
Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric

 ,
,
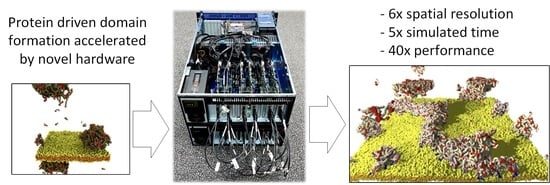
Abstract
:
1. Introduction
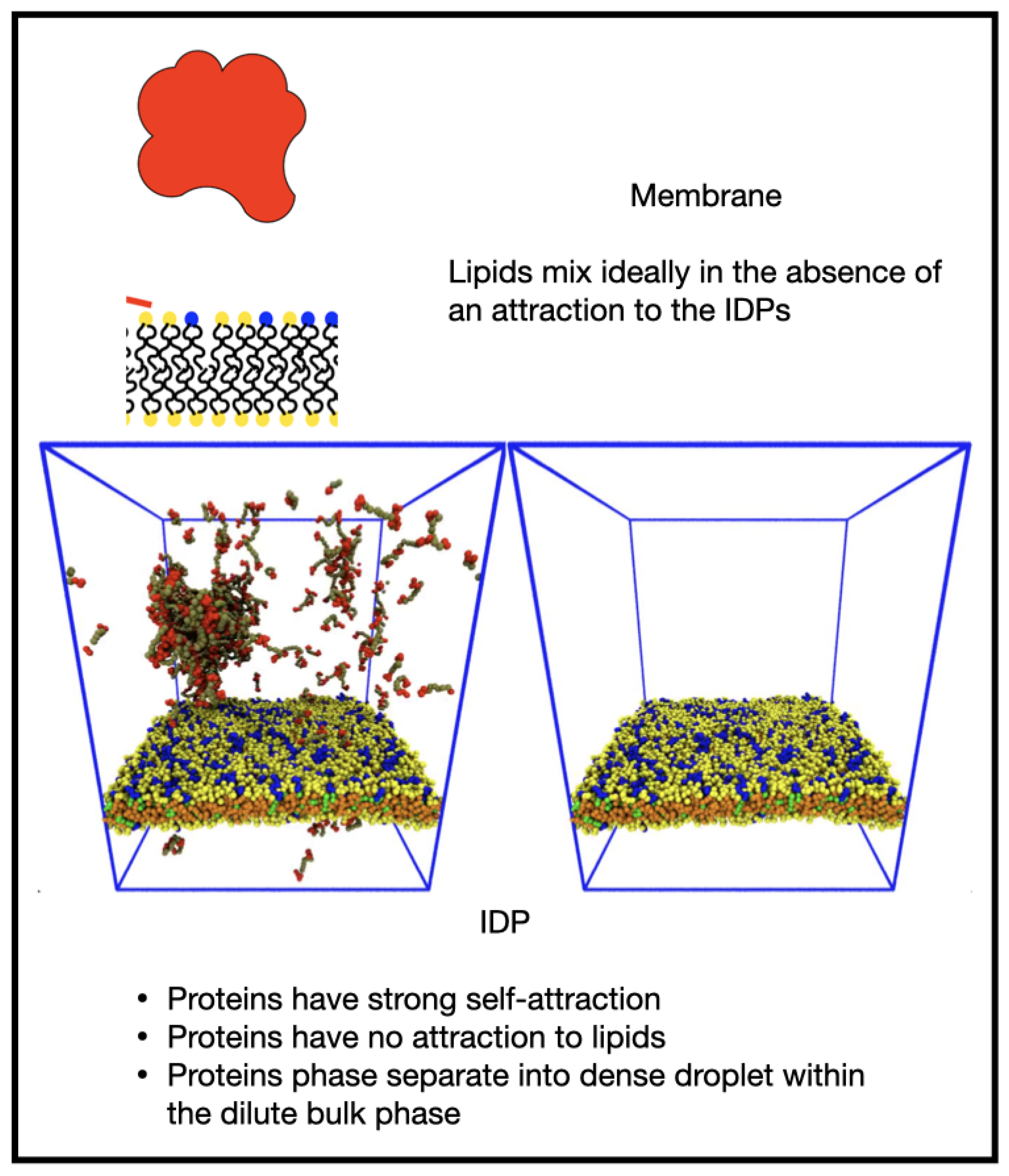
- We present a simulation-based exploration of the morphological outcomes resulting from the 3D phase separation of intrinsically-disordered proteins near a 2D lipid membrane;
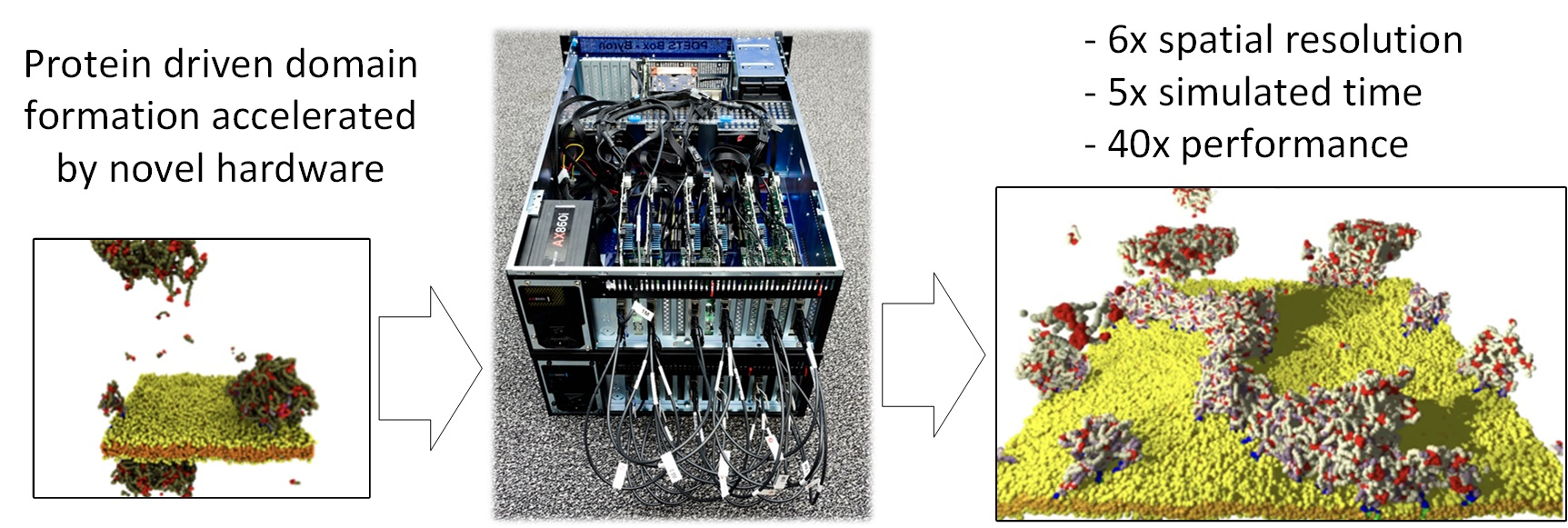
- We describe the novel POETS compute architecture and the algorithm used in POETS-DPD, and demonstrate the power of this approach by simulating selected systems at a fraction of the wall clock time: a 14 cpu-day conventional DPD simulation is completed by POETS-DPD in 8 h, a speedup of more than a factor of 40.
