1. Introduction
The use of artificial intelligence (AI) in bioinformatics and computational molecular biology research has been growing fast over the last two decades [
1,
2]. Bioinformatics methods attempt to model known biological structures and predict unknown ones. Versatile bioinformatics techniques are capable of storing the information processed in various biological and biophysical studies in the created databank, and calling and utilizing the information from the databank in pinpointing crucial molecular processes of an individual system or collective ones. The techniques thus help establish scientific links between various mechanisms and processes and produce concluding evidence that is otherwise often unattainable using conventional theoretical and experimental techniques. Besides, computational techniques are popularly found to model the biomolecular complexes in silico studies to mainly address their statics, dynamics, and energetics in an artificially constructed, yet mimicking the biological systems’ environment.
Although about just 2% of the protein structures that are experimentally identified are among the transmembrane proteins (many of which construct ion channels), genome studies suggest that these special proteins together make up about 30% of all of the coded proteins. While mapping the membrane proteome Almén and colleagues found 27% of the total human proteome to be α-helical transmembrane proteins [
3]. Bioinformatics method enables modeling of the unknown structure of the proteins, predicts their functions, their transmembrane location, and their ligand binding potency. Current in silico modeling tools use various computational methods, which are capable of providing results that may mimic nearly the biologically relevant functionality. General understanding of genetics, the gene-based mutations, emergence of disease, etc., as well as information on even evolution that concern ion channel structures and functions including both normal and abnormal biological systems’ status quos may be addressed using bioinformatics techniques. A huge amount of data from all this research contain information about certain biological systems, processes or mechanisms. These data and information are stored at various locations and sites utilizing random methods. Pulling them with the use of valid scientific ways and processing towards constructing any meaningful conclusions are challenging tasks. AI techniques appear as helpful tools to deal (extract, process and analyze) with such kind of big biological research data [
4]. Knowledge on computing models using AI, advanced analytics of data and various optimization approaches that are used in bioinformatics, bioengineering and biophysics research on designing drugs and related analysis, medical imaging data analysis, biologically inspired artificial learning and adaption for general analytics, etc. is very useful. This knowledge is often found applicable in understanding many specific aspects of ion channels.
Association of AI, ML, and DL Techniques with Ion Channel Bioinformatics
DL is a subset of ML and ML is a subset of AI: AI(ML(DL))). For ML, machines are supposed to learn and adapt through experience; for AI, machines can smartly execute specified tasks. DL is basically concerned with specific algorithms that are inspired by the human brain structure and function, known commonly as artificial neural networks (ANNs). The opportunity of using AI techniques in system biology is enormous [
5]. ML techniques appear as powerful tools with capability to extract information from any data sets which are massive in size and noisy in nature. A review has described approaches that are based on simultaneous use of the systems biology and the ML in order to access the gene and the protein druggability [
6]. It also elaborated on the sources of data, algorithms, and performance of different methods. The mathematical and computational methodologies underlying DL models appear quite challenging for interdisciplinary scientists, who may consult a recent review for being familiar with the techniques [
7]. This article has presented a review on introduction to DL approaches that include Convolutional Neural Networks (CNNs), Deep Feedforward Neural Networks (D-FFNN), Deep Belief Networks (DBNs), Autoencoders (AEs), Long Short-Term Memory (LSTM) networks; many (if not all) of which have already found applications in bioinformatics field dealing with biological structures and functions.
AI has long been found useful in bioinformatics, and computational molecular biology (e.g., especially in the field of DNA sequencing) [
1]. The main use of AI in these fields is in understanding of the organisms’ evolution, and slow growth of complexity of working with data having errors. AI softwares and modeling help to search, make classification and mine versatile biological databases; and especially simulate biological, physiological, biochemical, and biophysical experiments with and without errors. AI techniques are now found generally useful to handle (process, understand and create conclusion on specific aspects) partially the human genome data with billions of basepairs (bps), the necessity of what was rigorously addressed in ref. [
8].
In an ML paper on bioinformatics, two decades ago, Tan and Gilbert analyzed learning systems (7 individual ones) and methods (9 combined ones) using 4 data sets of biological systems, and provided a few crucial issues (which are still considered generally applicable) to follow while answering a few questions on choosing correct algorithm to best suit for a data set, possibility of having any combined method(s) which might be better than especially any singular approach, comparing the effectiveness of any particular algorithm over others, etc. [
9]. Even about three decades ago, people used ML approaches for gene recognition [
10].
ML techniques; the ANN and the support vector machine (SVM) have been recently found to help predict the secretory proteins that may not necessarily require the presence or absence of the N-terminal signaling peptides, which are commonly known as the classical and the non-classical secreted proteins [
11]. Here the methods have been trained and tested on a dataset of 3321 secretory and 3654 non-secretory proteins of mammals have been used to train the methods here with the use of a technique consisting of five-fold cross-validations. ANN-based modules were developed for mainly predicting the secretory proteins where 33 physicochemical properties, with compositions of the amino acids and the dipeptide, were considered. Considerable accuracies (73.1%, 76.1%, and 77.1%, respectively), were achieved. SVM-based modules used 33 physicochemical properties, with the compositions of amino acid, and the dipeptide and found similar accuracies (77.4%, 79.4%, and 79.9%, respectively). Basic Local Alignment Search Tool, commonly known as BLAST and the Position-Specific Iterative BLAST (PSI-BLAST) modules got designed for the purpose of predicting the secretory proteins considering similarity search which achieved 23.4% and 26.9% accuracy, respectively. A hybrid-approach that integrated amino acid and dipeptide composition-based modules SVM, and PSI-BLAST, which found increased accuracy 83.2% and sensitivity 60.4% having low 5% false positive predictions). This reflects a substantial increase than achieved using individual modules.
