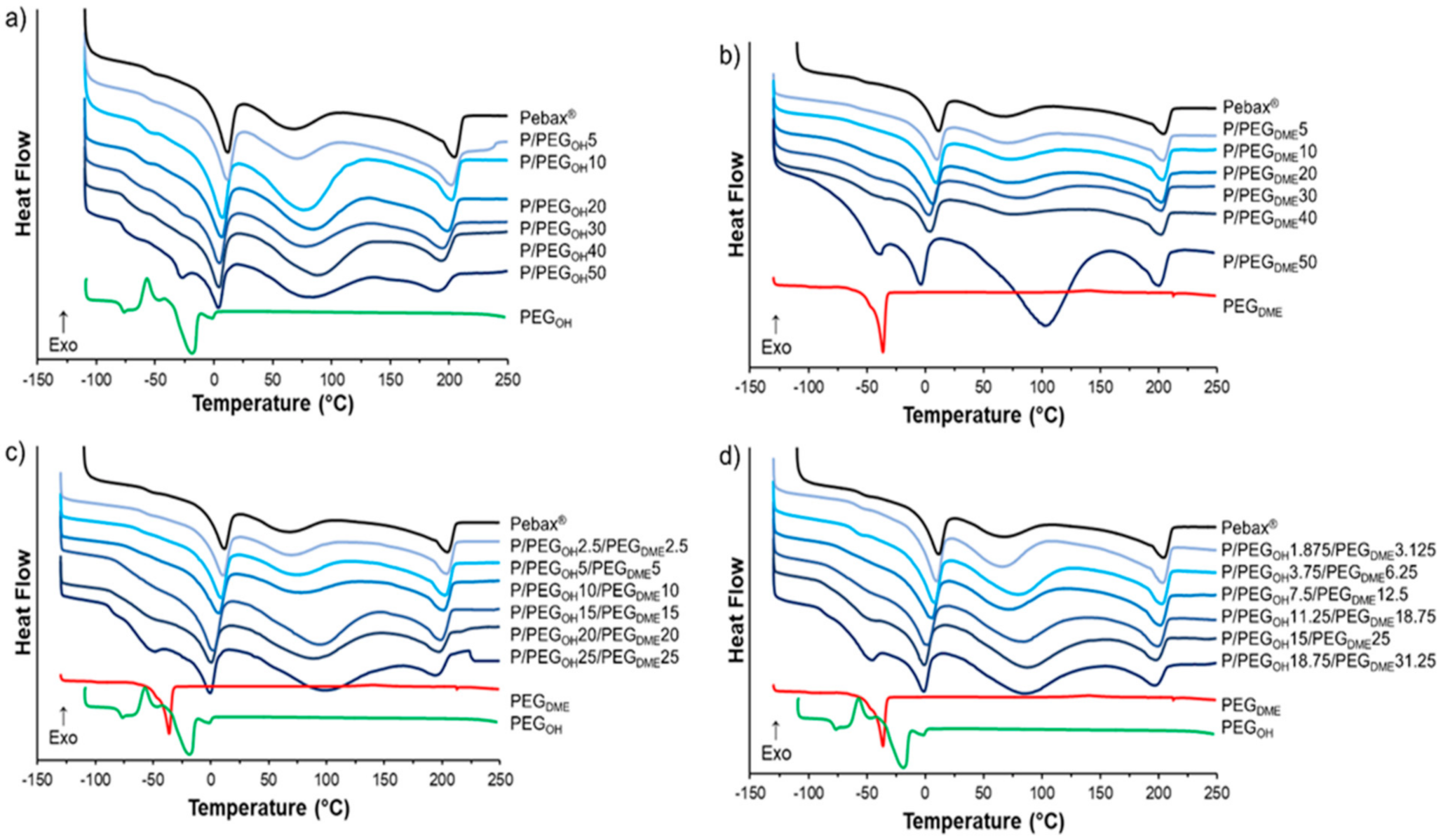
3.1. Microstructure Analysis
Thermal properties of the films composed of binary Pebax
®/PEGOH and Pebax
®/PEGDME as well as ternary Pebax
®/PEGOH/PEGDME blends were studied by DSC analysis with the aim of obtaining information about the miscibility of the PEG additives with the Pebax
® matrix. The thermograms corresponding to the first heating run of the blended Pebax
®/PEG films, the pristine Pebax
®, and the PEG additives are presented in
Figure 2.
The thermogram of the pristine Pebax
® exhibits a heat flow (heat capacity) jump at −56 °C corresponding to the glass transition temperature of the soft PE segments. Then, three endothermic peaks were observed. The first peak and the last peak, with values at the peak apex of around 11 °C and 205 °C, were attributed to the melting of the PE and PA crystalline phases, respectively, in agreement with the characteristic separated microphase structure found in block copolymers, such as poly(ether-bloc-amide) [
17]. The third broad peak between 50 °C and 125 °C was observed only for the first heating run. It could be attributed to the significant amount of water sorbed during the sample’s storage under ambient conditions due to the presence of polar chemical groups within both Pebax
® blocks [
29]. It has to be noticed that the glass transition of the PA segments was not detected in the thermograms in agreement with the low amount of PA amorphous phase in the Pebax
® MH1657 and the difficulty of determining the glass transition of PA by DSC analysis [
17]. The thermogram of PEG
OH exhibited a glass transition at −77 °C, followed by a cold crystallization phenomenon from −66 °C to −47 °C and a melting peak centered at −20 °C. For PEG
DME, the glass transition was observed at −100 °C followed by a melting peak centered at −36 °C. The obtained DSC data for the neat Pebax
® and the PEG additives were in good agreement with those reported in the literature [
17,
23,
24,
30].
The thermal transitions of the films based on the binary and ternary blends were analyzed with respect to those of Pebax
® and PEG references. Regardless of the additive, for additive content inferior to 30 wt.%, the thermograms of all blends were similar to the one obtained for the neat Pebax
®. No thermal transition due to the presence of the additive was observed. Above 30 wt.% of additive, a melting peak of the additive, which increased as the content of the additive increased, appeared at around −25 and −45 °C for the binary Pebax
®/PEG
OH and Pebax
®/PEG
DME blends, respectively (see
Figure 2a,b). Likewise, regarding both ternary blends, a shoulder appeared at around −25 °C corresponding to the melting point of the PEG
DME additive (see
Figure 2c,d).
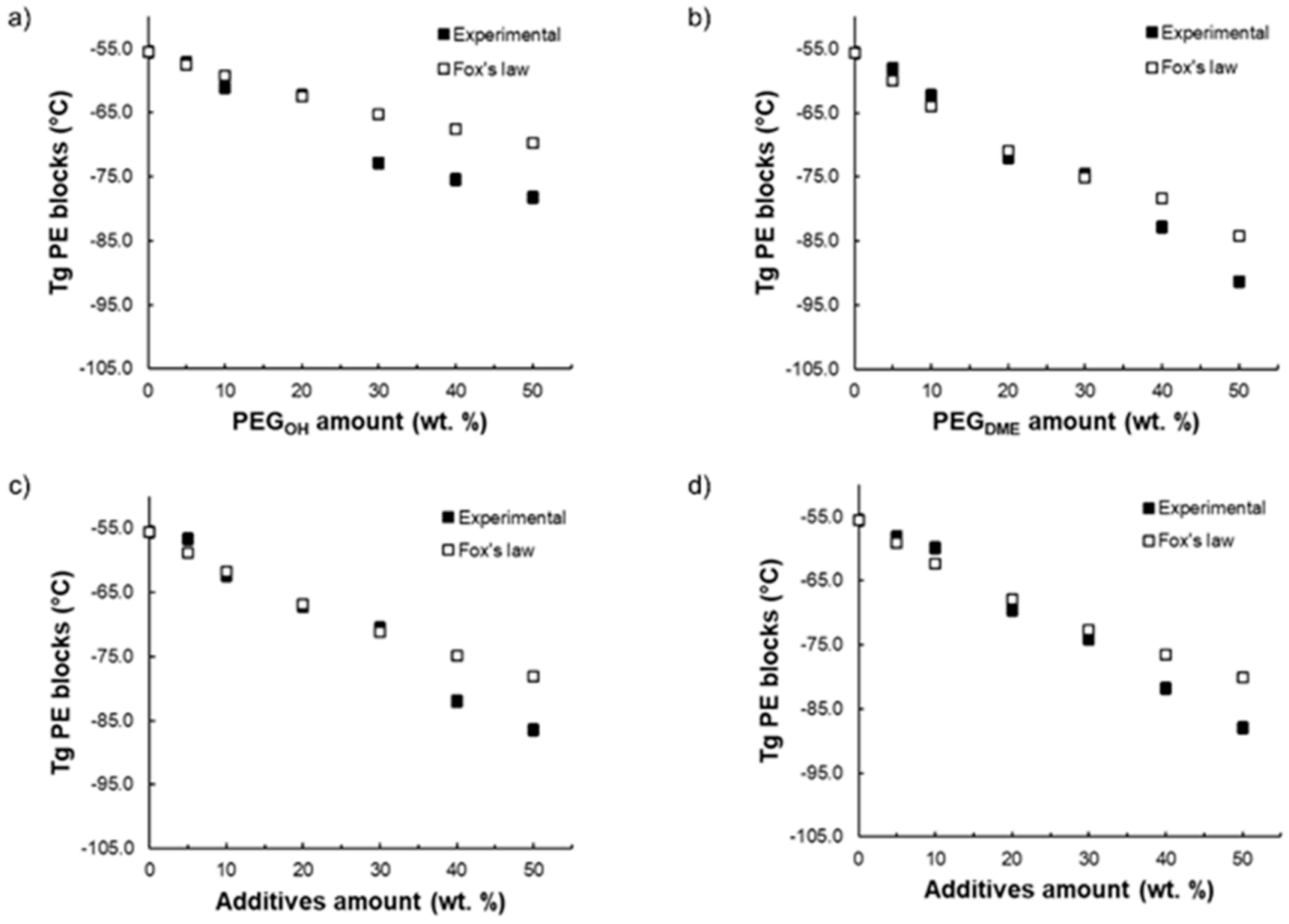
A partial miscibility of the additives within the copolymer matrix is to be expected since the chemical structures of both PEG additives and polyether blocks are similar. Thus, to confirm the miscibility degree of the PEG additives with the Pebax
® soft phase, the experimental
Tg values were compared with the glass transition temperature values calculated using Fox’s law (see
Figure 3). This law is often used to study the ability of two components to lead to a homogeneous blend:
where
Tg and
Tg,i are the glass transition temperature of the blend and the neat components, respectively.