2. Materials and Methods
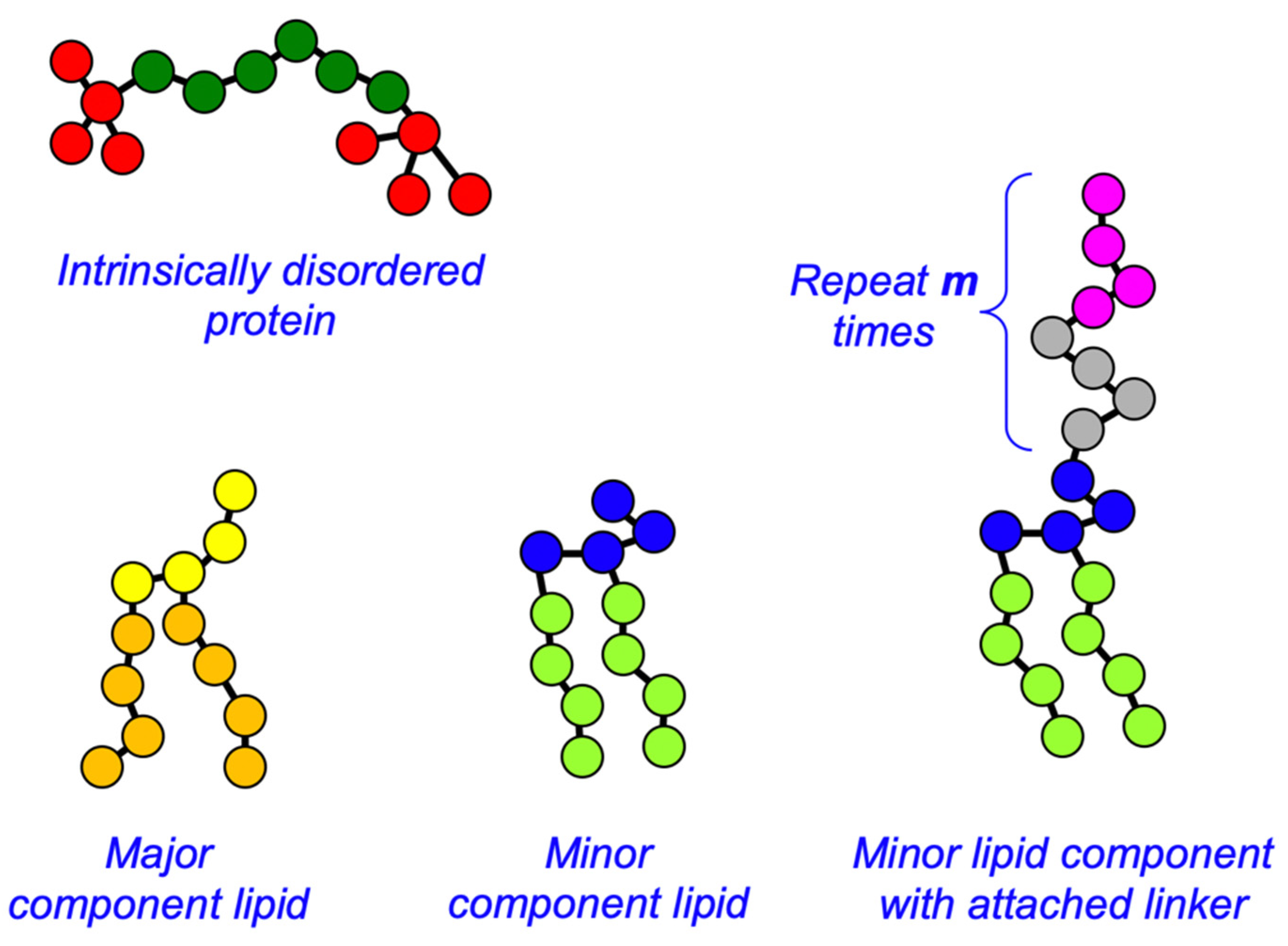
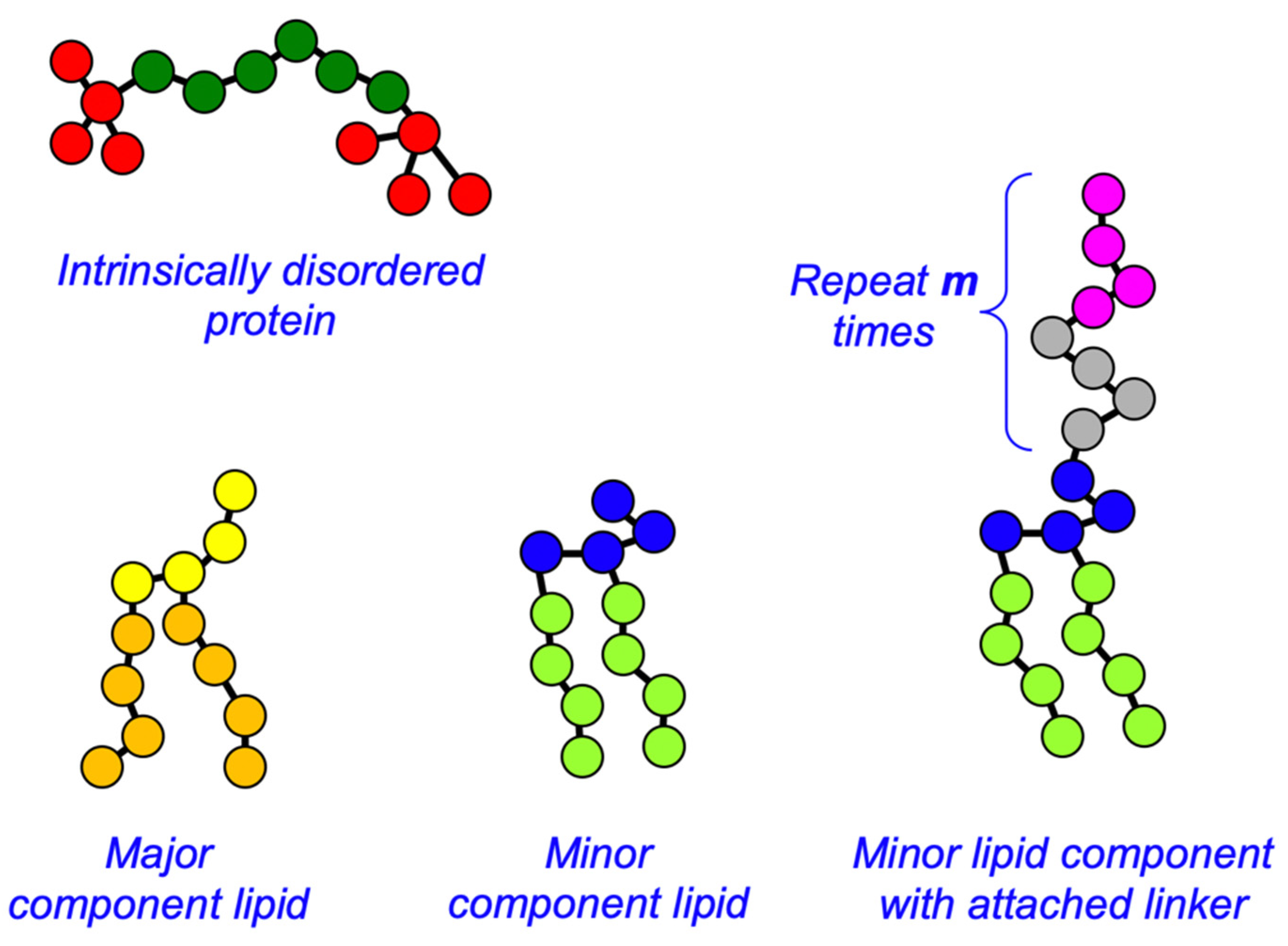
2.1. Dissipative Particle Dynamics Simulations
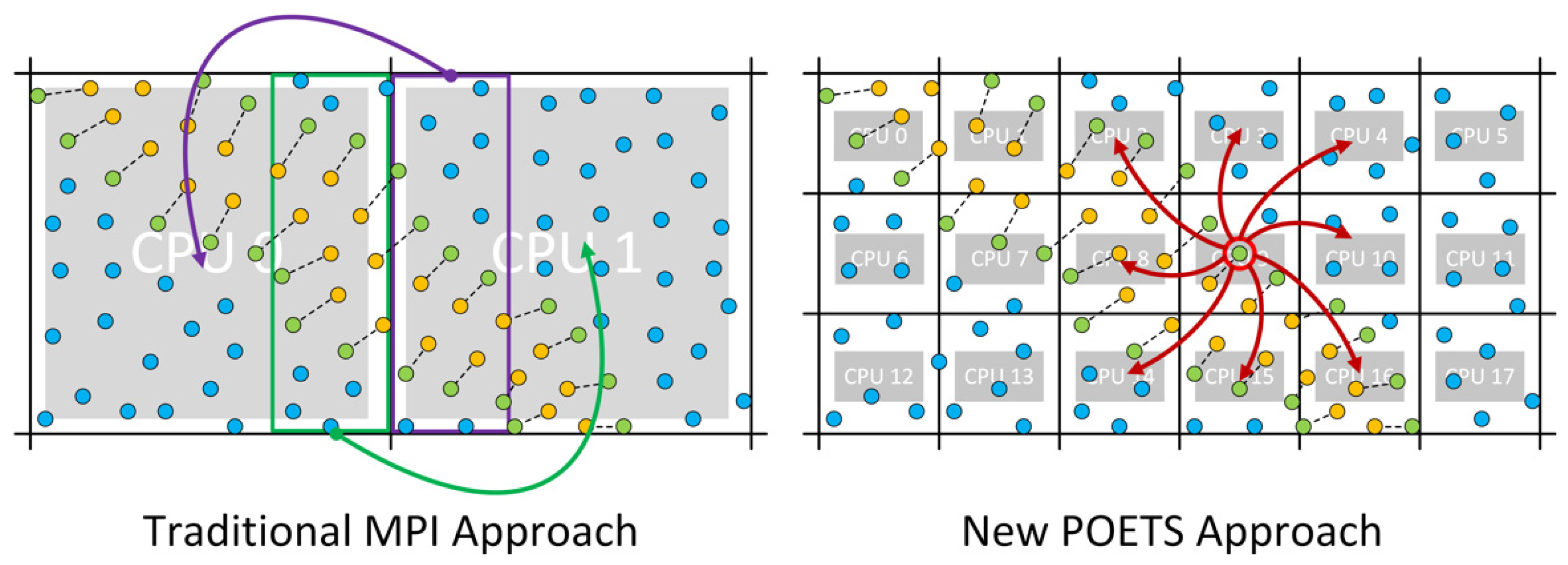
2.2. Massively Parallel Event-Based Simulations
- Force calculation
- a
- Sharing: Broadcast each resident bead position and velocity to neighbors;
- b
- Integration: For each received bead, calculate non-bonded DPD and Hookean bond interactions with resident cells;
- c
- AngleForces: Once head and tail beads for an angle bond are received by middle bead, calculate angle forces and broadcast to neighbors (which must include the owner of the angle bond’s head and tail;
- d
- AngleAddition: If an angle force is received and this cell contains the related head or tail bead, apply angle-bond forces to that head/tail.
- Bead movement
- a
- Movement: Apply equations of motion to all beads resident in the cell;
- b
- BeadExit: If any bead leaves the cell, broadcast it to neighbors and remove from resident set;
- c
- BeadEntrance: If a bead is received from a neighbor and its new position is in the current cell, add it to the resident set.