As presented here, versatile applications of AI, MI, and DL in various protein, gene structural, and functional aspects have been evident. Our goal in this article is to go beyond addressing these general features and pinpoint the membrane proteins structures and functions which are addressable using artificial modeling and algorithms. The use of AI, ML and DL techniques is popularly used to understand various features of ion channels. From understanding the amino acid properties and classifications to classifying specific channel subunits representing ion channel families, artificial techniques are utilized [
12]. We see that artificial techniques, such as the ML approach, can now capture crucial ion channel complexities related to channel protein expression, correct insertion and folding in membranes, and trafficking to proper locations inside the cell, thus help in further membrane protein engineering and artificial designing [
13,
14]. AI techniques help us track the early animal evolution by comparative genomics studies of ion channels that specifically help us understand the early evolution of animal nervous systems [
15]. ML has recently been used to analyze ion channel genes, especially to extract the feature vectors of various ion channels [
16].
It is clear that artificial techniques, models, and algorithms are utilized to program various ion channel features, including classification of channels, channel subunit proteins, or even amino acids and genes, which addresses evolution, modern engineering, and various other related aspects. AI, ML, and DL have a lot of involvement in this new area. Experimental address and their theoretical analysis have produced so much data that we now need these artificial techniques to grasp most about ion channels’ various features in a simplistic manner, using models and algorithms that are made possible using the power of AI, including its subfields ML and DL.
2. Bioinformatics Predictions of Ion Channel Structures and Functions
X-ray crystallography, NMR data, etc. on transmembrane proteins are generally used to predict the optimal protein structures. These techniques require the use of extremely expensive necessary ingredients and a tuned laboratory setup. Bioinformatics modeling utilizing appropriate techniques that may promote in silico mechanics and energetics of the protein structure considering the underlying mechanisms are often popularly considered in biophysical studies of proteins. Membrane proteins are generally studied specifically to address their ion channel-forming potency. Bioinformatics techniques play crucial roles when important molecular actions are to be inspected to explain the experimental facts obtained in vitro studies, such as their imaging in the interface of hydrophobic/hydrophilic regions, electrophysiology record of currents across membranes hosting the proteins, etc. Molecular dynamics (MD) simulations often appear as important computational techniques to detect energetics underlying biomolecular interactions. We have been quite successful in biophysical addressing, using MD simulations, of the channel energetics involving channel subunits and membrane lipids for small channels, such as gramicidin A, alamethicin, and chemotherapy drug-induced channels in model membrane systems [
17,
18,
19,
20,
21,
22]. In these publications altogether we could establish a single fundamental fact that the channel stability inside the membrane is due to nothing but molecular mechanisms depending on charge-based screened Coulomb interaction energetics among functional charge groups in the ion channel complex involving channel subunit peptides or drugs and membrane lipids. Our computational in silico assays (numerical computations and MD simulations) simply supported the experimental findings in the distance and time-dependent channel subunit-lipid interaction energetics theoretically. We could calculate the binding energies and evaluate the binding energetics in the channel complex and thus know of the statistical mechanical nature in the channel stability in a biological thermodynamic environment. The readers are invited to read directly from these articles to gain further insights.
Besides various computational assays addressing the general structure and function of channel proteins, bioinformatics templates that draw information from various databank on the channel protein structures, genomics of the proteins, mutations in genes of the ion channel proteins are found to produce crucial information about channel functions in both healthy cells and mutated (disease) conditions.
The aspects addressing the ion channel protein genetics and mutations are presented later in this article using a few example case studies. Here we wish to address the general aspects of ion channel structures and functions using bioinformatics techniques [
23] including various computational assays and in silico modeling.
Table 1 presents a set of ion channels that are addressed using various in silico computational techniques [
24].
A two-decade-old review provided analysis combining MD simulations and various associated calculations with modeling to provide approaches that help understand the structure/function relationships for channels in human cells [
25]. Here the modeling techniques were analyzed for potassium channels, the voltage-gated (Kv), and the inward rectifier (Kir) channels. The NMR structures of (the pore-lining) M2 helix were the basis on which the transmembrane region of the pore could be modeled.
What matters to understand the ion channel function is based mostly on two things: (i) ion channel pore region geometry, and (ii) energetics that controls the pore opening/closing phenomena. Direct and indirect experimental techniques usually can address them phenomenologically but underlying mechanisms largely rely on modeling of the channel using bioinformatics techniques [
24].
Taking the potassium channel as an example case, Heil and colleagues introduced an interesting bioinformatics method, the so-called ‘Property Signature Method’ (PSM), to address this issue of identification of the channel sequences [
12]. This technique relies on physicochemical amino acid properties, instead of amino acid building blocks. A pore region signature (including the selectivity filter) was created, representing the most common physicochemical properties of the known potassium channel, thus enabling the genome-wide screening for the sequences having similar features, despite having low degree of the amino acid similarity within any specific family of the protein.