is the weight fraction of each component
i. This law has already been used in numerous studies to discuss the miscibility of different additives within Pebax
® [
31,
32].
Figure 3 shows the evolution of the measured
Tg of the PE blocks for each blend as a function of the PEG additive amount and compares it to the theoretical
Tg calculated from Equation (4). For the binary Pebax
®/PEG
OH and Pebax
®/PEG
DME blends, respectively, a good agreement between the experimental and calculated
Tg values was obtained for a PEG
OH amount up to 20 wt.% (
Figure 3a) and a PEG
DME amount up to 30 wt.% (
Figure 3b). Thus, in these ranges of composition, the PEG additives were mainly dissolved within the polyether phase of Pebax
®. For higher additive contents, the experimental and theoretical
Tg values differed, showing the occurrence of a phase separation. Surprisingly, the measured
Tg values were lower than those calculated from Fox’s law. This result suggests that the
Tg values that were evidenced were the signature of the separated rich PEG additive phase. Likewise, for both ternary blends (see
Figure 3c,d), a good agreement between the experimental and calculated
Tg was obtained for PEG additive amounts up to 30 wt.% suggesting, here again, that the PEG additives were dissolved within the polyether phase of Pebax
® in this range of composition. Furthermore, it should be noted that, for the same Pebax
®/additive ratio, the
Tg value was lower when the blend contained a higher PEG
DME amount. This result is in agreement with the lower
Tg of the PEG
DME additive with respect to PEG
OH (see
Figure 3).
For all binary and ternary blends, it can be observed that the melting temperature of the PE segments (T
mPE) decreased as the amount of additive increased (see data in the
Supplementary Materials). This shift in T
mPE was more pronounced for the blends containing the PEG
DME additive. Indeed, the T
mPE varied from 11 °C for pristine Pebax
® to around 3 °C for Pebax
®/PEG
OH, to −3 °C for Pebax
®/PEG
DME, and to −1 °C for the ternary Pebax
®/PEG
OH/PEG
DME blends over the studied composition range. Three reasons could explain this trend: a contribution of the additive melting peak to the melting peak of the polyether block; a decrease in the size of the polyether block’s crystalline lamellas; and the formation of defects within the polyether crystalline domains. It can also be noticed that the enthalpy value related to this melting peak, denoted ∆H
mPE, first slightly increased for additive contents up to 20 wt.% and then decreased for higher contents (see data in the
Supplementary Materials). Thus, at a low PEG content, it seemed that the PEG species could contribute to the melting peak whereas at higher PEG contents, when a specific melting peak of the PEG species was detected, the enthalpy related to the melting peak of PE was mainly governed by the proportion of PE within the Pebax
®/PEG blend.
Likewise, the melting temperature of the PA blocks (T
mPA) for all binary and ternary blends decreased with the increase in the additive amount, suggesting that the size of the PA crystalline lamellas decreased or that the formation of defects was favored (see data in the
Supplementary Materials). It can also be noted that the melting enthalpy of the polyamide blocks (∆H
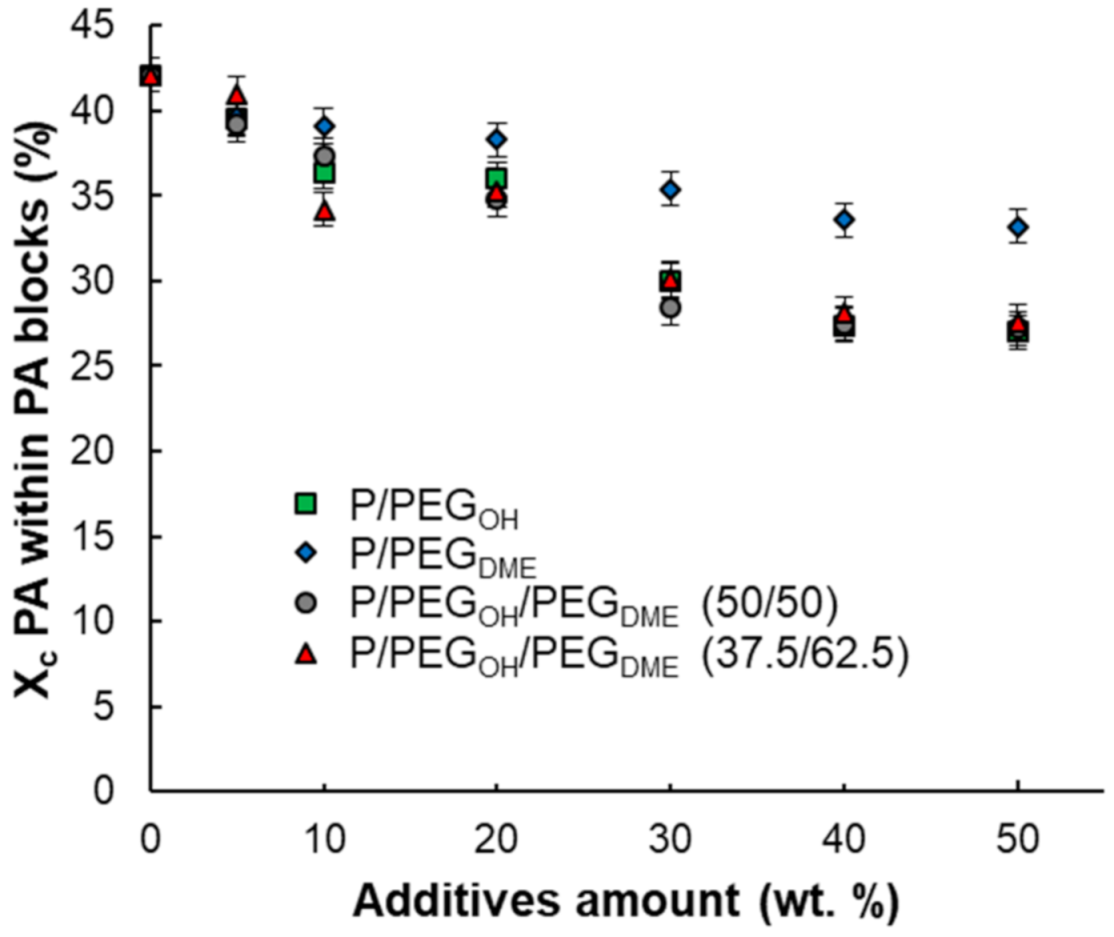
mPA) decreased with the increase in the PEG amount, which is consistent with the fact that the PA proportion decreased in the blend. In order to better assess the impact of the PEG additives on the PA crystallinity, the degree of PA crystallinity within PA blocks was calculated for each formulation and plotted as a function of the additive amount. As shown in
Figure 4, for all blends, the PA crystallinity within PA blocks decreased as the additive amount increased, leading to a system with a higher amount of content with an amorphous structure. The degree of PA crystallinity within the PA blocks remained higher for the binary Pebax
®/PEG
DME blends compared with the Pebax
®/PEG
OH blends. Moreover, the degree of PA crystallinity within the PA blocks was similar for the binary Pebax
®/PEG
OH blends and the ternary blends, suggesting that the PEG
OH additive played a major role in the ternary blends (see in
Figure 4).