- Go back to step 1 for next step of simulation
3. Results
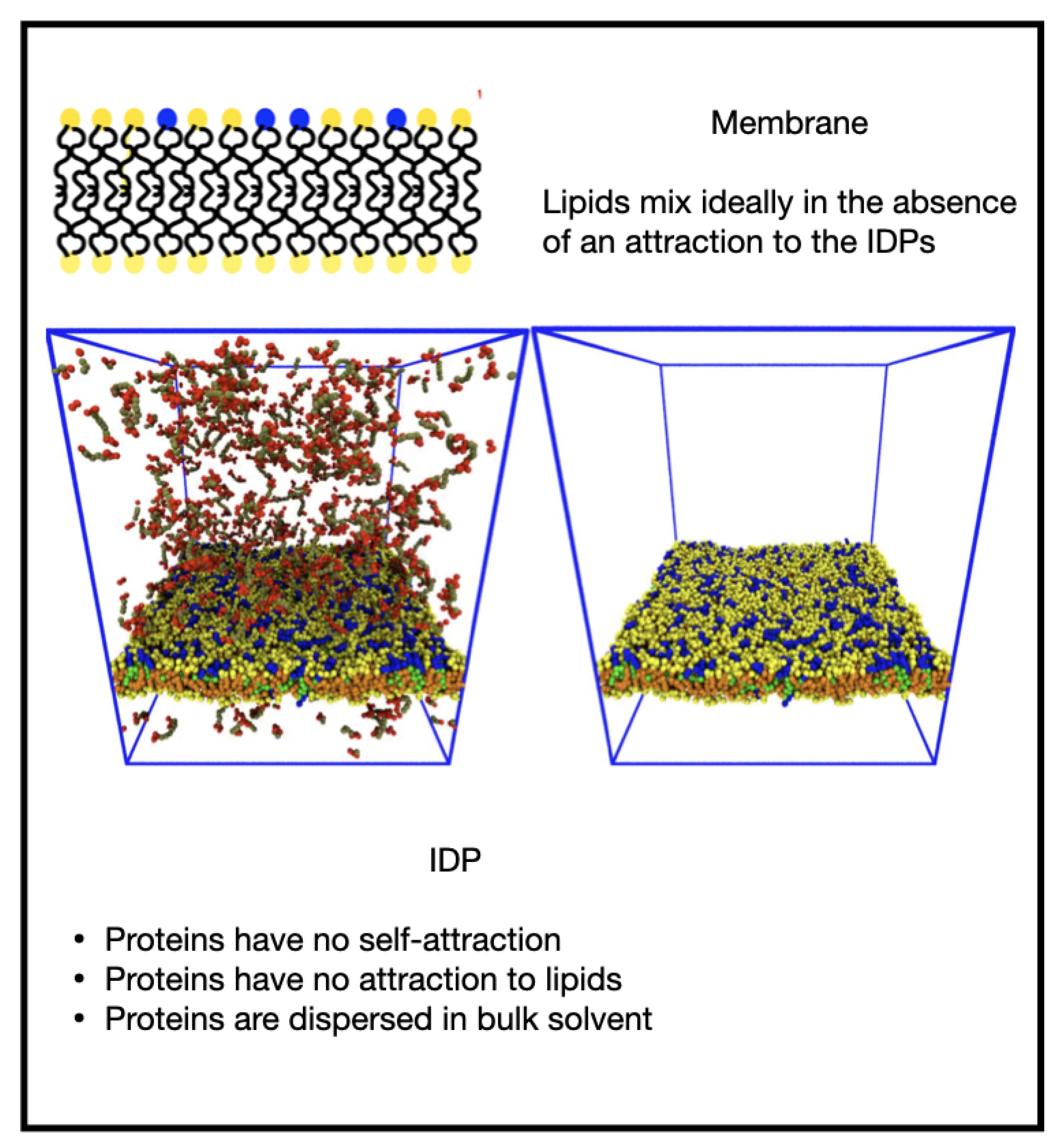
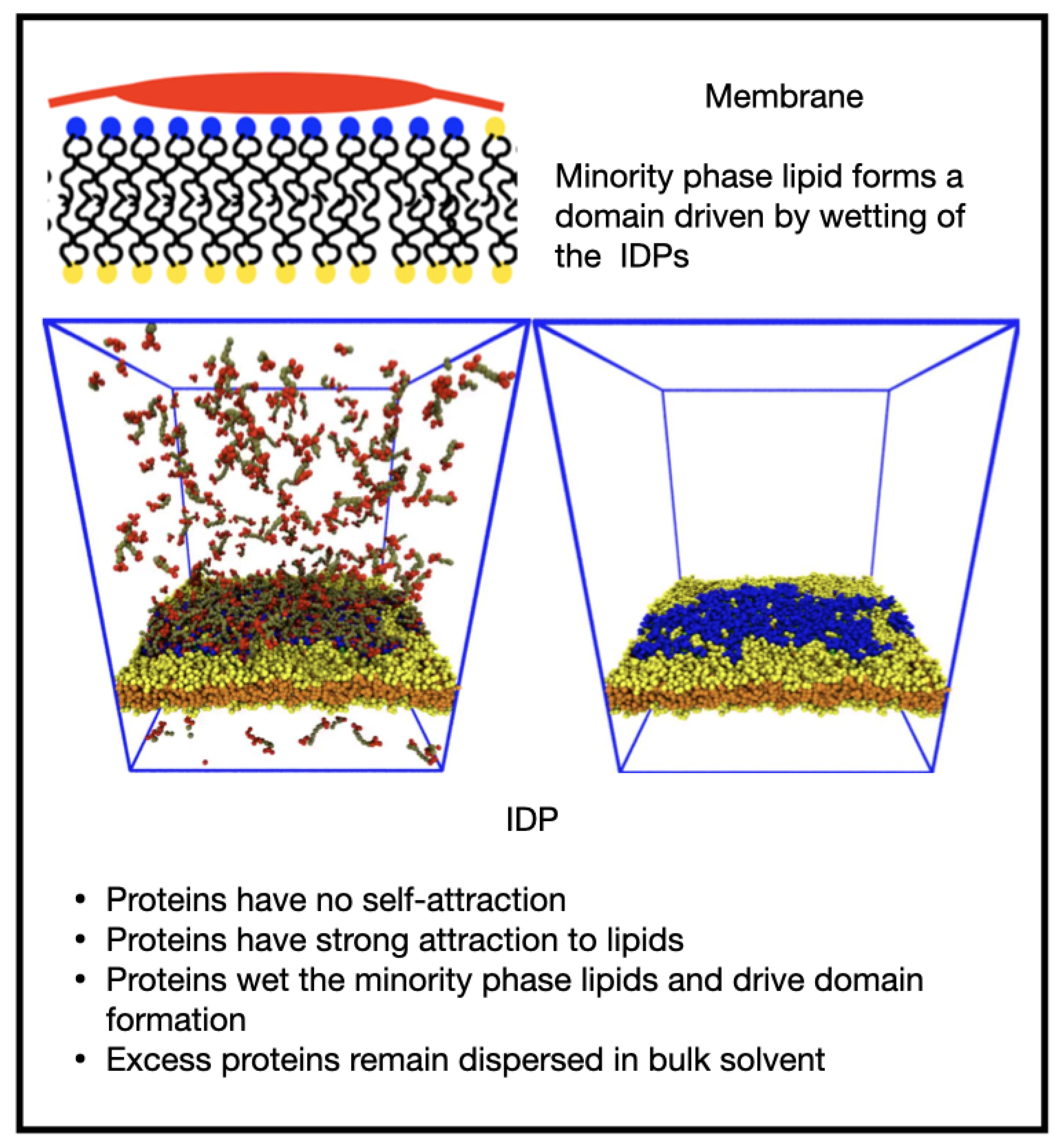
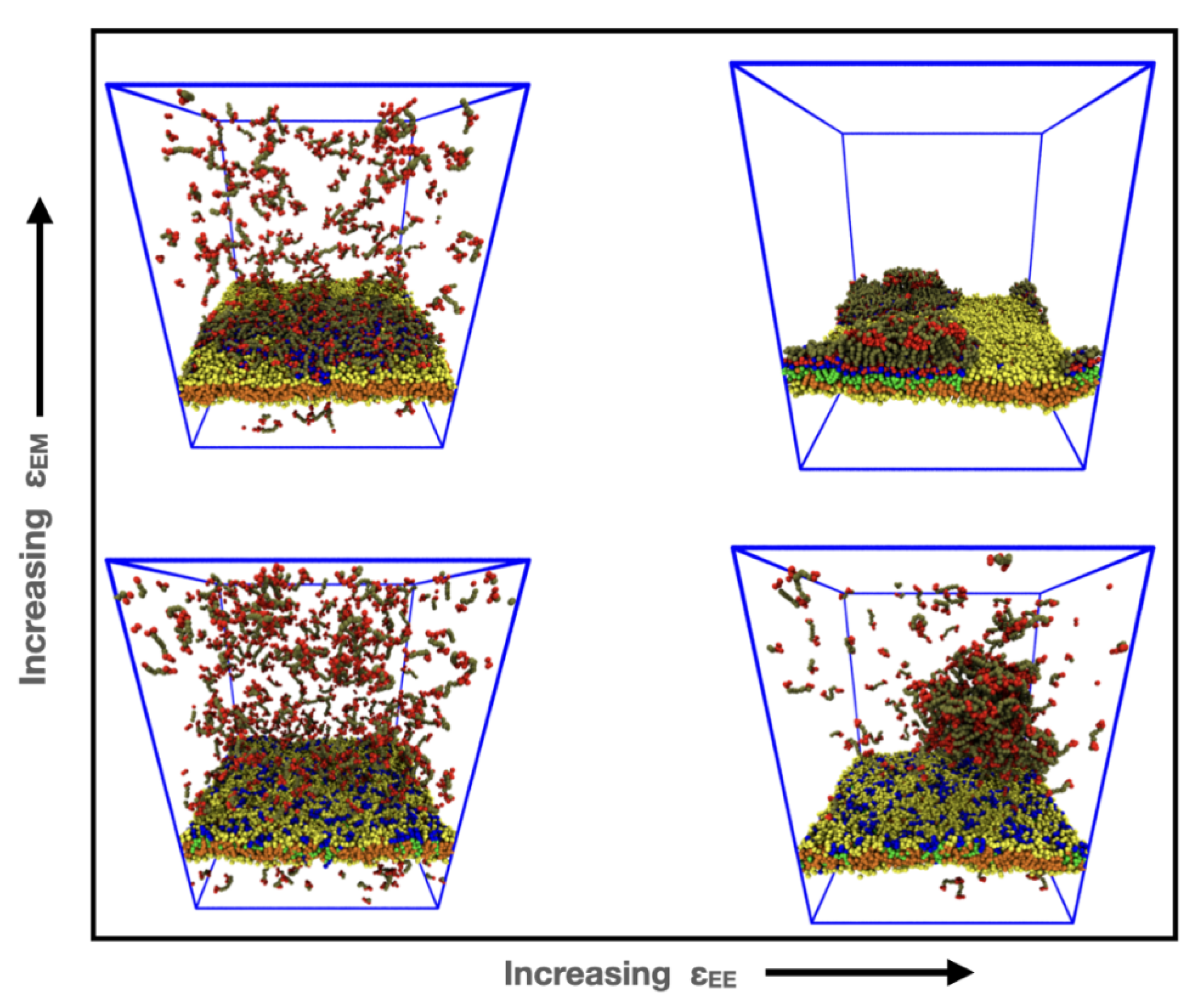
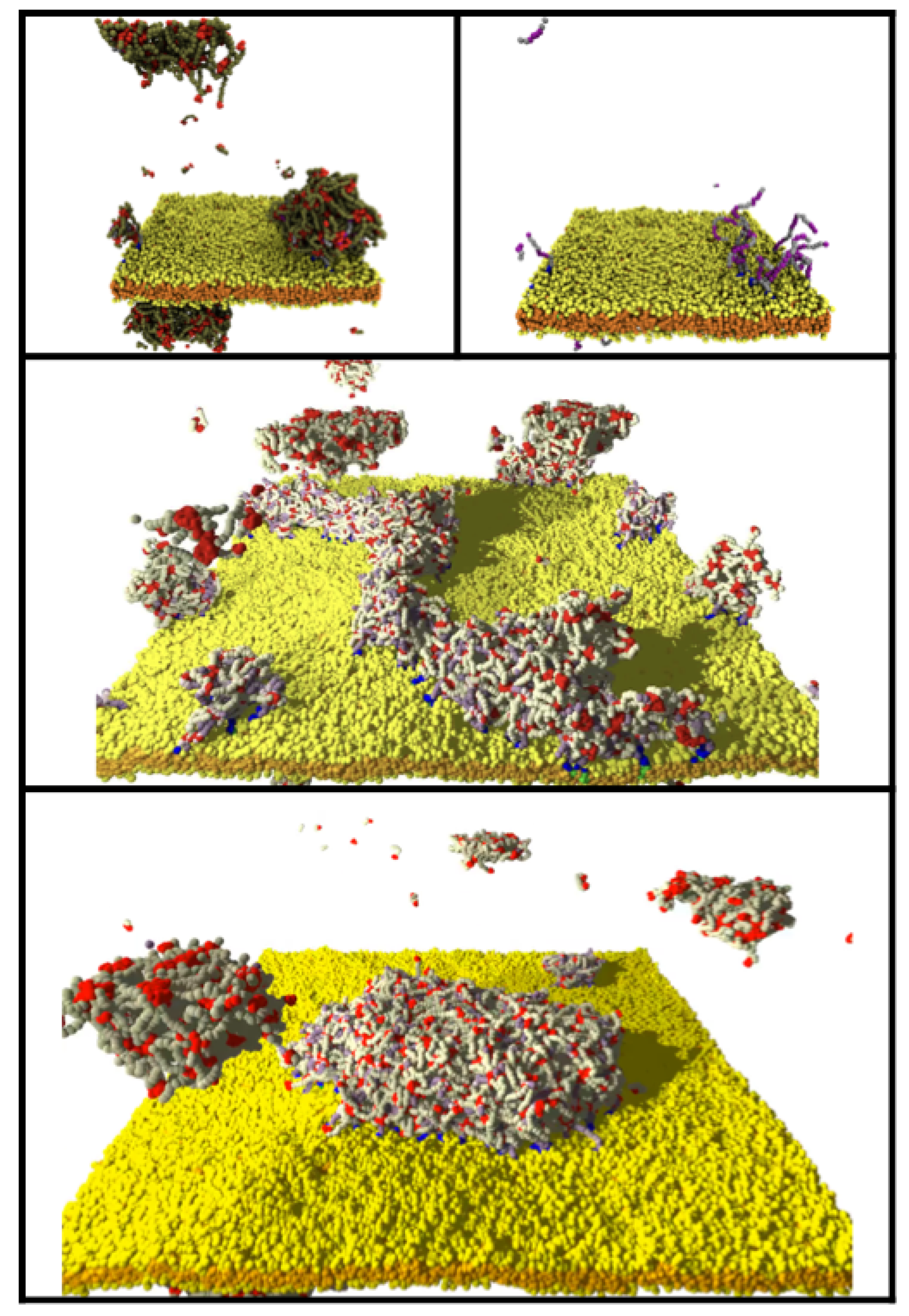
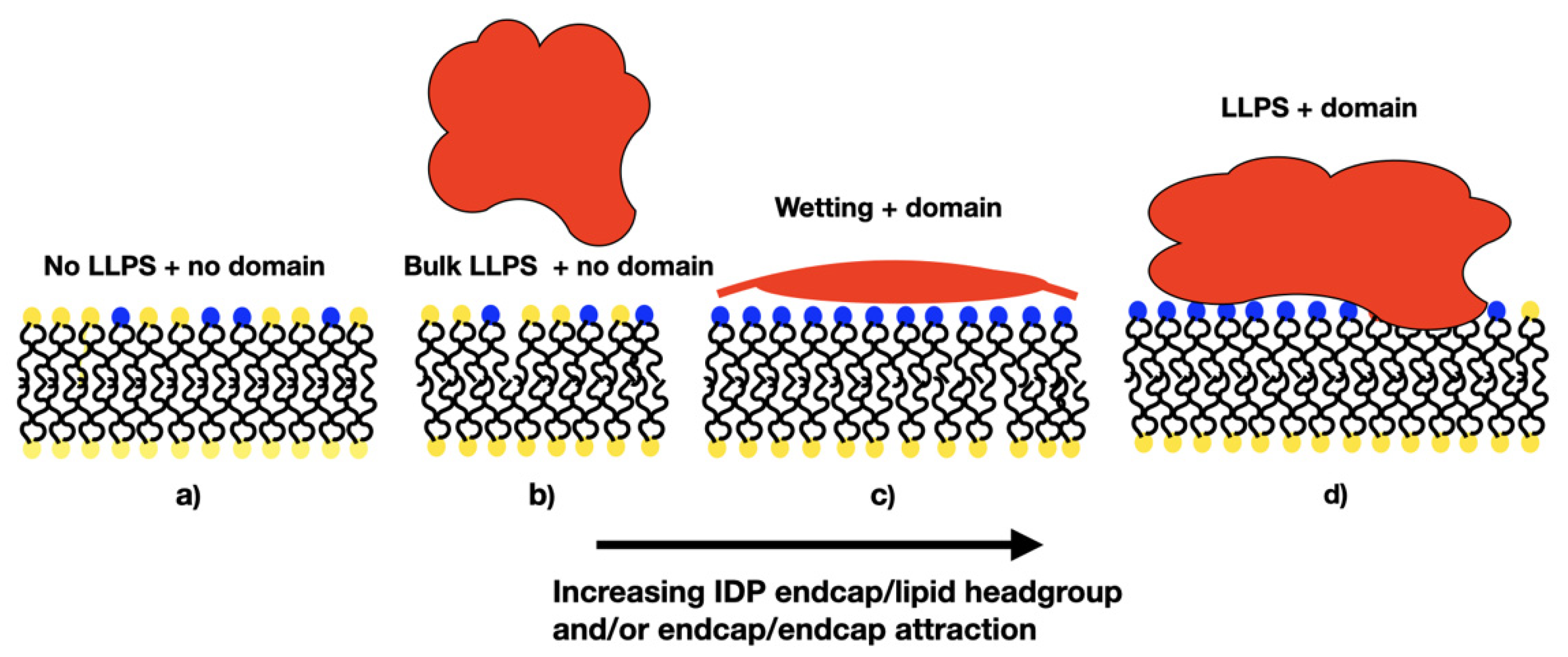
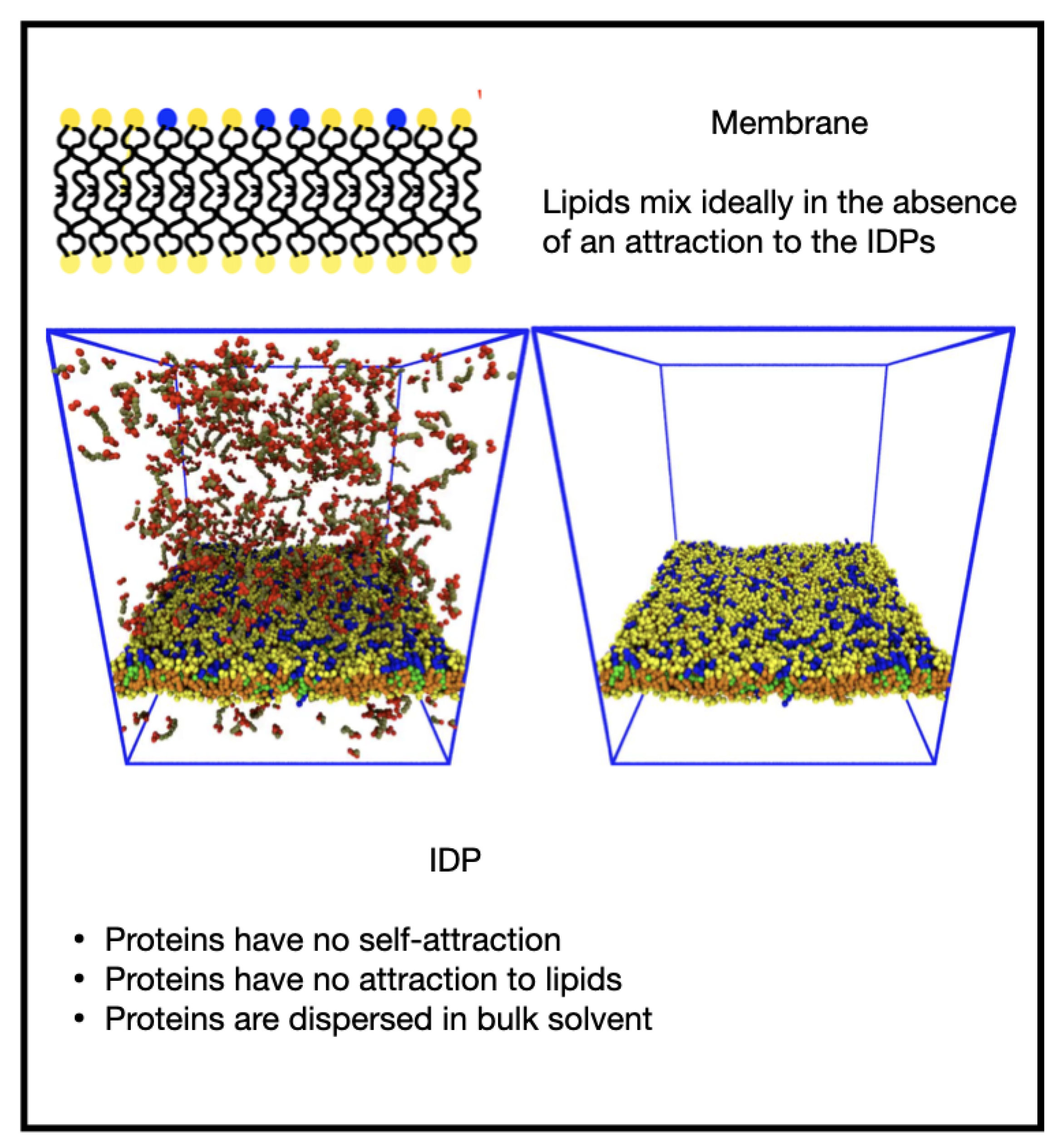
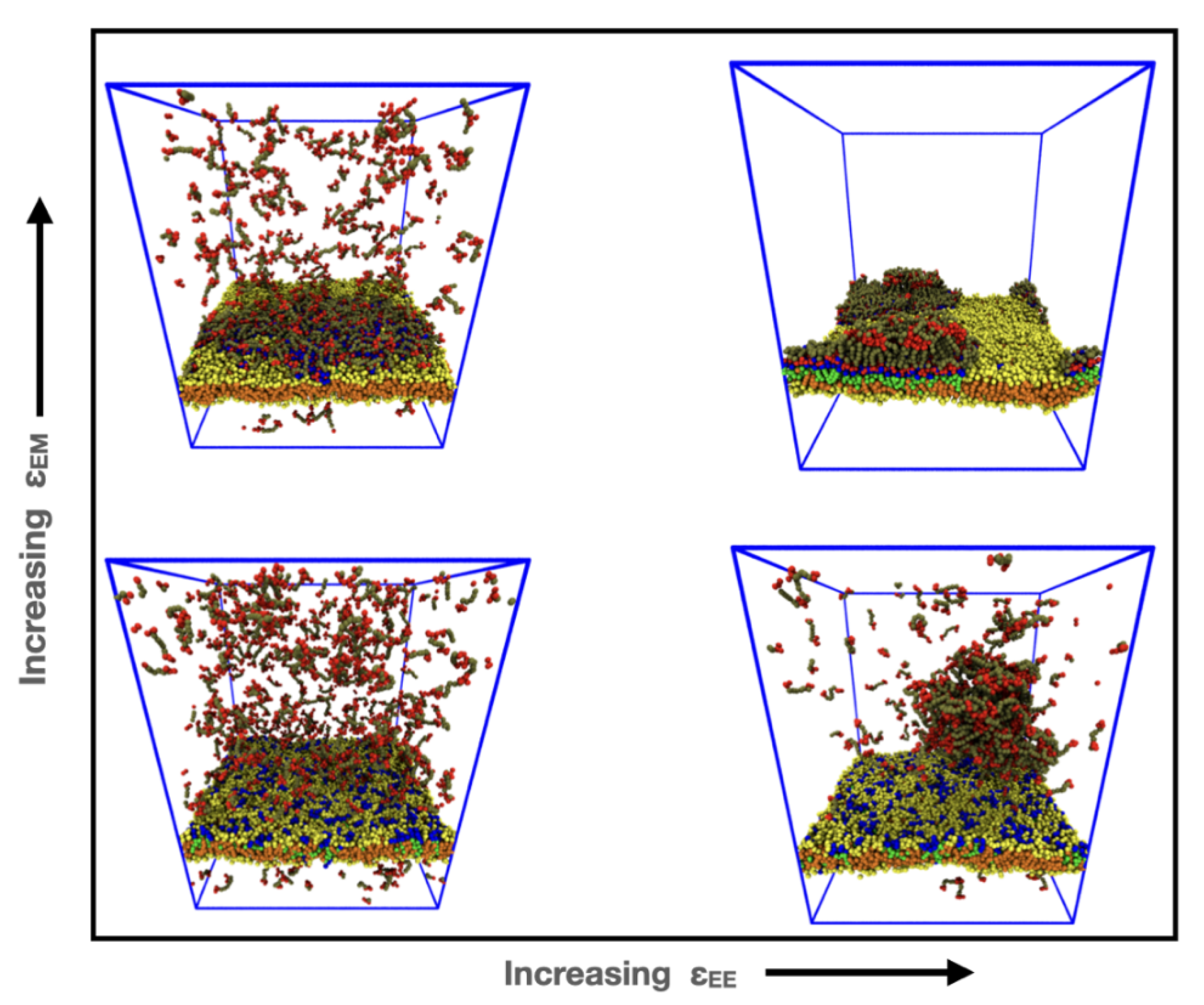
3.1. Influence of the IDP-Membrane Affinity on the Equilibrium Morphology
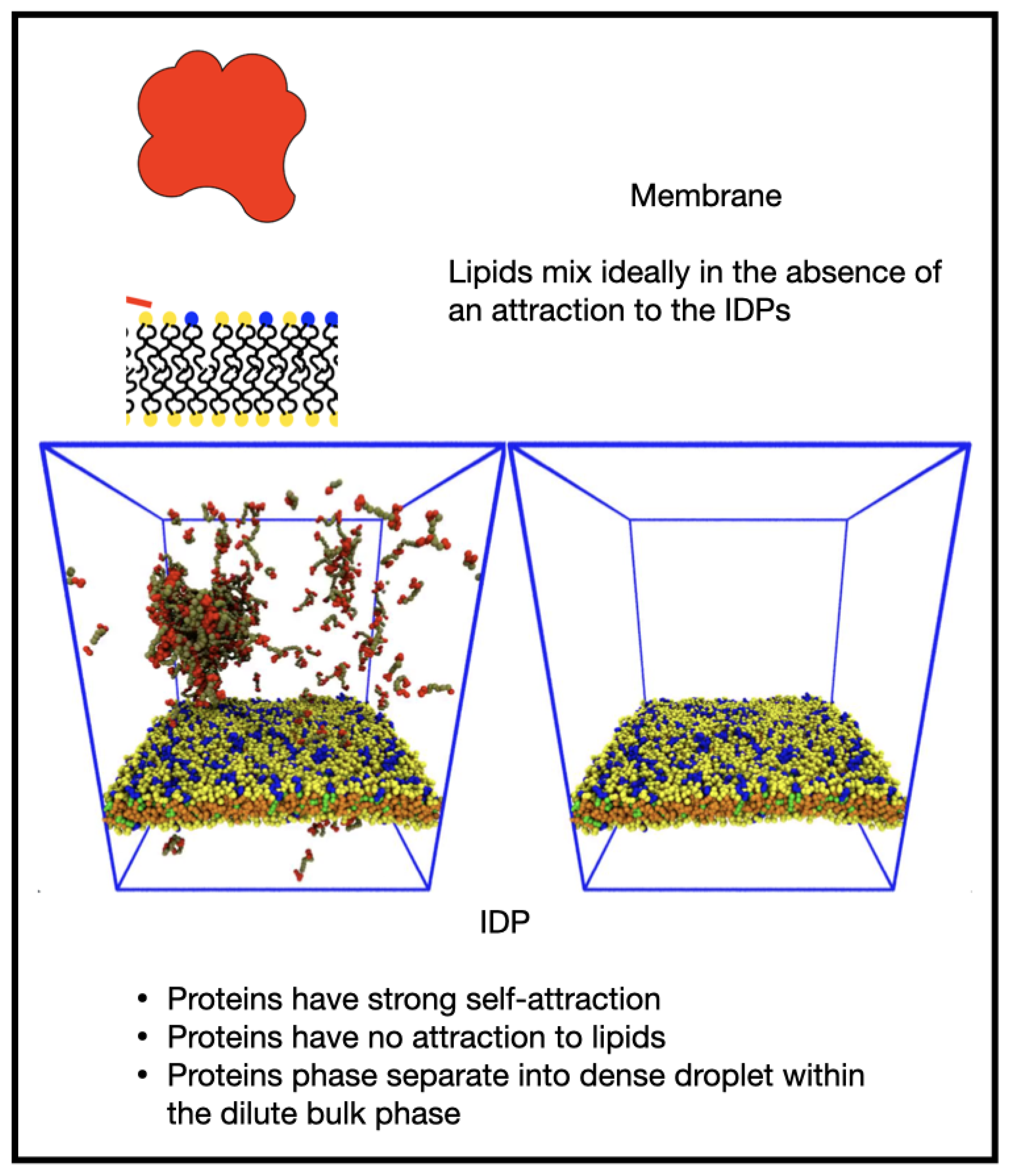
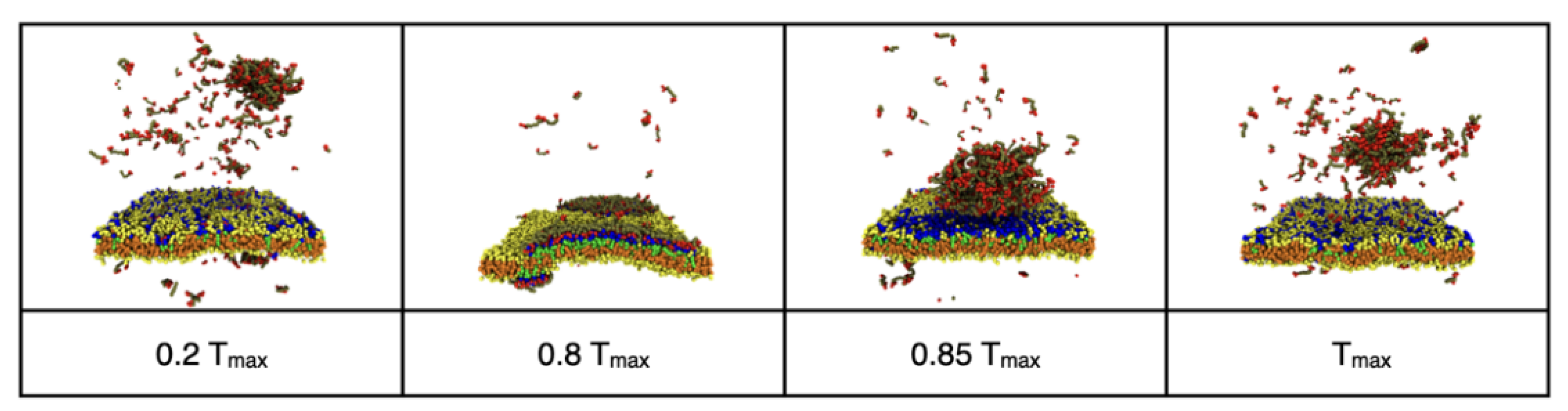
3.2. Reversible Coupling of Phase Separation and Domain Formation
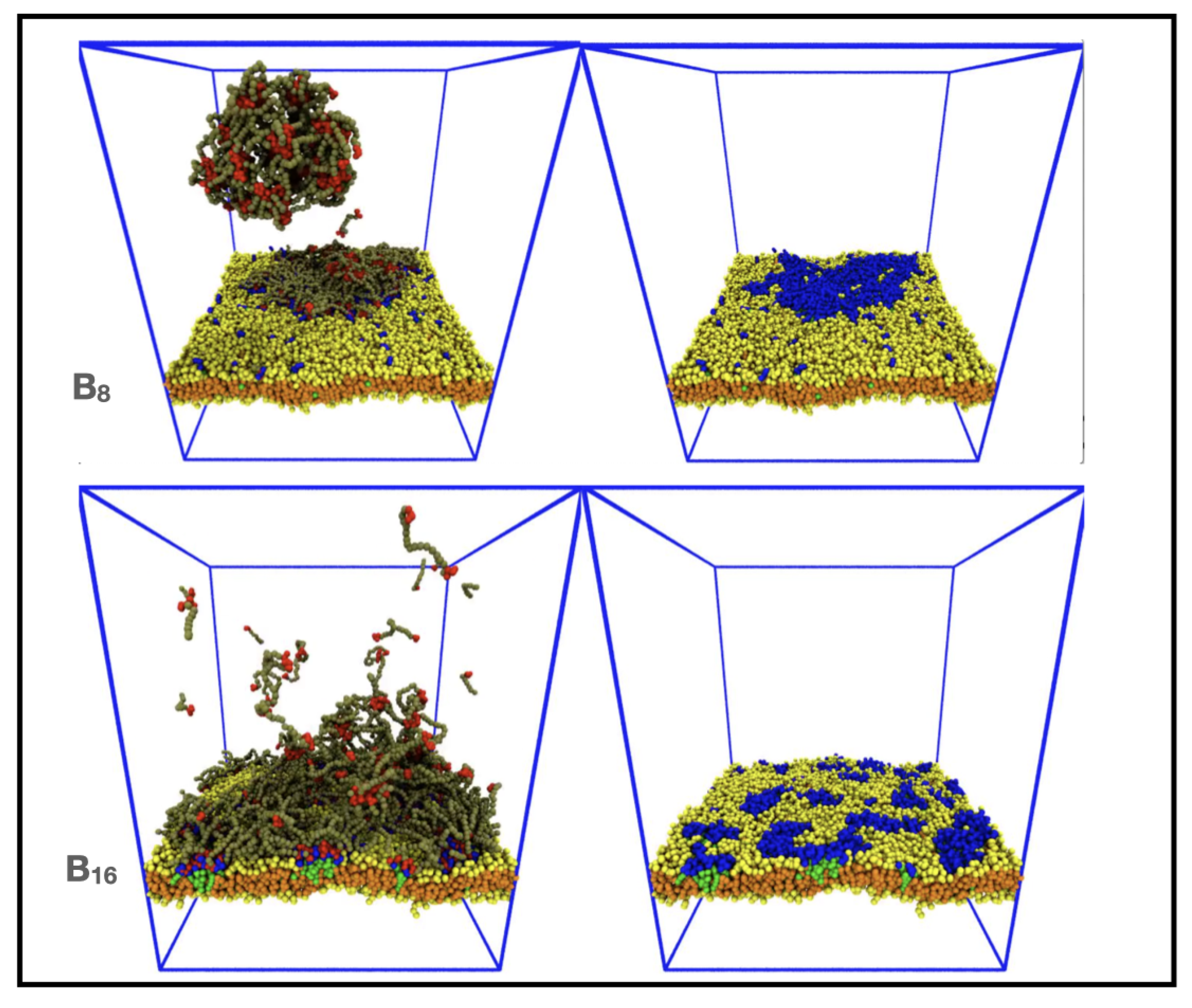
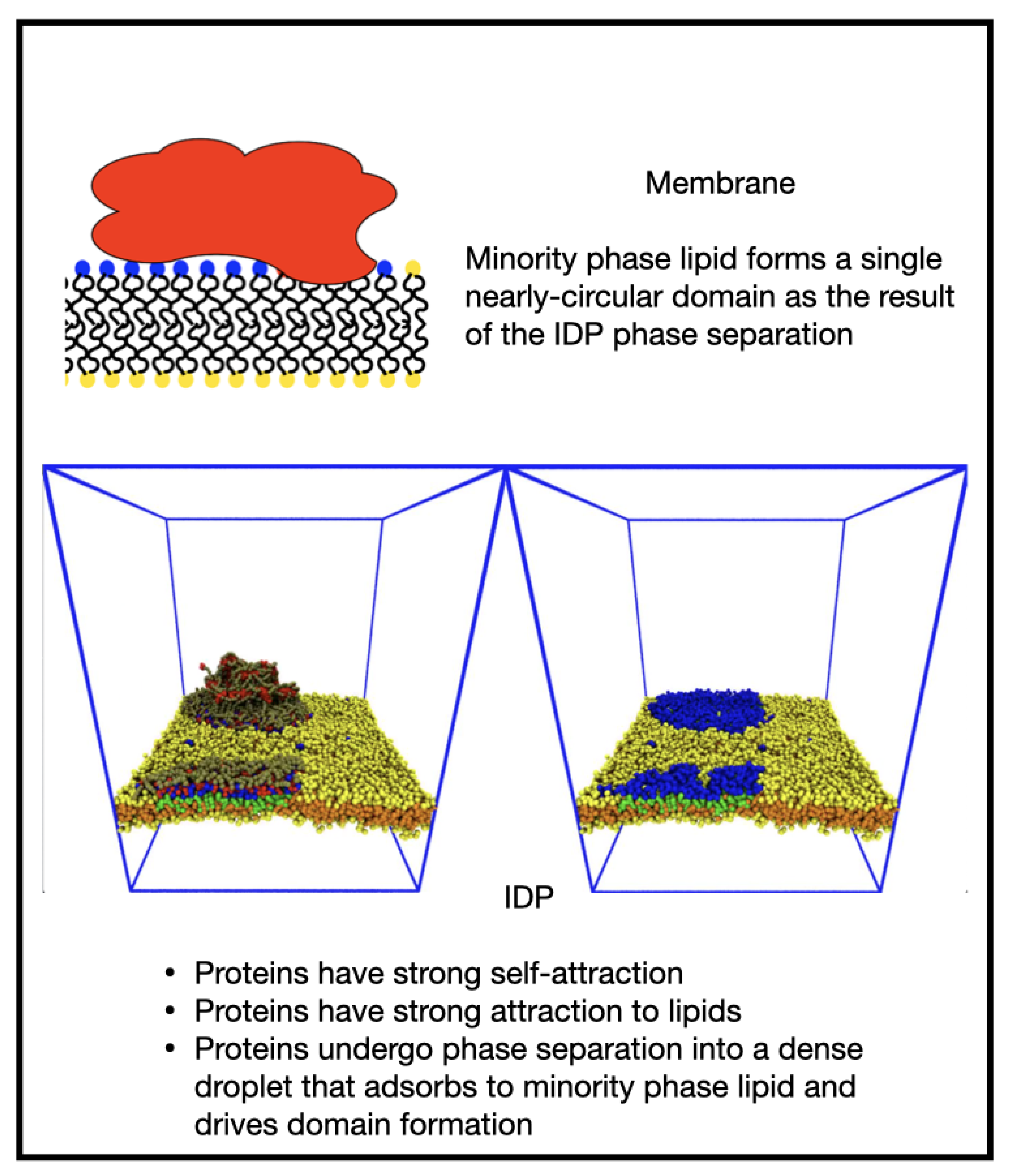
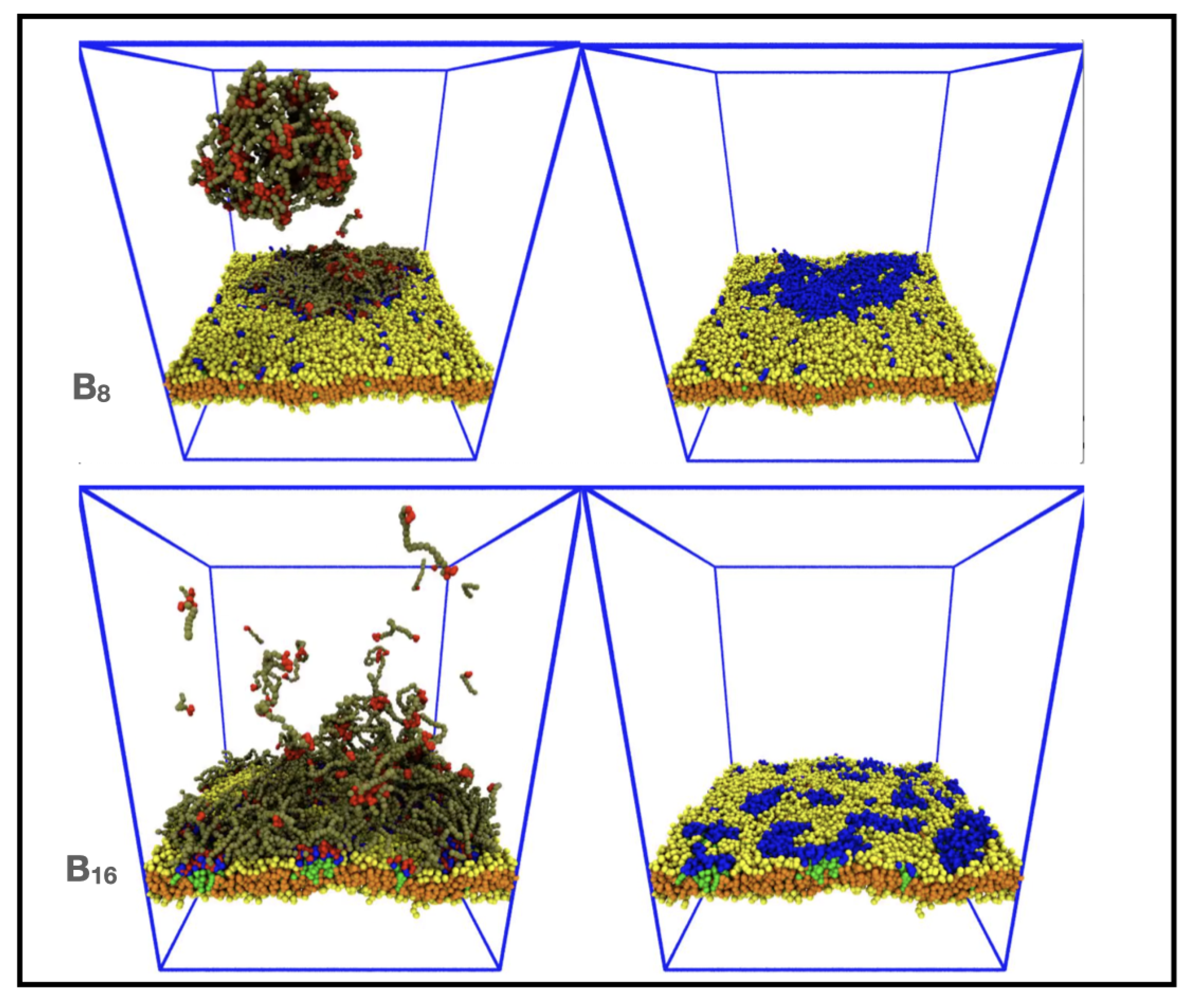
3.3. Increasing IDP Length Results in Patchy Domains
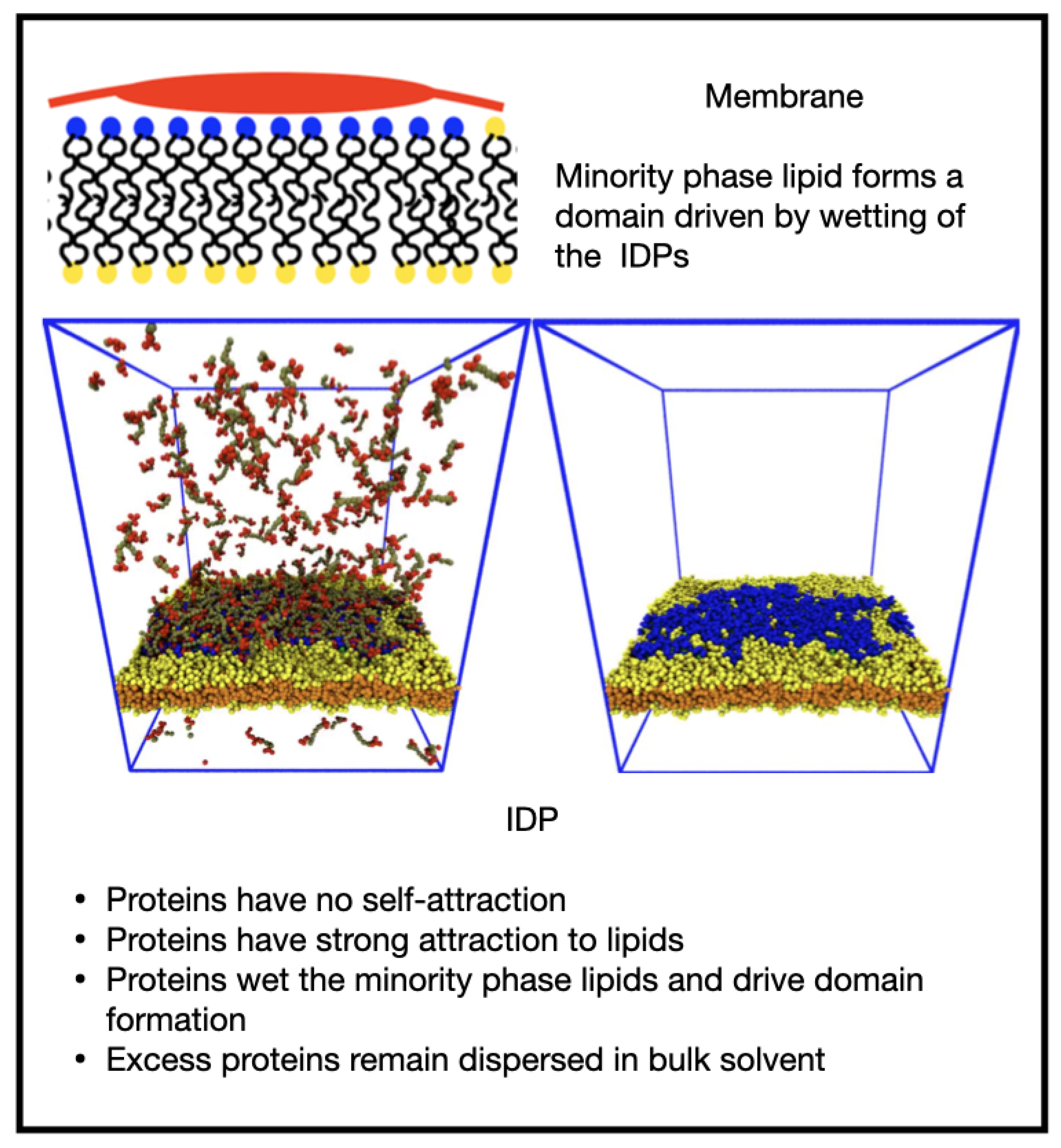
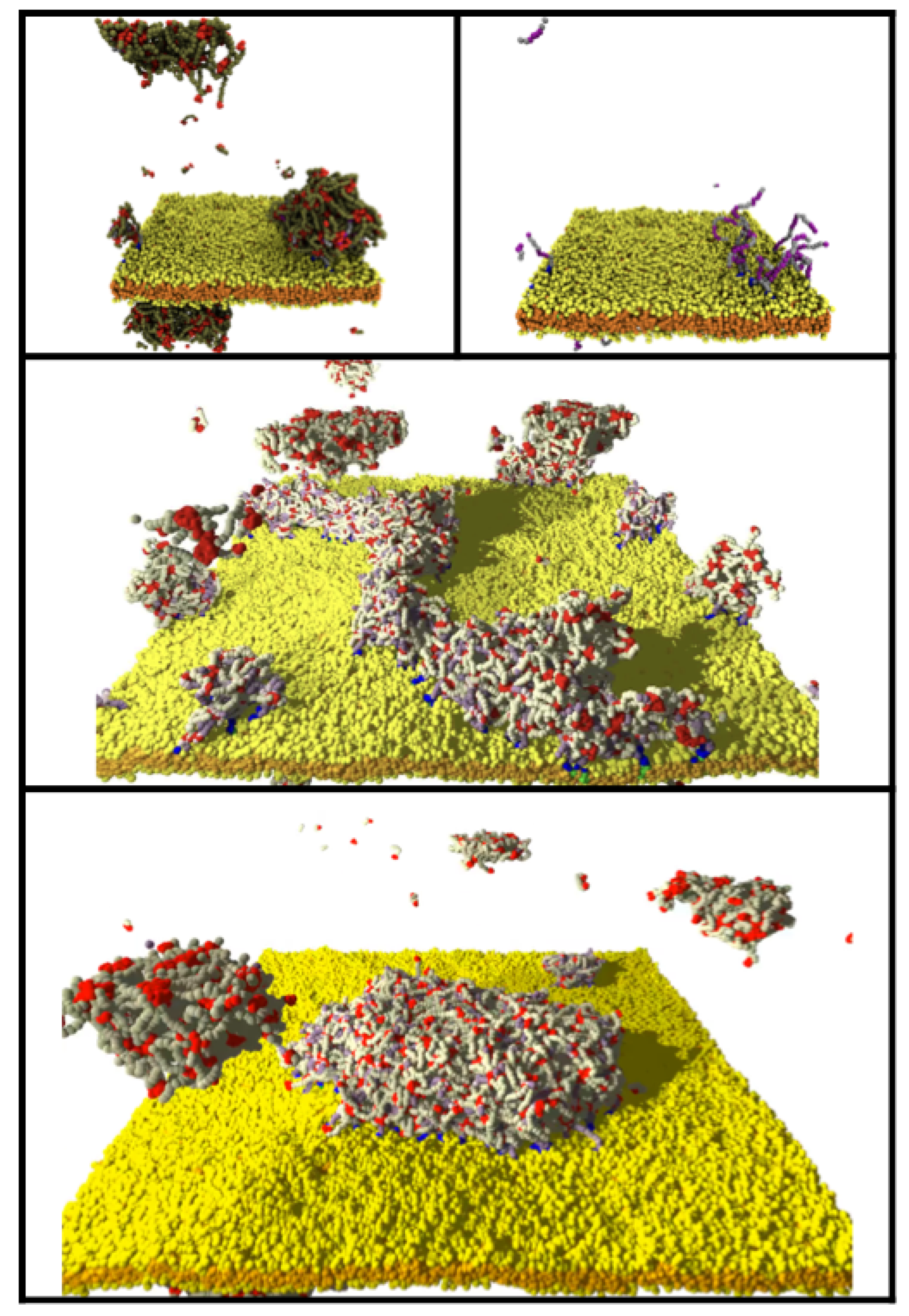
3.4. Clustering of Minority Lipids by IDP–Linker Attraction
4. Discussion
- -
- Produce reliable DPD results (agree with published DPD implementation) [53];
- -
- Are useful for practitioners;
- -
- Are competitive in performance with existing single-node systems.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell, 2nd ed.; Garland Publishing, Inc.: New York, NY, USA, 1989. [Google Scholar]
- Engelman, D.M. Membranes are more mosaic than fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef]
- Zeno, W.F.; Day, K.J.; Gordon, V.D.; Stachowiak, J.C. Principles and Applications of Biological Membrane Organization. Annu. Rev. Biophys. 2020, 49, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Lipowsky, R.; Brinkmann, M.; Dimova, R.; Haluska, C.; Kierfeld, J.; Shillcock, J. Wetting, budding, and fusion—Morphological transitions of soft surfaces. J. Phys. Condens. Matter 2005, 17, S2885. [Google Scholar] [CrossRef]
- Johannes, L.; Pezeshkian, W.; Ipsen, J.H.; Shillcock, J.C. Clustering on Membranes: Fluctuations and More. Trends Cell Biol. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Gao, H.; Arumugam, S.; Becken, U.; Bassereau, P.; Florent, J.