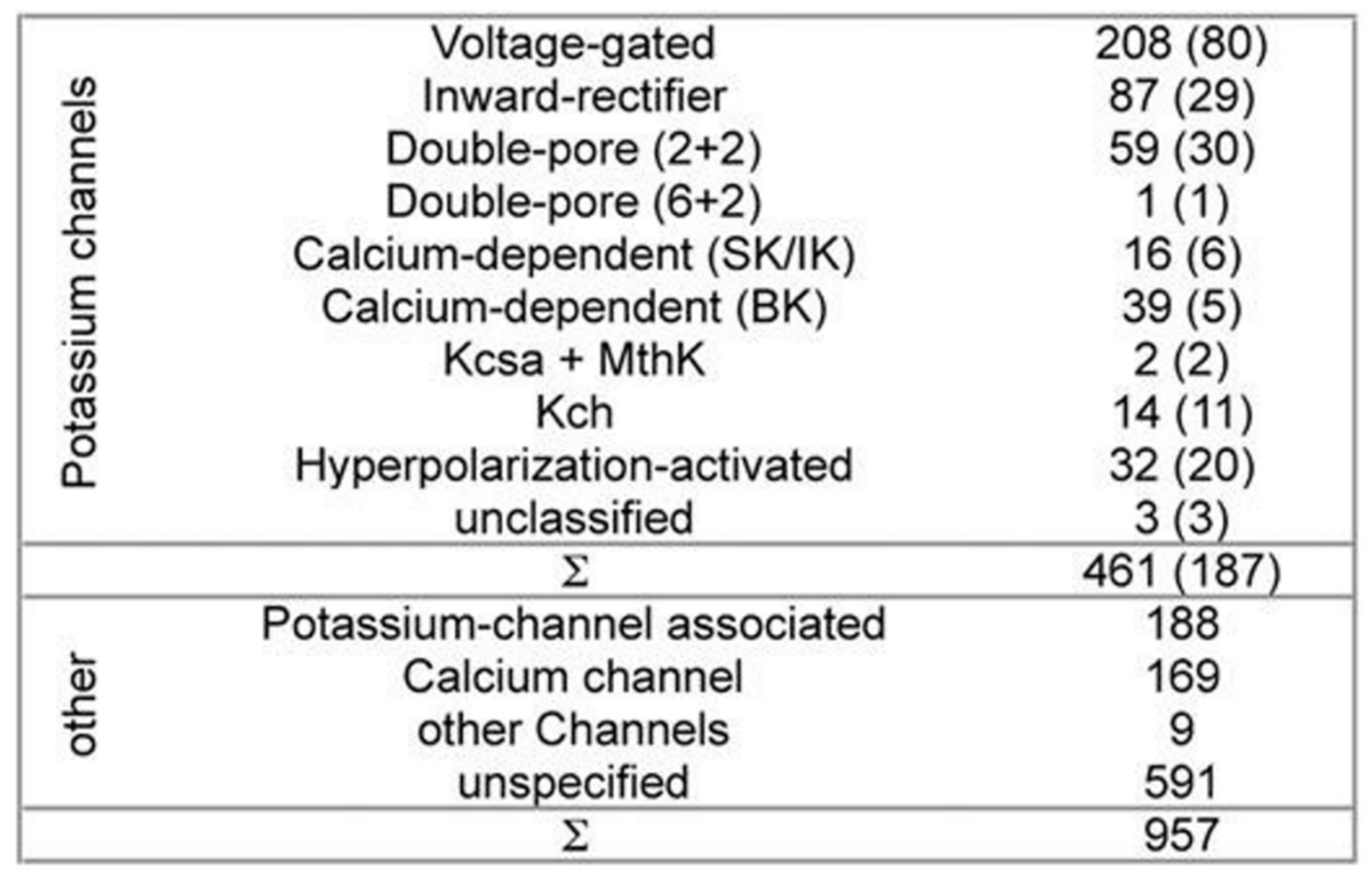
While developing PSM the dataset used 461 potassium channel α-subunits that represent different family types, see
Figure 1 [
12]. A pairwise similarity of the sequences <80% were considered (187 sequences). The set was considered to contain additional 957 non-α-subunits, so that false positive could be provided. The sequences included ion channels that are closely related. All of the sequences used here have been extracted from the Swiss-Prot [
26].
The pore region profile for a potassium channel was created with the use of the dataset. The profile wasn’t used for describing the conserved positions of the amino acids in the region. But it described all of the variations in various families of the potassium channels. The profile then was translated into creating a descriptor, which describes various sequence region properties. Each profile position located amino acids got analyzed, and the properties with conserved absence or presence were used in order to describe the mentioned position. Here the Hits were ranked following the properties that were found in the property descriptor and in the target sequence. The algorithm of screening was created in the C++ language of programming.