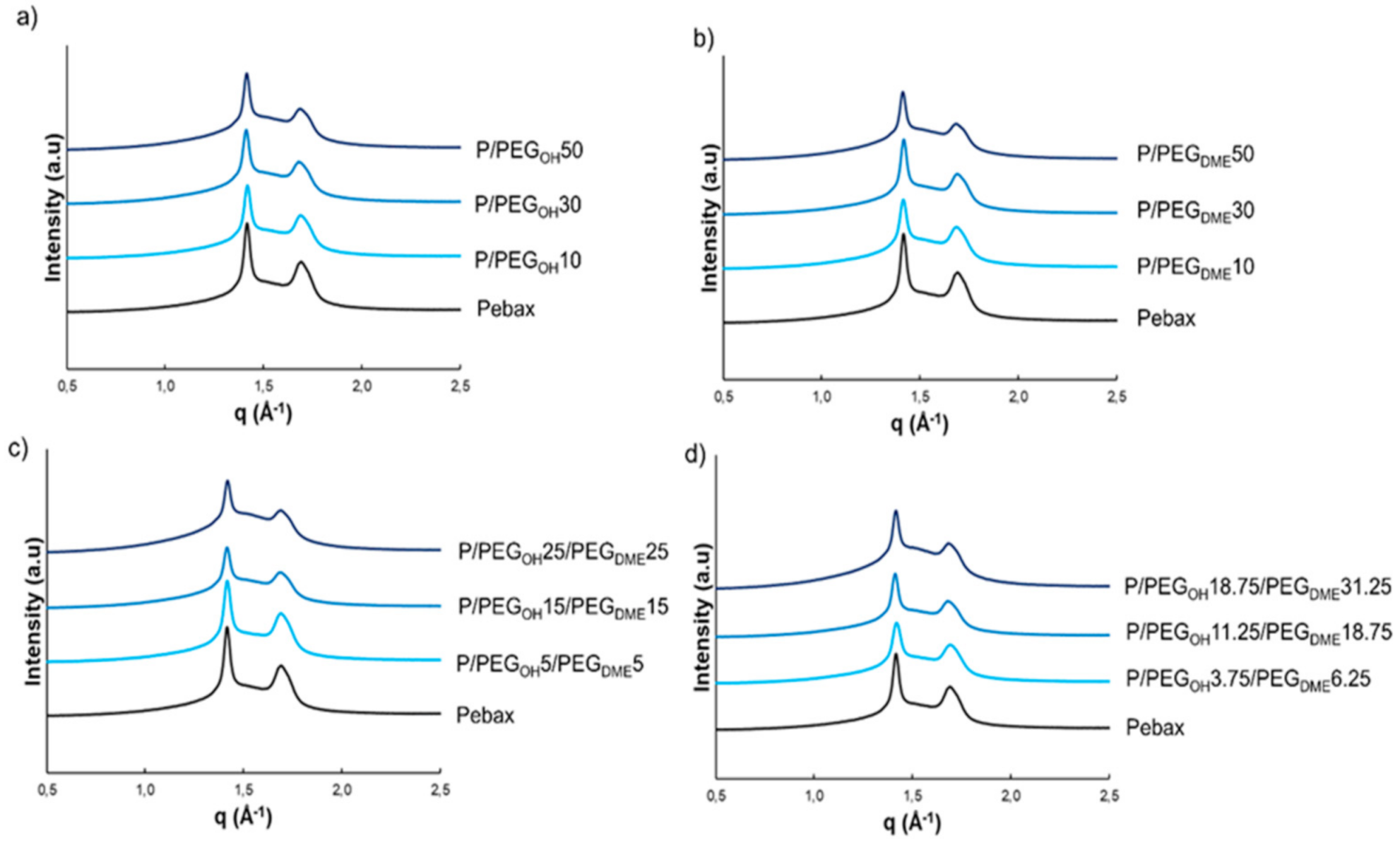
WAXS analysis was performed as a complementary method in order to obtain more information about the nature of the crystalline phases present at room temperature in the Pebax
® matrix and the binary and ternary blends.
Figure 5 displays the WAXS patterns of the studied systems.
WAXS patterns of the pristine Pebax
® and of the binary and ternary Pebax
®-based blends exhibited an amorphous halo and two reflection peaks centered at
q = 1.41 and 1.69 Å
−1. These peaks could be assigned to the
α form (monoclinic) of the PA crystalline phase of the Pebax
® copolymer in agreement with the WAXS analysis results obtained on Nylon 6 by Wu and coworkers [
33]. Moreover, these two peaks remained at the same
q-position for all blends, regardless of the nature of the additive or the composition (see
Figure 5). No diffraction peak related to a PEO-type crystalline microstructure was observed on WAXS patterns of the pristine Pebax
® as well as binary and ternary blends. Indeed, these peaks should be observed at
q = 1.35 and 1.64 Å
−1 according to the literature [
34,
35]. This confirmed that PE segments of Pebax
® and PEG additives were in the molten state at 23 °C, which is in accordance with the preliminary results obtained by DSC and the fact that the crystallinity at 23 °C (
Xc,23°C) resulted only from the PA segments. Moreover, it can be noted that the intensity of the diffraction peaks decreased as the PEG additive amount increased.
Xc,23°C was calculated according to the following equation:
where
Ac is the area of both peaks and
Aa the area attributed to the amorphous halo.
The obtained
Xc,23°C values are reported in
Table 1 and compared to the values calculated from the DSC experiments, taking into account only the contribution of the PA phase. The
Xc,23°C calculated from both WAXS and DSC decreased with the increase in the PEG additive amount. However, it should be noted that there were differences between the degrees of crystallinity obtained by the two methods. Such trends have already been observed in the literature [
36] and could be explained by the uncertainty related to the establishment of the amorphous halo for the WAXS analysis and the difficulty of detecting the melting of small crystalline regions using the DSC apparatus, which often lead to an underestimation of the degree of crystallinity determined by DSC with respect to WAXS [
37].
Both sets of analysis results reveal that the evolution of Xc,23°C was in the same order of magnitude as a function of the additive amount, regardless of the nature of the additive.
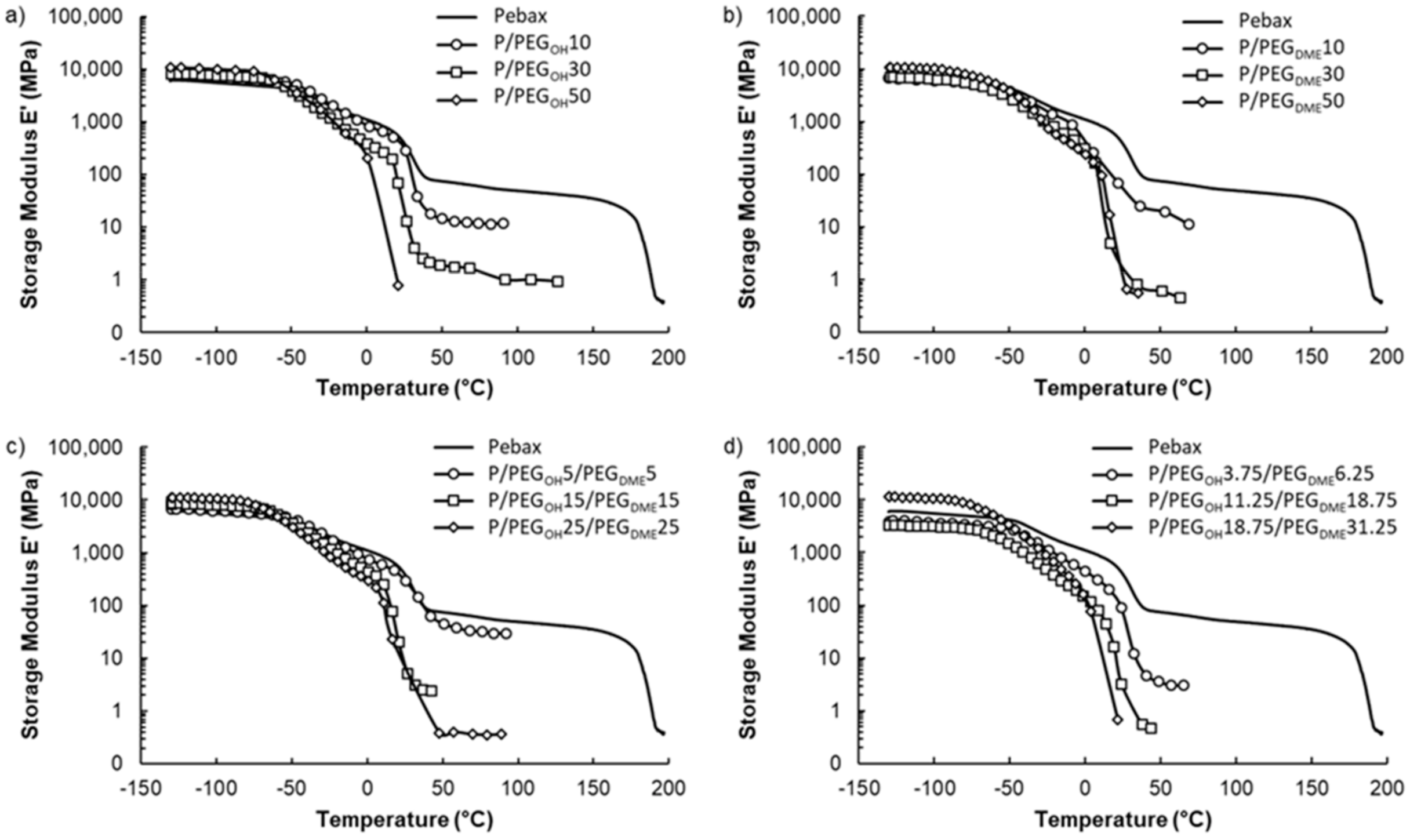
3.3. Thermal–Mechanical Properties
The thermal–mechanical properties of blended Pebax
® films were investigated by measuring their temperature-dependent storage modulus (
E’) in the temperature range from −130 to 200 °C. The evolution of
E’ as a function of temperature is displayed in
Figure 8 for each studied system.