-C.; Ipsen, J.H.; Johannes, L.; Shillcock, J.C. Mechanism of Shiga Toxin Clustering on Membranes. ACS Nano 2017, 11, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Römer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nature Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Bosch, L.V.D.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Mehta, S.; Zhang, J. Liquid–liquid phase separation: A principal organizer of the cell’s biochemical activity architecture. Trends Pharmacol. Sci. 2021, 42, 845–856. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer Physics of Intracellular Phase Transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Pappu, R.V. Functional Implications of Intracellular Phase Transitions. Biochemistry 2018, 57, 2415–2423. [Google Scholar] [CrossRef]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Chang, Y.-C.; Lee, D.S.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 2018, 175, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Wu, X.; Zhang, M. Presynaptic bouton compartmentalization and postsynaptic density-mediated glutamate receptor clustering via phase separation. Neuropharmacology 2021, 193, 108622. [Google Scholar] [CrossRef]
- Söding, J.; Zwicker, D.; Sohrabi-Jahromi, S.; Boehning, M.; Kirschbaum, J. Mechanisms for Active Regulation of Biomolecular Condensates. Trends Cell Biol. 2019, 30, 4–14. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Lowe, A.; Saibil, H.R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell 2018, 70, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Are Aberrant Phase Transitions a Driver of Cellular Aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell. 2021, 39, 174–192. [Google Scholar] [CrossRef]
- Spanni, S.; Tereshchenko, M.; Mastromarco, G.J.; Ihn, S.J.; Lee, H.O. Biomolecular condensates in neurodegeneration and cancer. Traffic 2019, 20, 890–911. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Akimitsu, N. Aberrant phase separation and cancer. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bartolucci, G.; Honigmann, A.; Jülicher, F.; Weber, C.A. Wetting and Prewetting Phase Transitions facilitated by Surface Binding. arXiv 2021, arXiv:2106.12565. [Google Scholar]