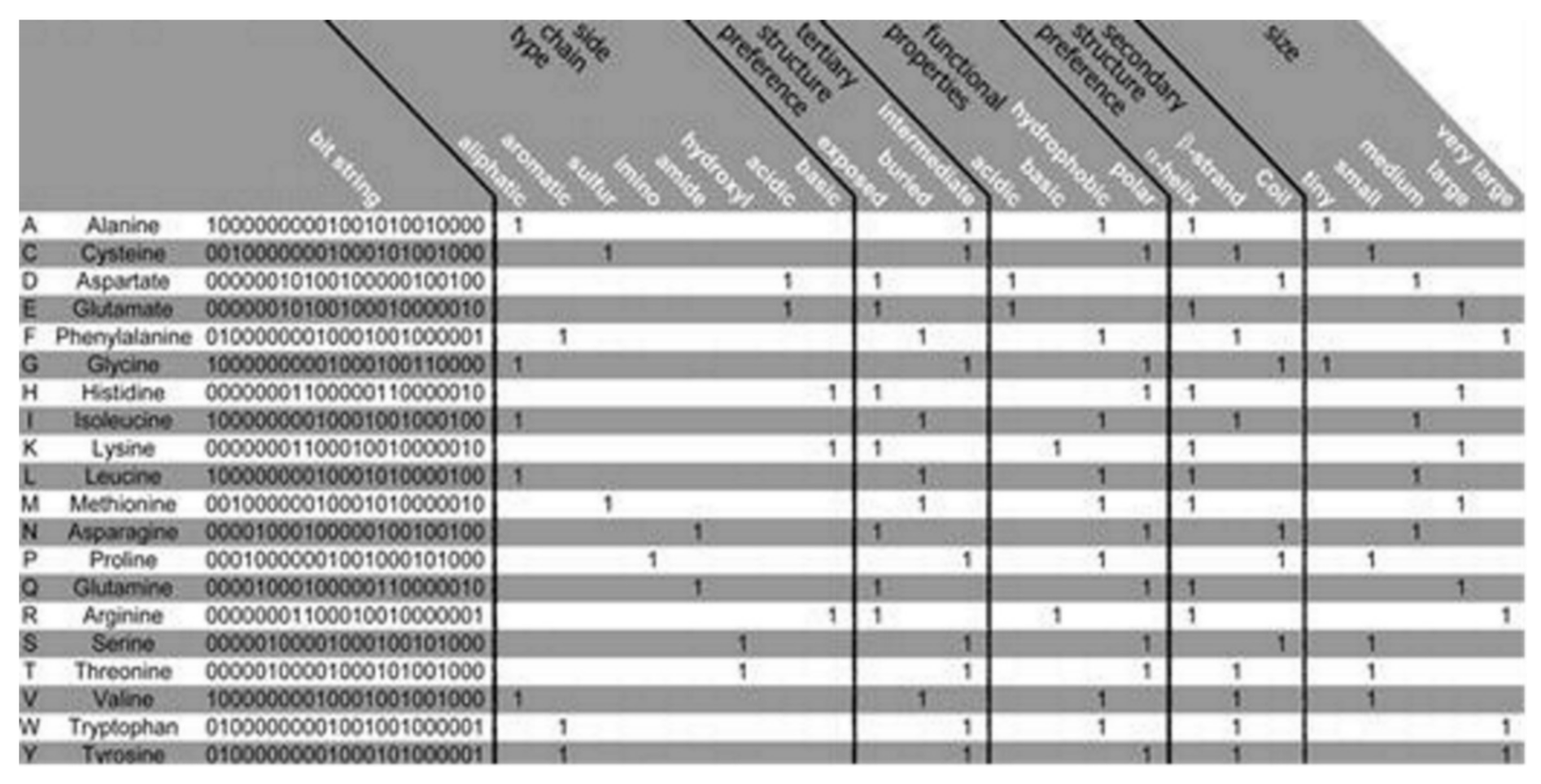
The PSM is found to use the representation of the amino acid via consideration of a binary signature that was derived using varieties of physicochemical properties. Altogether, 23 properties were used, combined into 5 groups as follows: the side chain type, the functional properties, the secondary and the tertiary structure (preference), and size, see
Figure 2 [
12]. Each amino acid is represented by a created binary string, where a bit has been set to 1 for a corresponding property found to apply to the considered amino acid. Five bits have been set, one for every group of the property. Zero is assigned for all of the bits that are remaining. Thus, 20-bit strings (unique) have been found, 1 for every amino acid, which was used in this algorithm. Two steps are considered in the method as follows:
- (i)
an aligned pore domain profile was created including all of the amino acids that were present (in >3% of the investigated total 461 potassium channels),
- (ii)
the profile was translated into a representing string consisting of the sequences’ physicochemical properties.
Large-scale potassium channel sequence analysis confirms the requirement of identifying the potassium channel α-subunit proteins [
27]. As the family of the potassium channel is found to be highly diverse and also closely related to many other ion channels, the use of the amino acids in order to classify the potassium channels in PSM has been found imprecise. PSM is found superior over Markov models and the BLASTp, see refs. [
27,
28,
29]. Moreover, the PRINTS Database provided potassium channel motifs are used [
30]. These approaches are found to utilize multiple methods to overcome a method’s limitations of recognizing the potassium channel family’s subset sequences. These issues are indeed resolved in PSM. Because it can detect properties representing all of the subsets of the family of the potassium channels. Moreover, PSM is able to analyze amino acids’ physicochemically relevant properties and enables pretty sensitive extraction of the information that is coded in the sequences of the amino acids. For details, readers may consult the original article [
12].
The
Saccharomyces cerevisiae genome was well screened applying PSM [
12]. Two hits were found, the domains in the pore in the two-pore potassium channel, TOK1, which is the only one known as the S.cerevisiae potassium channel. Despite having a strong relationship including high homology among the potassium transporters, TRK1 and TRK2, to the potassium selective domains of the pore of TOK1, the mentioned two are classified as nothing but the non-potassium channels.
Heil and colleaques also performed another test with Caenorhabditis elegans having a complete genome sequence [
31]. Its genome regarding the sequences of the potassium channels is well understood; almost 40 double-pore domains have been annotated. PSM helped recover all. Additionally, a new (potential) pore domain was identified.