Four domains were visible on the storage modulus curve of the pristine Pebax
® film. For temperatures inferior to −50 °C, the Pebax
® copolymer was in the glassy state. From −50 °C, the significant gain in mobility of the amorphous polyether segments resulted in a sharp decrease in the storage modulus, which was due to the alpha transition of the PE block of the copolymer. Thereafter, an even more pronounced second decrease in
E’ was observed between 15 °C and 40 °C, which was attributed to the melting of the polyether crystalline phases. Finally, a plateau only depending on the rigid polyamide phase of the Pebax
® was observed until the PA crystalline phase melted. This thermo-mechanical behavior is typical of the Pebax
® materials family [
39].
For all studied film series, the thermo-mechanical properties evolved in a similar way as a function of the additive amount, independently of the PEG chain end’s nature. Indeed, the storage modulus values measured on the glassy plateau (between −130 and around −50 °C) were of the same order of magnitude (around 9 GPa) and did not vary significantly as a function of the additive structure and amount. However, a shift in the first drop in the storage modulus towards lower temperatures occurred as the PEG content increased. This behavior was in agreement with the decrease in the glass transition temperature of the polyether blocks evidenced by the DSC analysis and can be explained by the plasticization of the Pebax® polyether chains. The second sharp decrease in the storage modulus was also shifted toward lower temperatures with the PEG amount. This behavior can be assigned to the decrease in the melting temperature of the polyether crystalline phases as shown by DSC. Finally, it has to be noticed that the storage modulus values at the plateau (above 50 °C) were considerably decreased with the PEG amount. This result can be attributed to the presence of these low-molar-mass additives, which have poor mechanical properties, and also to the decrease in the amide phase proportion in the total blend, which contributes to the mechanical resistance properties of the film. As a consequence, the plateau range was narrowed. It was even no longer observable for high additive contents due to the complete collapse of the film. Thus, adding a PEG amount above 30 wt.% appeared to be detrimental to the mechanical properties of the films regardless of the nature of the PEG chain ends.
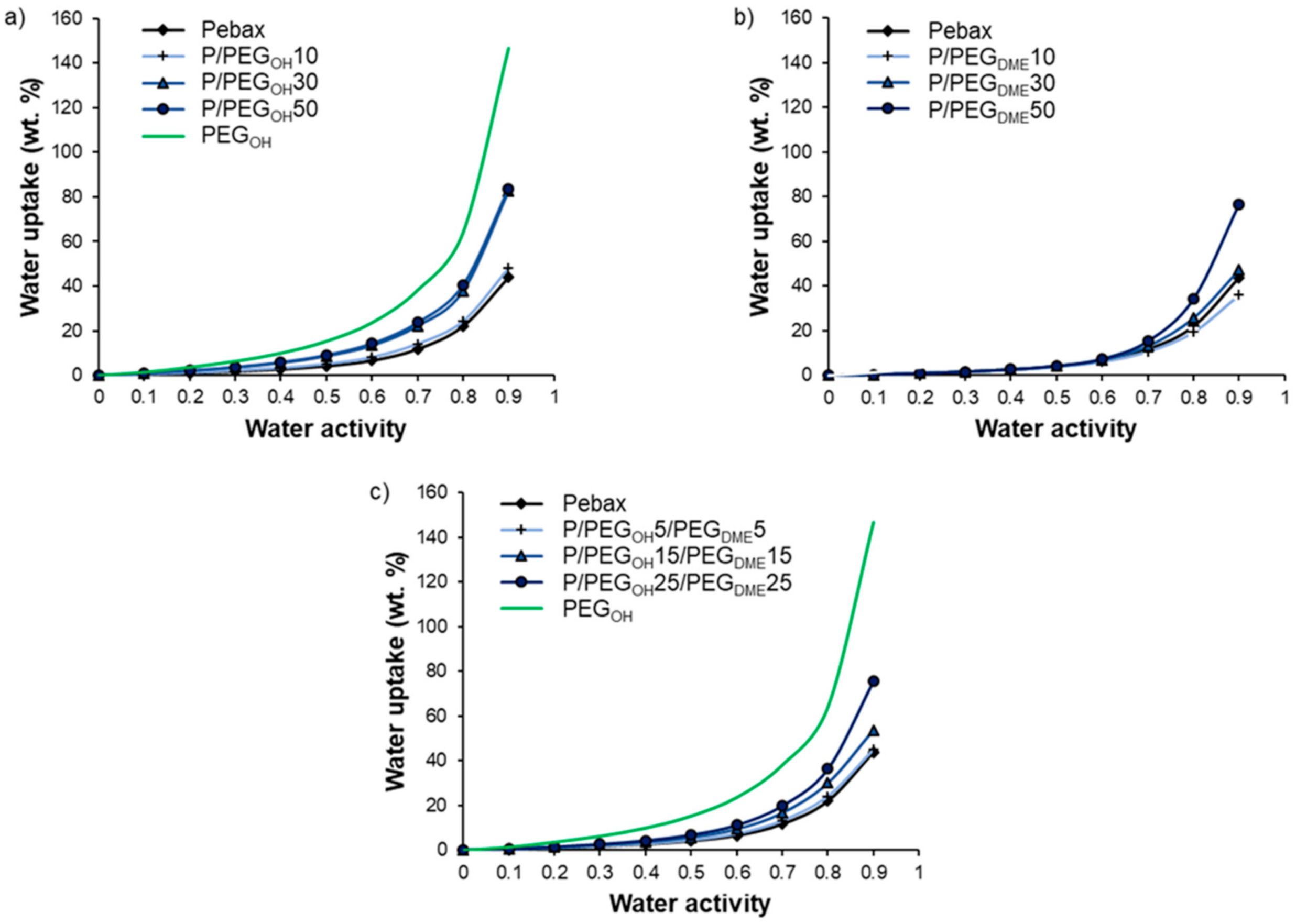
3.4. Hydration Properties
Water vapor sorption properties were investigated for three different systems through a thermodynamic analysis (sorption isotherms) and a kinetic analysis (diffusion coefficients).
Figure 9 displays the water vapor sorption isotherms of Pebax
®/PEG
OH blends (
Figure 9a), Pebax
®/PEG
DME blends (
Figure 9b), and Pebax
®/PEG
OH/PEG
DME blends with a 50/50 PEG
OH/PEG
DME weight ratio (
Figure 9c). The water sorption isotherms of the pristine Pebax
® and the PEG
OH additive are reported as references. The water uptake values for different activities are detailed in
Table 2.
First of all, the isotherm curve of Pebax
® had a BET III shape according to the classification of Brunauer, Emmett, and Teller [
40]. A similar isotherm shape has been observed for other Pebax
®-type copolymers [
32]. The water uptake was important at a low water activity level (4% for an activity level of 0.5) due to the presence of ether and amide polar groups in the Pebax
® structure. Moreover, the chain flexibility allowed for important swelling that led to an important increase in the water uptake at a high activity level (44% for an activity level of 0.9). The isotherm curve of PEG
OH also had a BET III shape with even higher water uptake values (146.7% for an activity level of 0.9). Not surprisingly, the isotherm curves of binary and ternary systems had a BET III shape.