- Botterbusch, S.; Baumgart, T. Interactions between Phase-Separated Liquids and Membrane Surfaces. Appl. Sci. 2021, 11, 1288. [Google Scholar] [CrossRef] [PubMed]
- Kusumaatmaja, H.; May, A.I.; Knorr, R.L. Intracellular wetting mediates contacts between liquid compartments and membrane-bound organelles. J. Cell Biol. 2021, 220, e202103175. [Google Scholar] [CrossRef]
- Snead, W.T.; Gladfelter, A.S. The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell 2019, 76, 295–305. [Google Scholar] [CrossRef]
- Banjade, S.; Rosen, M.K. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife 2014, 3, e04123. [Google Scholar] [CrossRef]
- Jaqaman, K.; Ditlev, J.A. Biomolecular condensates in membrane receptor signaling. Curr. Opin. Cell Biol. 2021, 69, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Muller, J. Liquidity in immune cell signaling. Science 2016, 352, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.A.; Forman-Kay, J.D. Liquid–liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.K.; Huang, W.Y.; Carbone, C.B.; Nocka, L.M.; Parikh, A.N.; Vale, R.D.; Groves, J.T. Coupled membrane lipid miscibility and phosphotyrosine-driven protein condensation phase transitions. Biophys. J. 2020, 120, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Ji, Z.; Zhou, M.; Wu, J.; Lin, Y.; Qiao, Y. Membrane-confined liquid-liquid phase separation toward artificial organelles. Sci. Adv. 2021, 7, eabf9000. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Espagnol, P.; Warren, P.B. Statistical Mechanics of Dissipative Particle Dynamics. Europhys. Lett. 1995, 30, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Groot, R.D.; Warren, P.B. Dissipative Particle Dynamics: Bridging the Gap Between Atomistic and Mesoscopic Simulations. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Shillcock, J.; Lipowsky, R. Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys. 2002, 117, 5048–5061. [Google Scholar] [CrossRef] [Green Version]
- Laradji, M.; Kumar, P.B.S. Dynamics of Domain Growth in Self-Assembled Fluid Vesicles. Phys. Rev. Lett. 2004, 93, 198105. [Google Scholar] [CrossRef] [Green Version]