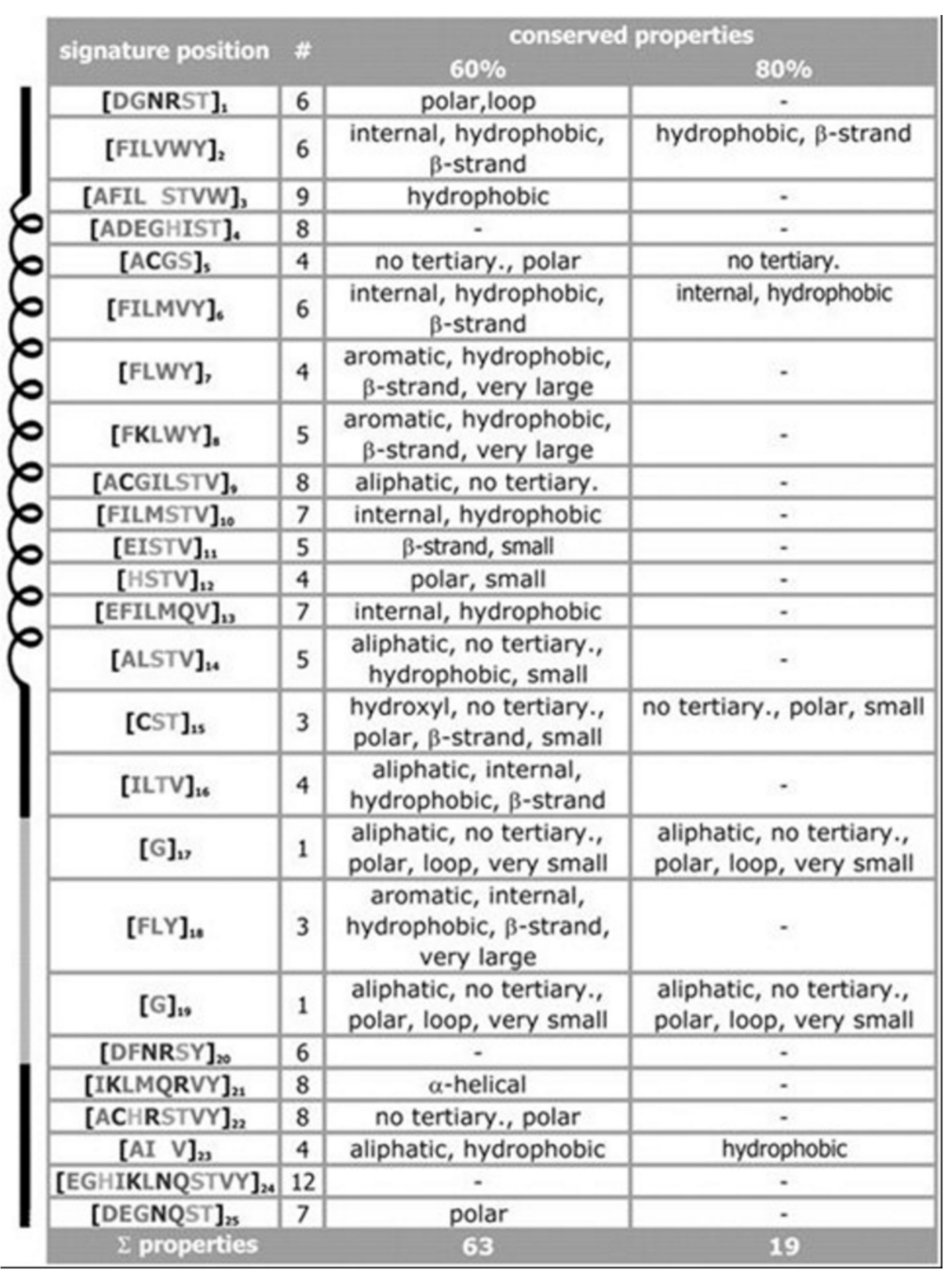
For the signature of the potassium channel, a summary of the conserved properties (at 60% with 80% threshold of conservation) is presented, see
Figure 3. Despite considerable sequence set divergency, as many as 63 properties are found conserved with as high as 60% level of significance, and 19 properties are found conserved with as high as 80% level of significance. Unusual properties (not shown) coded in signature; almost 350 properties with 60% level of significance and 330 properties with 80% level of significance. The method specificity draws significant contributions from these mentioned properties.
PSM is considered superior to other conventional methods while searching for the sequences having a pretty low level of conservation. PSM has an important advantage. For every amino acid position, the signature describes the frequent properties (selected and uncommon ones) in the α-subunit portion of the potassium channel. The use of the position-bound signature properties has additional advantages, interpretation of the results appears pretty simple. Next to the missing and unusual number properties, this method is found to return, for every sequence, the display of a vector whose sequence positions are found to contain the untypical and missing residues, respectively, thus facilitating the fast sequence analysis.
3. Ion channel Genomes Track the Early Animal Evolution
A comparative study of genomics provides novel windows into the (confusing) past that may be applied for the understanding of the early nervous systems evolution of the animal kingdom [
15]. There is a controversy on nervous systems whether they got evolved just once, or independently being distinctive in various animal lineages. Liebeskind and colleagues explored the historical aspects of the gene families of the ion channels, central to the function of the nervous system. They tracked the timeline when the families of the genes expanded in the evolution of the animal and discovered the gene families to be radiated on multiple occasions, occasionally, they underwent various periods of contraction. Multiple gene family origins may be considered to signify considerably the large-scale evolution convergence for the complexity of the nervous system.
The ancestral gene content reconstruction helped was used in tracking the gene family’s expansion timing. Here the majority of the ion-channel protein families that may drive nervous system functions are used. Animals having nervous systems are found broadly to have identical complements of the types of ion channels. But it was also found that these complements could have been evolved independently. Ion channel gene family evolution was found to experience a large amount of loss events, among those two were found to immediately be followed by a few rounds of duplications. Ctenophores, cnidarians, and bilaterians have been found to undergo independent bouts of the gene expansion in the involved channel families connected to the synaptic transmission and the shaping of the action potential, suggesting the genomic signature of the expanding complexity in the nervous system. Ancestral nodes, where the nervous systems probably originated, were found to experience not-so-large expansions. This suggests for the origin of nerves not to experience any immediate complexity bursts, instead, the complexity of evolution perhaps experienced a rather slow fuse in the stem animals, which got followed by gene gains and losses independently.