For Pebax
®/PEG
OH blends, the water uptake values were higher than those obtained for the pristine Pebax
®, especially at high activity levels (
aw > 0.5). Moreover, a shift in the water isotherm towards higher water uptake values was observed as the PEG
OH amount increased (see
Figure 9a). This behavior can be explained by the hydrophilic character of PEG
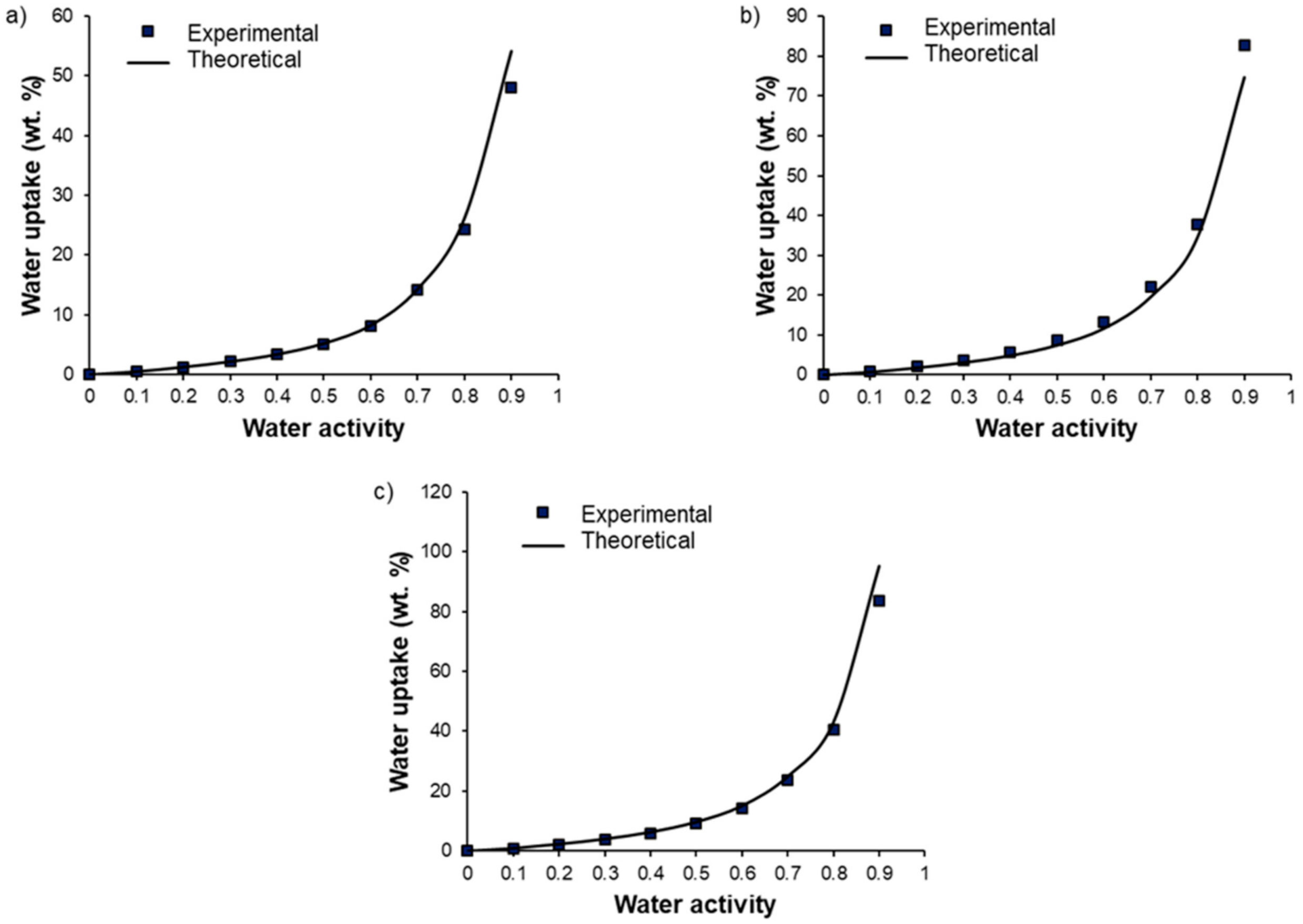
OH and the plasticizing effect of the additive. The experimental water uptake values determined at different activities for each blend composition (90/10, 70/30, and 50/50 wt.%) were then compared to theoretical values calculated by an additive model (see
Figure 10):
where
,
, and
are the water uptake values of the considered blend, Pebax
® and PEG
OH, respectively, for the same activity level.
and
are the weight fraction of Pebax
® and PEG
OH in the blend, respectively.
In all cases, the experimental isotherms exhibited very close water uptake values compared to those calculated by the additive model, showing that each component made its own contribution to the water uptake.
For Pebax
®/PEG
DME blends, no significant difference was observed in the water uptake values compared to those obtained for neat Pebax
® up to an activity level of 0.5. The water uptake then increased with the PEG
DME amount, showing that the plasticization provided by the PEG
DME additive played a key role in water sorption at high activity levels (see
Figure 9b). Moreover, it should be noted that the water uptake values obtained for Pebax
®/PEG
DME blends were lower than those obtained with the Pebax
®/PEG
OH blends in the whole range of water activities for the same PEG amount. This can be explained by the more hydrophilic character of the chain ends of PEG
OH.
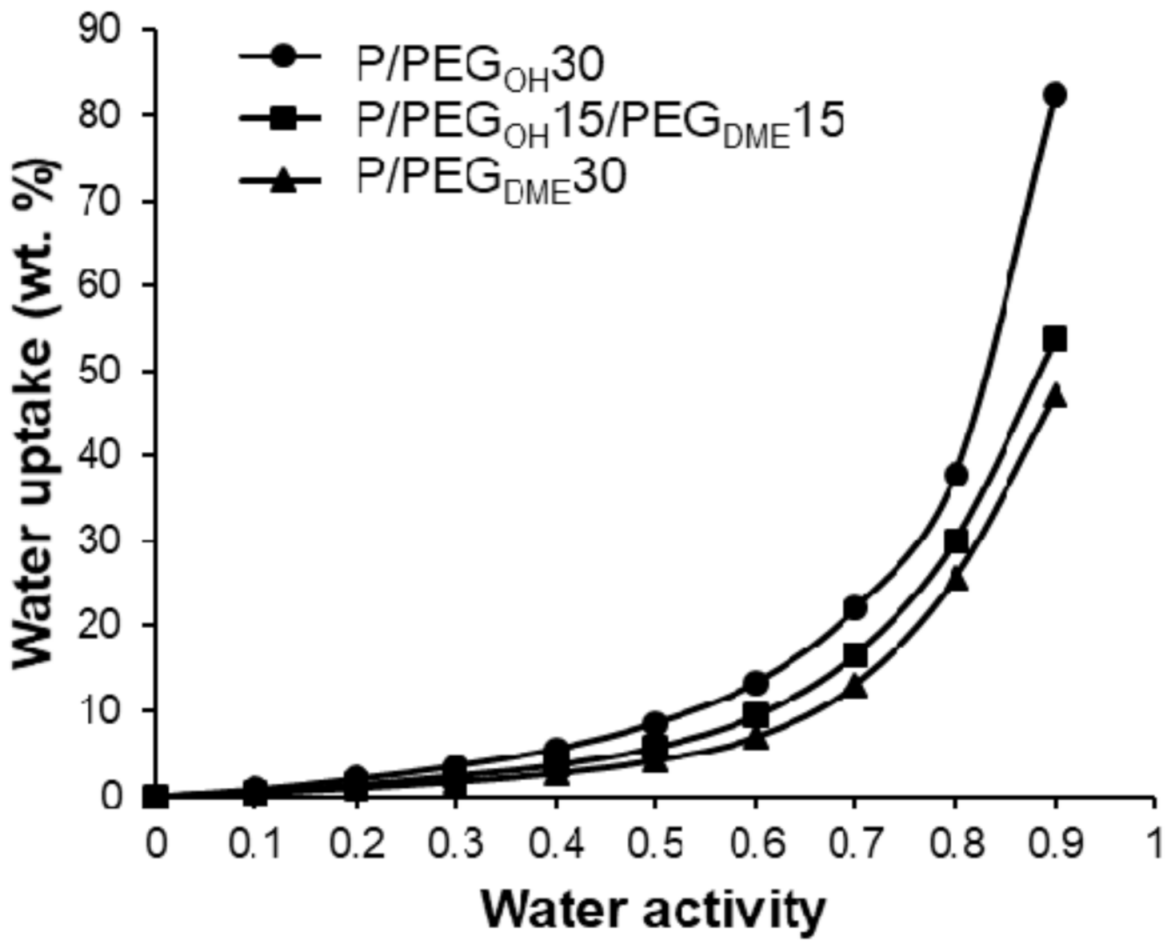
For the ternary Pebax
®/PEG
OH/PEG
DME blends with a 50/50 PEG
OH/PEG
DME weight ratio, the water uptake values ranged between those measured on the binary systems in the whole range of water activities as shown in
Figure 11.