- Espagnol, P.; Warren, P.B. Perspective: Dissipative Particle Dynamics. J. Chem. Phys. 2017, 146, 150901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Q.; Colby, R.H. Dynamics of associative polymers. Soft Matter 2018, 14, 2961–2977. [Google Scholar] [CrossRef]
- Li, L.; Hu, J.; Shi, X.; Różycki, B.; Song, F. Interplay between cooperativity of intercellular receptor–ligand binding and coalescence of nanoscale lipid clusters in adhering membranes. Soft Matter 2021, 17, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkian, W.; Marrink, S.J. Simulating realistic membrane shapes. Curr. Opin. Cell Biol. 2021, 71, 103–111. [Google Scholar] [CrossRef]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale Simulations of Biological Membranes: The Challenge To Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezeshkian, W.; König, M.; Wassenaar, T.A.; Marrink, S.J. Backmapping triangulated surfaces to coarse-grained membrane models. Nat. Commun. 2020, 11, 2296. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Karniadakis, G.E.; Kirby, R.M.I. Parallel Scientific Computing in C++ and MPI; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Naylor, M.; Moore, S.W.; Thomas, D. Tinsel: A Manythread Overlay for FPGA Clusters. In Proceedings of the 29th International Conference on Field Programmable Logic and Applications (FPL), Barcelona, Spain, 8–12 September 2019; pp. 375–383. [Google Scholar]
- Shillcock, J.C. OSPREY-DPD. Open Source Polymer Research Engine—Dissipative Particle Dynamics. 2020. Available online: https://github.com/Osprey-DPD/osprey-dpd (accessed on 18 December 2021).
- Beaumont, J.R.; Brown, A.D.; Thomas, D.B.; Shillcock, J.C.; Naylor, M.F.; Bragg, G.M.; Vousden, M.L.; Moore, S.W.; Fleming, S.T. An event-driven approach to Dissipative Particle Dynamics. ACM Trans. Parallel Comput. (not published). 2021. [Google Scholar]
- Shillcock, J.C.; Brochut, M.; Chénais, E.; Ipsen, J.H. Phase behaviour and structure of a model biomolecular condensate. Soft Matter. 2020, 16, 6413–6423. [Google Scholar] [CrossRef]