A custom bioinformatics pipeline [
15] was used for collecting and annotating proteins that are predicted in a group of 16 families of ion channels, see
Table 2 where 41 sample opisthokonts (this group includes animal, fungi, and related protest members), and an apusozoan outgroup are presented. The channels’ families are found to be playing diversified roles in the nervous systems. Some families (e.g., the families of the voltage-gated ion channels) are found to solely be associated with the function of the nervous systems in the animals, while others (e.g., P2X receptors) are found to play relatively diverse types of roles. Only a handful of isoforms are expressed in the nervous systems. The dataset then got used in order to infer the ancestral contents of the genome and understand the timing of the happening of the gene duplications with the help of EvolMap [
32].
These gene families were found ancient [
33,
34]. All except for two, acid-sensing channel (ASC) and the Cys-loop receptor (LIC), are found in the most recent common ancestor (MRCA) of the examined taxa [
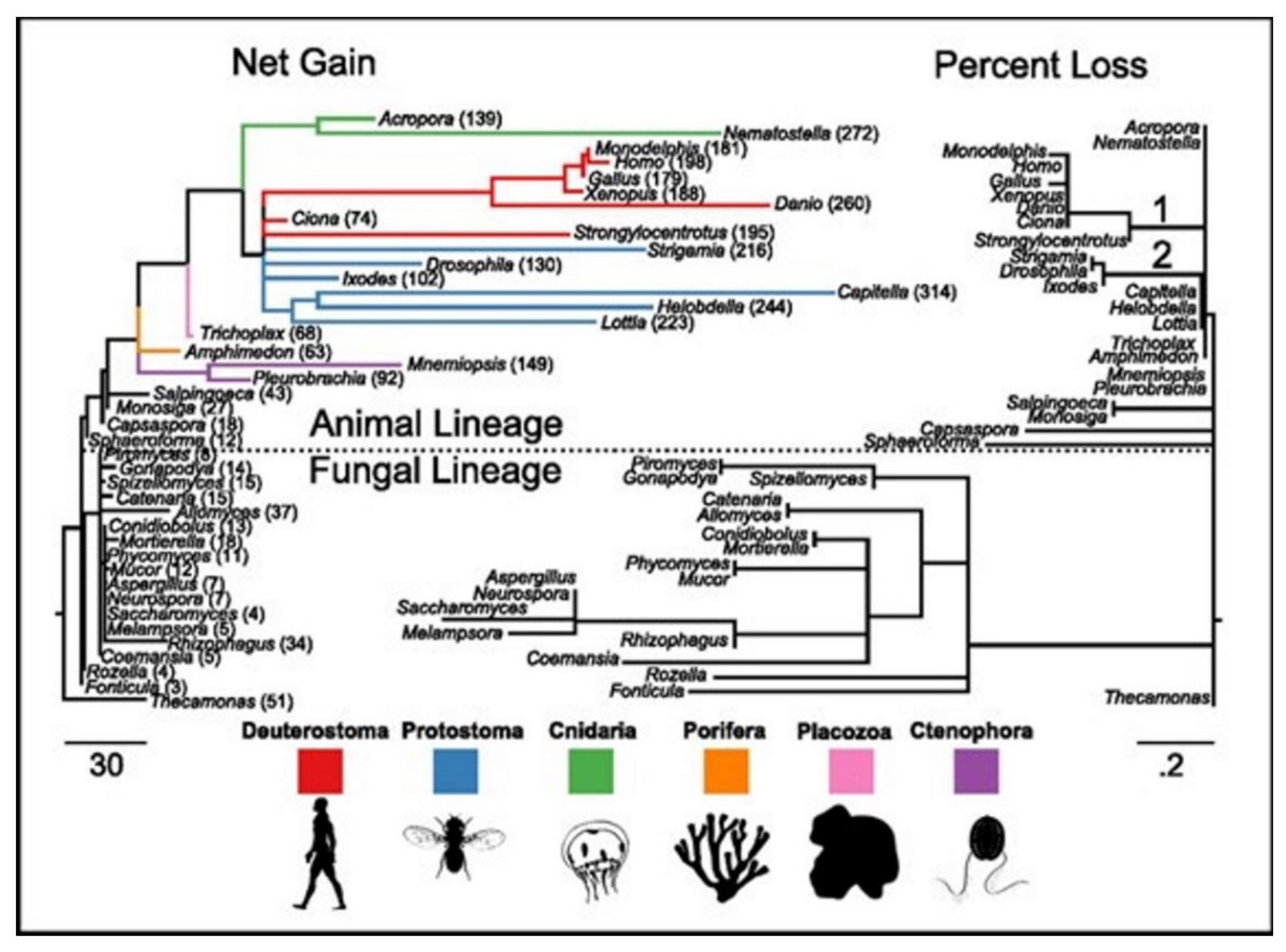
15]. ASC family was the only one found as the metazoan-specific. The families were pulled together and they then plotted the net gains and the percent losses (on the species tree), see
Figure 4 [
15]. The animal lineage was dominated by the gains but losses led to the fungal lineage.
In phylogenetic gain and loss patterns for all of 16 families of ion channels (
Table 2), large expansions of LIC, voltage-gated potassium channel (Kv), and glutamate-gated channel (GIC) families at multiple places were reported, see details in ref. [
15]. This independent gene-(family) expansions lead to MRCAs of the bilaterians, the vertebrates, and the cnidarians [
34].