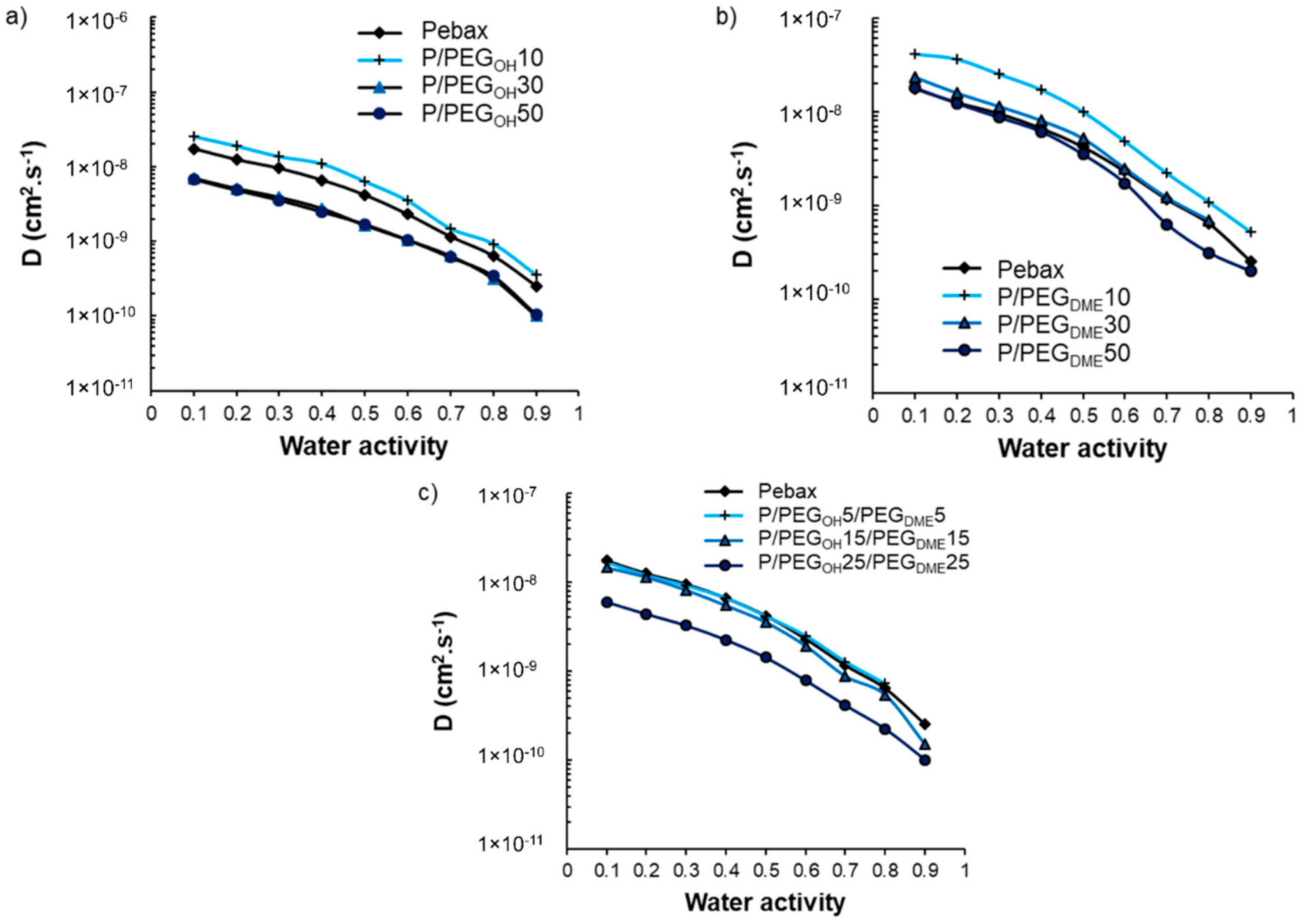
The impact of PEG additives on the water sorption kinetics was also investigated. The water diffusion coefficients (
D) were determined after having checked that the water uptake curves obeyed a Fickian diffusion mechanism. The evolution of
D as a function of water activity is displayed in
Figure 12.
As one can observe in
Figure 12, all curves presented the same shape, consisting of a slow-down of the water diffusion as the water activity increased. This trend is in agreement with a BET-III-type sorption mechanism and the formation of water aggregates at high activity levels [
40,
41]. For all studied film series, the evolution of the water diffusion as a function of the PEG additive amount was similar and not monotonic. Indeed, the values of the kinetic parameter increased as the PEG amount increased from 0 to 10 wt.% and then decreased and became similar or lower than the values obtained on the pristine Pebax
® for blends comprising additive amounts of 30 and 50 wt.%.
As discussed previously, the PEG additives were totally dissolved within the polyether phase of Pebax
® at a low PEG amount (10 wt.%), which led to a decrease in water–Pebax
® as well as water–PEG interactions, making the water’s diffusion easier. However, a new PEG-rich phase was formed at high PEG amounts (30 and 50 wt.%) in which strong water–PEG interactions could be established due to the polar nature of PEG, thus explaining the decrease in the diffusion coefficient. A similar trend was also observed for Pebax
®/ionic liquid blends [
32].
The antiplasticization phenomenon observed at activities above 0.5 was more deeply analyzed using the following equation:
where
Dagg is the limit diffusion aggregation coefficient at nil concentration,
β is the antiplasticization coefficient, and
M is the water uptake in the material. The antiplasticization coefficient
β was determined for each film from the linear regression of the decreasing part of the curve ln(
D) = f(
M).
β values are summarized in
Table 2. The
β of the pristine Pebax
® and the binary as well as ternary Pebax
®/PEG systems was very similar with values comprised between 3 and 5, showing that the addition of PEG species within the Pebax
® matrix did not significantly modify the impact of the water clustering mechanism at high activities.
3.5. Gas Transport Properties
The CO
2 and O
2 permeability coefficients as well as the ideal CO
2/O
2 selectivity were determined on the pristine Pebax
® and the different Pebax
®/PEG film series (
Table 3). The experimental values of permeability measured on the pristine Pebax
® MH1657 are in accordance with those reported in the literature [
42,
43]. As a reminder, according to the DSC and WAXS results, the flexible polyether blocks of the Pebax
® copolymer were totally amorphous at the temperature of the permeation experiments (23 °C), whereas the rigid polyamide blocks were semi-crystalline. Therefore, gas transport was mainly governed by the polyether phase under these conditions. As one can observe from the permeability values reported in
Table 3, the CO
2 permeability coefficient of the pristine Pebax
® was considerably higher than the one measured for O
2 (84 and 3.9 Barrer, respectively). Thus, the pristine Pebax
® film possessed a high CO
2/O
2 selectivity of 21.5, which could be assigned to the presence of CO
2-philic ether groups within the copolymer that increase the CO
2 solubility [
19,
44].
The impact of PEG addition on the gas transport properties of Pebax
® was investigated for the binary and ternary blends. The data presented in
Table 3 and
Figure 13 clearly show that all blended Pebax
®/PEG films exhibited superior CO
2 and O
2 permeability coefficients and higher ideal CO
2/O
2 selectivity than the pristine Pebax
®. We have shown that the PEG additives led to a plasticizing effect that increased as the additive amount increased. Moreover, a decrease in density and a decrease in the PA blocks’ crystallinity was evidenced upon PEG addition. All these changes are favorable to an increase in gas diffusion, explaining the general increase in gas permeability. Moreover, the presence of CO
2-philic groups within both Pebax
® and PEG additives should contribute to high CO
2 solubility and therefore to a reinforcement of selectivity.