- Venturoli, M.; Sperotto, M.M.; Kranenburg, M.; Smit, B. Mesoscopic Models of Biological Membranes. Phys. Rep. 2006, 437, 1–54. [Google Scholar] [CrossRef] [Green Version]
- Laradji, M.; Sunil Kumar, P.B. Domain growth, budding, and fission in phase-separating self-assembled fluid bilayers. J. Chem. Phys. 2005, 123, 224902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illya, G.; Lipowsky, R.; Shillcock, J.C. Two-component membrane material properties and domain formation from dissipative particle dynamics. J. Chem. Phys. 2006, 125, 114710. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Jasnow, D.; Balazs, A.C. Designing synthetic vesicles that engulf nanoscopic particles. J. Chem. Phys. 2007, 127, 084703. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Ma, Y.-Q. Computer simulation of the translocation of nanoparticles with different shapes across a lipid bilayer. Nat. Nanotechnol. 2010, 5, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Grafmuller, A.; Shillcock, J.; Lipowsky, R. The fusion of membranes and vesicles: Pathway and energy barriers from dissipative particle dynamics. Biophys. J. 2009, 96, 2658–2675. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Vousden, M.; Rast, A.; Bragg, G.M.; Thomas, D.; Beauont, J.; Naylor, M.; Mokhov, A. POETS: Distributed event-based computing—Scaling behaviour. In Proceedings of the International Conference on Parallel Computing, Prague, Czech Republic, 10–13 September 2019. [Google Scholar]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Chen, X.; Guan, D.; Xu, J.; Wu, H.; Tong, P.; Zhang, M. Reconstituted Postsynaptic Density as a Molecular Patform for Understanding Synapse Formation and Plasticity. Cell 2018, 174, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Cai, Q.; Shen, Z.; Chen, X.; Zeng, M.; Du, S.; Zhang, M. RIM and RIM-BP Form Presynaptic Active-Zone-like Condensates via Phase Separation. Mol. Cell 2019, 73, 971–984. [Google Scholar] [CrossRef] [Green Version]