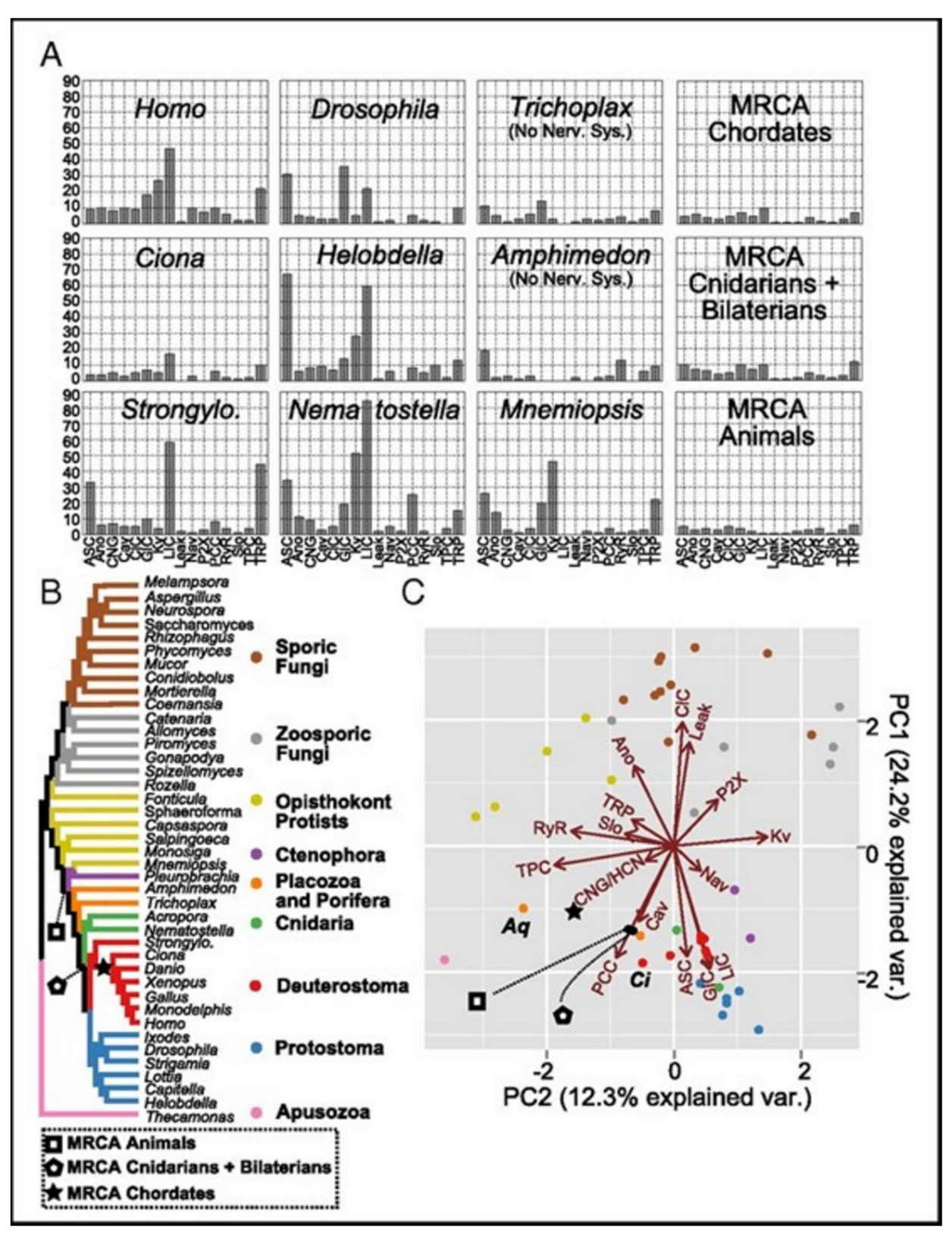
Ecdysozoans and lophotrocozoans were found to have large expansions in LIC, GIC, and Kv channels. A huge expansion in ASC family was also observed, see
Figure 5A [
15]. These expansions were observed to have happened in terminal lineages that led to every species, see
Figure 5.
Figure 5A presents the family count of ion channels from species of the major lineage. All taxa with nervous systems, with the notable exception of the tunicate Ciona, were enriched for similar gene families. Two taxa (without the nervous systems), Trichoplax and Amphimedon, were found to have smaller complements of ion channels. MRCAs of the chordates, the cnidarians plus the bilaterians, and the animals each were found to have ion-channel complements resembling the extant animals having no nervous systems more than animals having nervous systems.
6. Deep Learning Models Explain Ion Channel Features
Earlier we have addressed how ML can help understand crucial ion channel aspects. Here we wish to familiarize the role of another popular technique Deep Learning (DL) in understanding ion channels. Application of ML algorithms (e.g., in ion channel understanding) almost always requires structural (e.g., ion channel protein) data, while DL networks rely on layers of artificial neural networks. Both ML and DL are actually forms of AI, although DL is considered a specific kind of ML. Both of these AI techniques start with the training, and test the data and a model, then proceed with the process of optimization to ultimately search for the weights which make this model fit best to the data. In this section, we wish to see how DL may assist us in a developed understanding of ion channels. We must keep in mind that ion channel understanding using this new AI technique is just celebrating its beginning. So readers, though will get an introduction, may not get any fully conclusive scenario related to crucial ion channel structural and functional aspects.
6.1. Deep Learning Model Idealizes Single Molecular Activity of Ion Channels
A DL model considering the convolutional neural networks and the long short-term memory architecture has just been found. It automatically idealizes the complex activity of the single-molecule with enhanced accuracy and that the process is pretty fast, for details see ref. [
64]. The critical first step in understanding the electrophysiology technique recorded ion channel current traces lies in event detection, which is the so-called “idealization”. Here the (noisy) raw data have been are turned to the discrete protein movement trends [
65,
66]. But till today enormous practical limitations are faced in the idealization of the patch-clamp data. The highly acceptable, or quality idealization is found typically quite laborious, becomes infeasible, subjective with the complex biological data that contain various distinct native (single-ion channel) proteins’ simultaneous gatings. In the DL model of Celik and colleagues, there are no parameters to set; baseline, channel amplitude or numbers of channels for example. This DL model may therefore be useful in getting an unsupervised and automatic detection of the transition events of the single molecules.