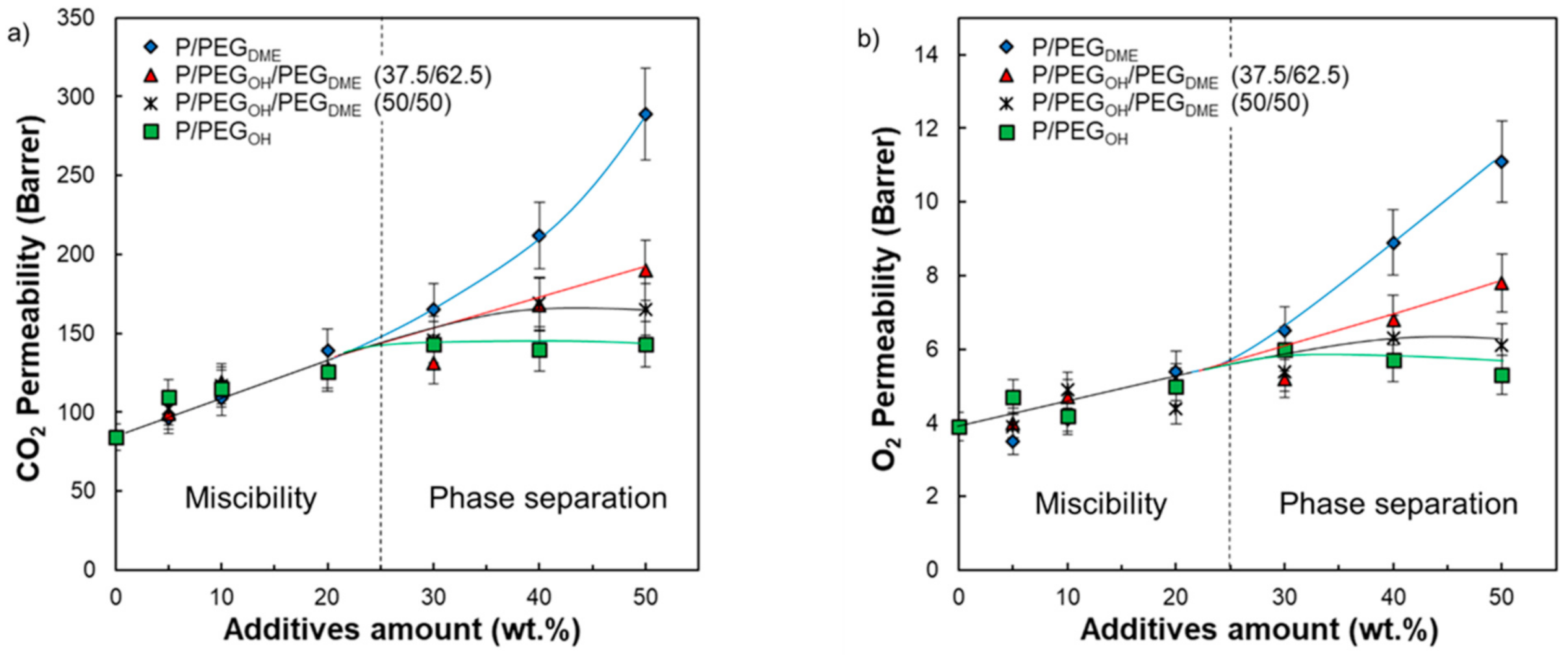
Looking more carefully at the evolution of the CO
2 and O
2 permeability coefficients as a function of the system’s composition, two domains were distinguished (see
Figure 13). In the first domain (for an additive amount inferior to 30 wt.%), the CO
2 and O
2 permeability coefficients increased and remained quite similar for the same additive content regardless of its nature. It is worth noting that the composition domain ranging from 0 to 30 wt.% of additives corresponded to the miscibility domain between Pebax
® and the PEG additives. Then, it seems that, in this domain, the effect of PEG chain ends was negligible. For additive contents above 30 wt.%, different behaviors were observed depending on the chain ends of the PEG additives (
Figure 13).
For Pebax
®/PEG
OH blends, the CO
2 and O
2 permeability coefficients reached a plateau around 143 and 5.5 Barrer, respectively. Thus, the CO
2 and O
2 permeability coefficients were improved by around 70% and 40%, respectively, compared with the neat Pebax
® matrix. It is worth noting that the plateau reached for the experimental CO
2 permeability coefficients corresponds to the intrinsic CO
2 permeability coefficient of a completely amorphous hydroxyl-terminated PEO precisely determined to be 143 Barrer by Lin and Freeman [
19]. Thus, the transport properties seemed to be limited by the intrinsic permeability of the most permeable component (PEG
OH in this case).
The CO
2 and O
2 permeability coefficients measured on Pebax
®/PEG
DME films with a PEG
DME amount superior to 30 wt.% were higher than those obtained for Pebax
®/PEG
OH blends for a similar additive amount. Indeed, both the CO
2 and O
2 permeability coefficients did not reach a plateau value but steadily increased up to 289 and 11.1 Barrer for a PEG
DME amount of 50 wt.%, respectively (see
Figure 13). Thus, the CO
2 and O
2 permeability coefficients were improved by a factor of 3 compared with the pristine Pebax
®. The observed deviation in the gas transport properties between Pebax
®/PEG
OH and Pebax
®/PEG
DME blends can only be explained by a chain end effect. Indeed, the presence of a bulky methyl end group probably hinders hydrogen-bonding interactions between polymer chains and allows for a higher free volume available for the transport of small gas molecules [
23]. It has to be noticed that, to the best of our knowledge, the intrinsic CO
2 permeability of a completely amorphous PEG
DME has not been referenced in the literature and so could not be compared to the one of a completely amorphous hydroxyl-terminated PEO.
The CO2 and O2 permeability coefficients measured on both ternary Pebax®/PEGDME/PEGOH blends ranged between those of Pebax®/PEGOH and Pebax®/PEGDME blends for PEG amounts superior to 30 wt.%. Higher gas permeability coefficients were obtained for ternary blends having the highest PEGDME proportion. Indeed, the P/PEGOH18.75/PEGDME31.25 blend had CO2 and O2 permeability coefficients of 190 and 7.8 Barrer, respectively, with respect to the P/PEGOH25/PEGDME25 blend, which had CO2 and O2 permeability coefficients of 169 and 6.3 Barrer, respectively. Moreover, it seemed that the molar fraction of PEGDME/PEGOH within the blend needed to exceed the ratio 1/1 in order to avoid a plateau effect for the studied range of compositions. Thus, the PEGDME additive was particularly interesting to introduce into the blend in order to increase the gas permeability coefficients.
For the three studied series, an average increase in the ideal CO
2/O
2 selectivity of between 10% and 20% was also observed regardless of the film’s composition (
Table 3). This increase in both permeability and selectivity is not common for polymer materials. Indeed, a natural intrinsic trade-off exists between these parameters for dense polymeric films as described by Robeson via the establishment of an upper bound for numerous gas pairs [
45,
46]. For our systems, the increase in CO
2/O
2 selectivity could be attributed to the increase in the proportion of CO
2-philic ether groups within the material. Therefore, the developed Pebax
®/PEG films have high potential as breathable films for food packaging applications as a result of their interesting gas transport properties. Among them, the Pebax
®/PEG
DME films with high PEG
DME contents offer the best gas transport properties.
