Albelda, S.M.; Mette, S.A.; Elder, D.E.; Stewart, R.; Damjanovich, L.; Herlyn, M.; Buck, C.A. Integrin distribution in malignant melanoma: Association of the beta 3 subunit with tumor progression. Cancer Res. 1990, 50, 6757–6764. [Google Scholar]
- Gingras, M.C.; Roussel, E.; Bruner, J.M.; Branch, C.D.; Moser, R.P. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J. Neuroimmunol. 1995, 57, 143–153. [Google Scholar] [CrossRef]
- Natali, P.G.; Hamby, C.V.; Felding-Habermann, B.; Liang, B.; Nicotra, M.R.; Di Filippo, F.; Giannarelli, D.; Temponi, M.; Ferrone, S. Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res. 1997, 57, 1554–1560. [Google Scholar]
- Malm, M.; Saghaleyni, R.; Lundqvist, M.; Giudici, M.; Chotteau, V.; Field, R.; Varley, P.G.; Hatton, D.; Grassi, L.; Svensson, T.; et al. Evolution from adherent to suspension: Systems biology of HEK293 cell line development. Sci. Rep. 2020, 10, 18996. [Google Scholar] [CrossRef]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Whitford, W.; Guterstam, P. Exosome manufacturing status. Future Med. Chem. 2019, 11, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.K.A.; Morille, M.; Piffoux, M.; Arumugam, S.; Mauduit, P.; Larghero, J.; Bianchi, A.; Aubertin, K.; Blanc-Brude, O.; Noel, D.; et al. Development of extracellular vesicle-based medicinal products: A position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs-EVOLVE France”. Adv. Drug Deliv. Rev. 2021, 179, 114001. [Google Scholar] [CrossRef]
- Burnouf, T.; Agrahari, V.; Agrahari, V. Extracellular Vesicles As Nanomedicine: Hopes And Hurdles In Clinical Translation. Int. J. Nanomed. 2019, 14, 8847–8859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, N.; Grant, L.; De Oliveira, T.M.; Rowlinson, R.; Osteikoetxea, X.; Dekker, N.; Overman, R. Rapid isolation and enrichment of extracellular vesicle preparations using anion exchange chromatography. Sci. Rep. 2018, 8, 5730. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, E.H.; Street, J.M.; Star, R.A.; Yuen, P.S. Quantification of Exosomes. J. Cell. Physiol. 2017, 232, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Banizs, A.B.; Shi, W.; Klibanov, A.L.; He, J. Size Exclusion HPLC Detection of Small-Size Impurities as a Complementary Means for Quality Analysis of Extracellular Vesicles. J. Circ. Biomark. 2015, 4, 6. [Google Scholar] [CrossRef]
- Wang, X.; Hunter, A.K.; Mozier, N.M. Host cell proteins in biologics development: Identification, quantitation and risk assessment. Biotechnol. Bioeng. 2009, 103, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Busatto, S.; Vilanilam, G.; Ticer, T.; Lin, W.L.; Dickson, D.W.; Shapiro, S.; Bergese, P.; Wolfram, J. Tangential Flow Filtration for Highly Efficient Concentration of Extracellular Vesicles from Large Volumes of Fluid. Cells 2018, 7, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.Q.; Almughlliq, F.B.; Vaswani, K.; Peiris, H.N.; Mitchell, M.D. Exosome enrichment by ultracentrifugation and size exclusion chromatography. Front. Biosci. 2018, 23, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wu, C.; Lin, X.; Zhou, J.; Zhang, J.; Zheng, W.; Wang, T.; Cui, Y. Establishment of a simplified dichotomic size-exclusion chromatography for isolating extracellular vesicles toward clinical applications. J. Extracell. Vesicles 2021, 10, e12145. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.E.; Korbie, D.; Trau, M.; Hill, M.M. Optimizing Size Exclusion Chromatography for Extracellular Vesicle Enrichment and Proteomic Analysis from Clinically Relevant Samples. Proteomics 2019, 19, e1800156. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Sztandera, K.; Gorzkiewicz, M.; Klajnert-Maculewicz, B. Gold Nanoparticles in Cancer Treatment. Mol. Pharm. 2019, 16, 1–23. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neurooncol. 2011, 103, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Bonvalot, S.; Rutkowski, P.L.; Thariat, J.; Carrere, S.; Ducassou, A.; Sunyach, M.P.; Agoston, P.; Hong, A.; Mervoyer, A.; Rastrelli, M.; et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): A multicentre, phase 2-3, randomised, controlled trial. Lancet Oncol. 2019, 20, 1148–1159. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Yim, H.; Park, C.; Ahn, S.-H.; Ahn, Y.; Lee, A.; Yang, H.; Choi, C. Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics. Membranes 2022, 12, 85. https://doi.org/10.3390/membranes12010085
Choi H, Yim H, Park C, Ahn S-H, Ahn Y, Lee A, Yang H, Choi C. Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics. Membranes. 2022; 12(1):85. https://doi.org/10.3390/membranes12010085
Chicago/Turabian StyleChoi, Hojun, Hwayoung Yim, Cheolhyoung Park, So-Hee Ahn, Yura Ahn, Areum Lee, Heekyoung Yang, and Chulhee Choi. 2022. "Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics" Membranes 12, no. 1: 85. https://doi.org/10.3390/membranes12010085
APA StyleChoi, H., Yim, H., Park, C., Ahn, S.-H., Ahn, Y., Lee, A., Yang, H., & Choi, C. (2022). Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics. Membranes, 12(1), 85. https://doi.org/10.3390/membranes12010085