- Castagna, J.; Guo, X.; Seaton, M.; O’Cais, A. Towards extreme scale dissipative particle dynamics simulations using multiple GPGPUs. Comput. Phys. Commun. 2020, 251, 107159. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol Graphics. 1996, 14, 33–38. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W | E | B | HA | TA | HB | TB | L | S | |
|---|---|---|---|---|---|---|---|---|---|
| W | 25 | ||||||||
| E | 25 | aEE | |||||||
| B | 23 | 25 | 25 | ||||||
| HA | 30 | 30 | 30 | 30 | |||||
| TA | 75 | 35 | 35 | 35 | 10 | ||||
| HB | 30 | aEM | 30 | 30 | 35 | 30 | |||
| TB | 75 | 35 | 35 | 35 | 10 | 35 | 10 | ||
| L | 30 | 30 | 30 | 30 | 35 | 30 | 35 | 30 | |
| S | 30 | aES | 30 | 30 | 35 | 30 | 35 | 30 | 30 |
| System | Year | Software | Watts | Nodes | Perf/106 | Perf/Watt |
|---|---|---|---|---|---|---|
| CPUx1 | 2020 | Osprey-DPD | 250 | 1 | 0.5 | 2000 |
| CPUx32 | 2020 | LAMMPS | 500 | 1 | 19.0 | 38,000 |
| GPUx1 | 2016 | DL-MESO | 400 | 1 | 23.7 | 59,195 |
| GPUx8 | 2016 | DL-MESO | 400 | 32 | 285.7 | 22,321 |
| POETS Gen1 | 2011 | POETS-DPD | 200 | 48 | 24.0 | 2500 |
| POETS Gen2 | 2022 | POETS-DPD | 250 | 48 | 360.0 | 30,000 |
| Target parameters for next generation POETS system: | ||||||
| POETS Gen3 | 2025 | POETS-DPD | 250 | 48 | 3600.0 | 300,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shillcock, J.C.; Thomas, D.B.; Beaumont, J.R.; Bragg, G.M.; Vousden, M.L.; Brown, A.D. Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric. Membranes 2022, 12, 17. https://doi.org/10.3390/membranes12010017
Shillcock JC, Thomas DB, Beaumont JR, Bragg GM, Vousden ML, Brown AD. Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric. Membranes. 2022; 12(1):17. https://doi.org/10.3390/membranes12010017
Chicago/Turabian StyleShillcock, Julian C., David B. Thomas, Jonathan R. Beaumont, Graeme M. Bragg, Mark L. Vousden, and Andrew D. Brown. 2022. "Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric" Membranes 12, no. 1: 17. https://doi.org/10.3390/membranes12010017
APA StyleShillcock, J. C., Thomas, D. B., Beaumont, J. R., Bragg, G. M., Vousden, M. L., & Brown, A. D. (2022). Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric. Membranes, 12(1), 17. https://doi.org/10.3390/membranes12010017