3.6. Film Stability
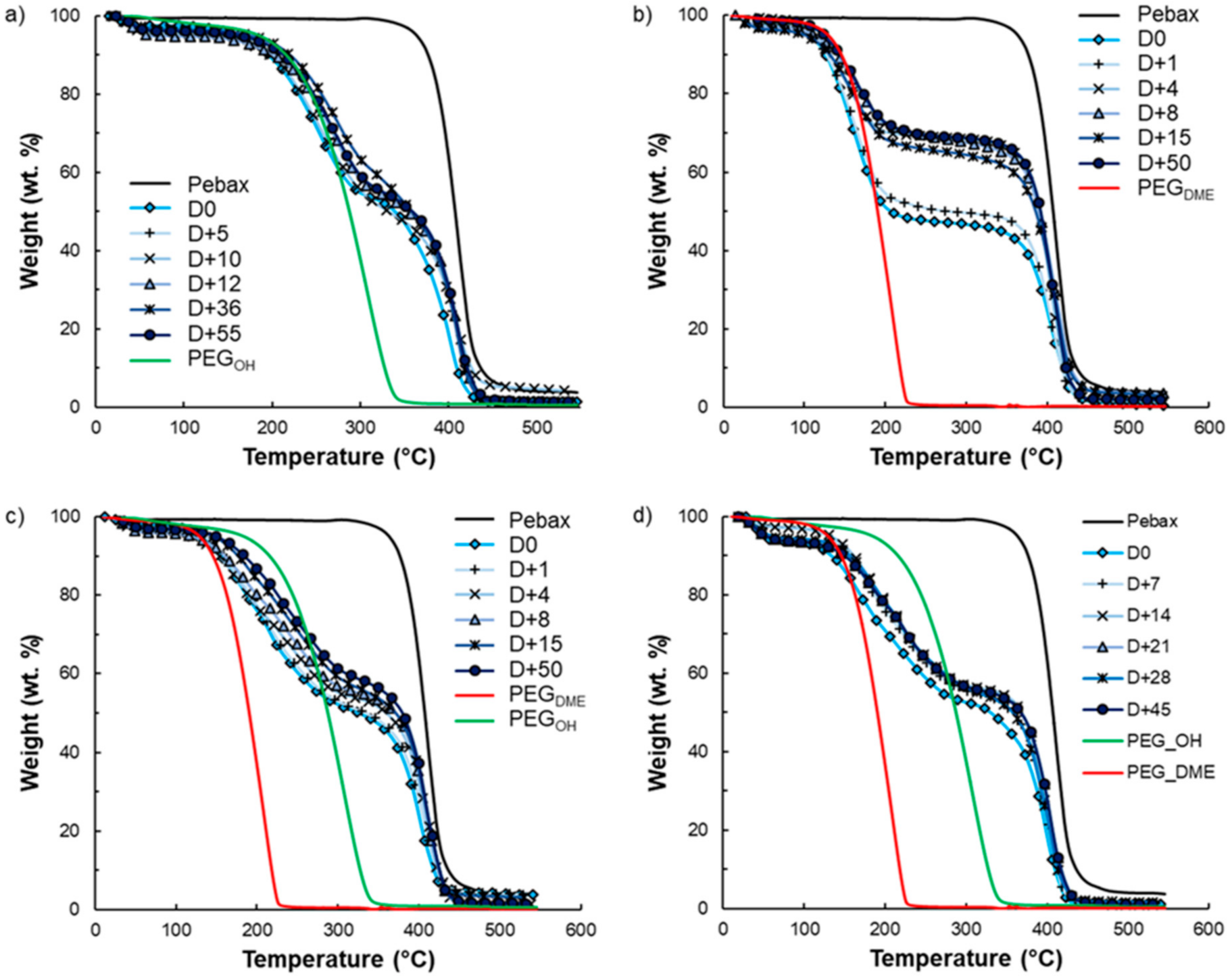
Film stability is an important criterion for long-term use. It is known that exudation phenomena may occur following the addition of low-molar-mass additives to a polymer matrix depending on the polymer–additive interactions and the additive amounts. That is why film stability in terms of composition over time was investigated by TGA analysis for the highest amount of additive studied in this work. TGA curves of the pristine Pebax
®, the PEG
OH and PEG
DME additives, and binary and ternary Pebax
®/PEG blends with an additive amount of 50 wt.% are displayed in
Figure 14. During heating from room temperature to 550 °C, degradation in one step occurred between 350 and 500 °C for pristine Pebax
®. For PEG additives, an average weight loss of 4 wt.% was observed between 50 and 100 °C, which was attributed to water evaporation, followed by total weight loss between 150 °C and 350 °C for PEG
OH and between 100 and 250 °C for PEG
DME.
Thus, the degradation temperatures of the pure components (Pebax
® vs. PEG additives) can be clearly distinguished, allowing us to follow the composition of the binary films over time as shown in
Figure 12b and
Figure 14a. Indeed, weight loss in two steps was observed on each binary Pebax
®/PEG blend due to the degradation of the additive and the Pebax
®, in agreement with the results obtained on pure components. Concerning the ternary blends, the degradation temperatures of PEG
OH and PEG
DME were too close to differentiate their signatures on the TGA curves. However, the first instance of weight loss observed in the temperature range 100–350 °C could clearly be assigned to the additives’ degradation, whereas the weight loss at a higher temperature (around 400 °C) was the signature of the Pebax
®’s degradation (see
Figure 14c,d). It is also worth noting that an average water loss of 4 wt.% was also observed on the TGA curves of all Pebax
®/PEG blends below 100 °C.
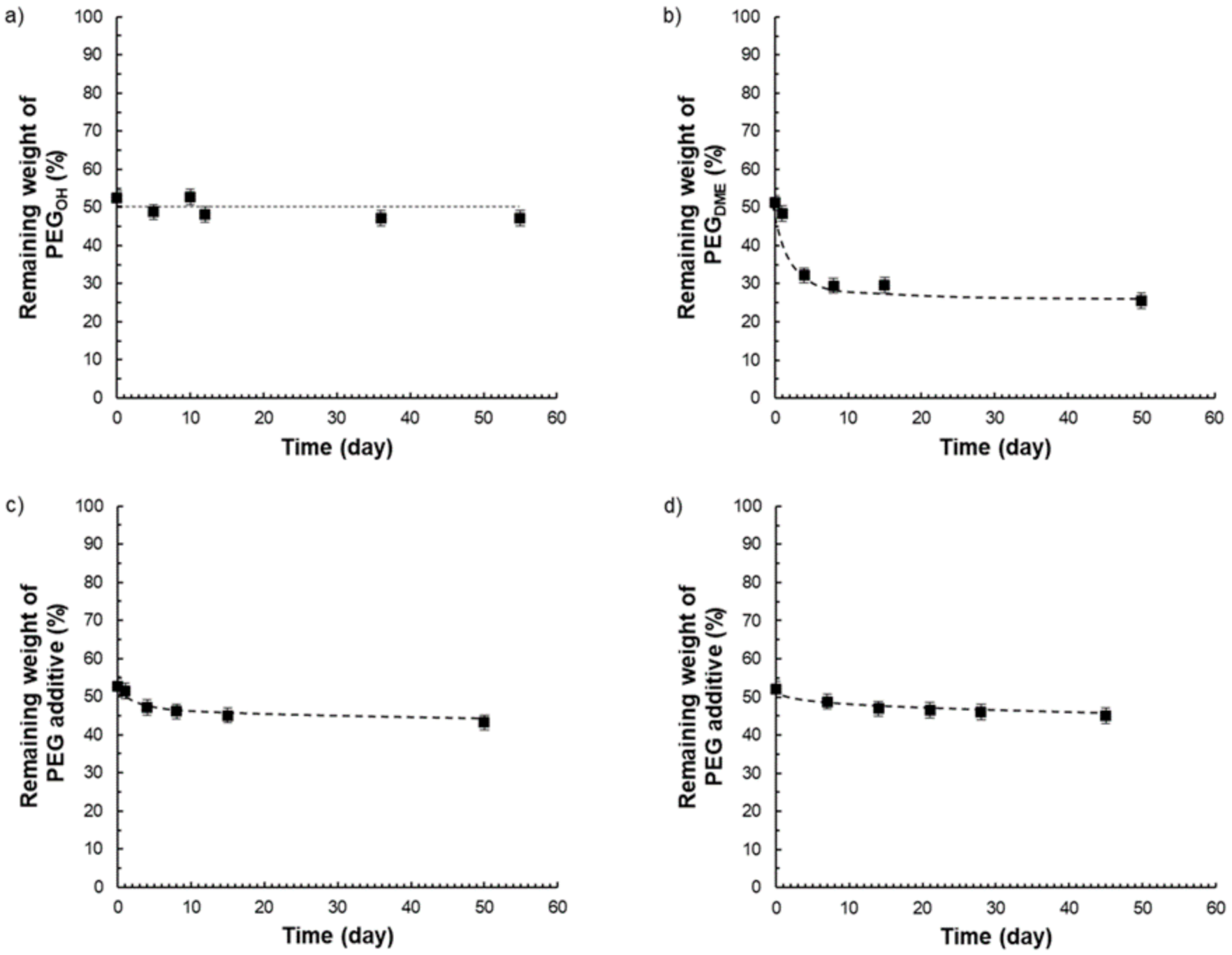
Thus, from the different TGA curves, it was possible to determine the amount of additive that remained in the films after different storage times under atmospheric conditions (23 °C, 30% RH) (see
Figure 15).
Pebax
®/PEG
OH films with a PEG
OH amount of 50 wt.% were stable in composition over time (
Figure 15a), whereas Pebax
®/PEG
DME films containing an additive amount of 50 wt.% were not stable in composition over time as illustrated in
Figure 15b. For this latter system, the matrix/additive proportion evolved from a 50/50 weight ratio to a constant 75/25 weight ratio after 50 days due to the exudation of the PEG
DME additive. Further studies carried out on Pebax
®/PEG
DME films with additive amounts of 30 and 40 wt.% confirmed a stabilization of the film composition towards a 75/25 matrix/PEG
DME weight ratio after a testing period of 50 days (
Supplementary Materials). For the ternary blend P/PEG
OH25/PEG
DME25, a slight weight loss (lower than 6%) was observed after 50 days (see
Figure 15c). Likewise, a slight weight loss (lower than 6%) was obtained after 45 days for the ternary blend P/PEG
OH18.75/PEG
DME31.25 (
Figure 15d). From the results obtained on binary blends, it could be supposed that the weight losses observed on both ternary blends could be assigned to the PEG
DME’s exudation.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}