Both the fluorescence resonance energy transfer (FRET) and the patch-clamp electrophysiology on single-molecule research are known to provide real-time data on the molecular protein state with high resolution. But the data analysis is usually very time-consuming, laborious, and requires expert-level supervision. Celik and colleagues have demonstrated that an automated event detection in patch-clamp data is possible using the deep neural network, and Deep-Channel, combining recurrent and convolutional layers. This relatively easier method is found to work with enhanced accuracy over a considerable amount of input datasets.
A hybrid recurrent convolutional neural network (RCNN) model [
64,
67] is introduced to idealize the records of ion channels, up to 5 channel events that occur simultaneously. For training and validating the models, another analog-synthetic ion channel record system generator was developed and it has been found that the our Deep-Channel model, involving long short-term memory (LSTM) and convolutional neural network (CNN) layers, idealizes rapidly with high accuracy, or detects the experimentally recorded single molecular events without the necessity of the human supervisions.
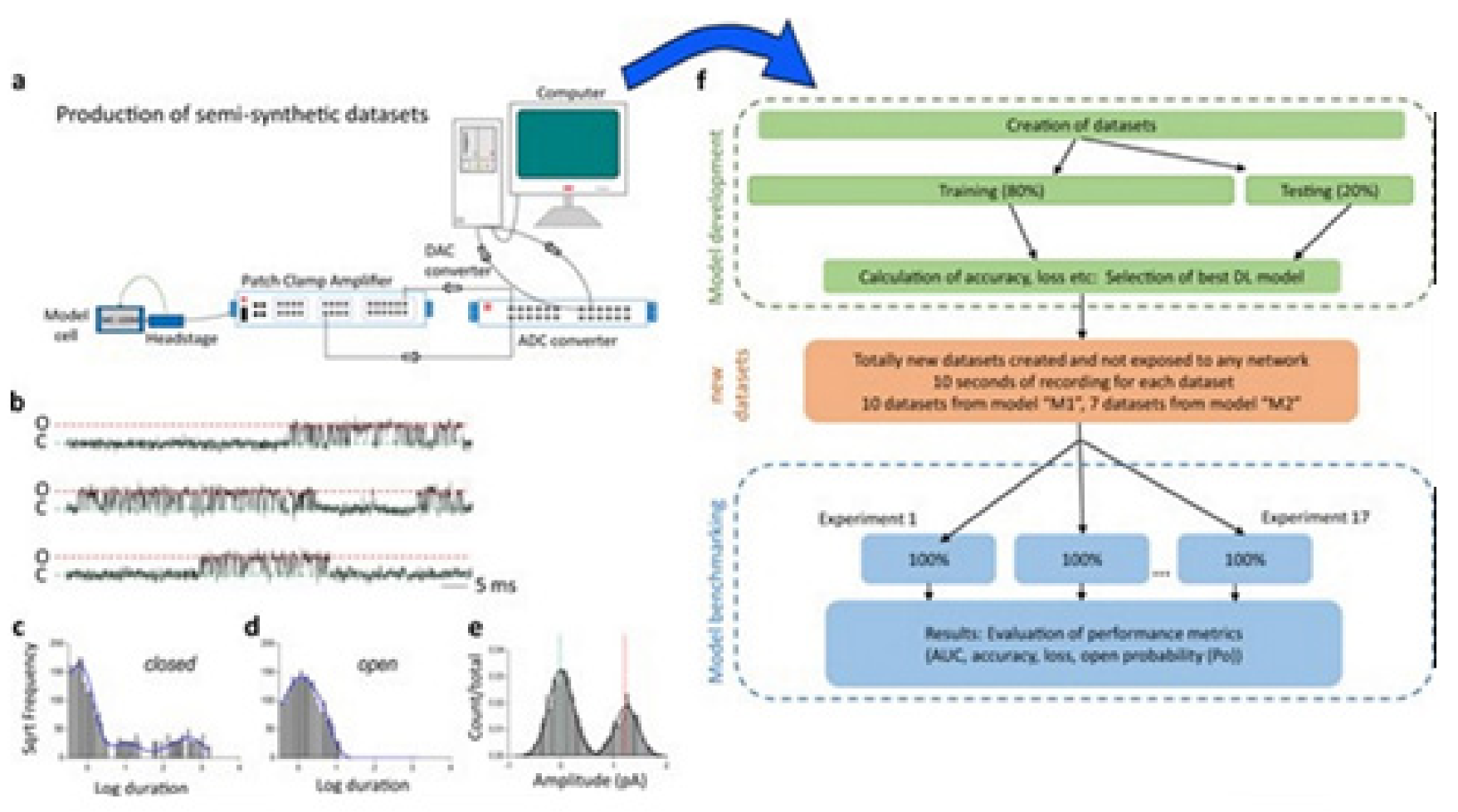
Figure 10 illustrates the data generation workflow and
Figure 11 illustrates the Deep-Channel architecture [
64]. Whilst the LSTM models were found to give a good level of performance, its combination with the time-distributed CNN was found to give higher or increased performance. This RCNN was so-called Deep-Channel by Celik and colleagues. Following the training and the model development, see methods in ref. [
64], 17 generated new datasets were used, unseen previously by the Deep-Channel, thus uninvolved in training processes. Authentic channel data, see
Figure 10b, have been generated. Two kinetic schemes, the so-called first (M1) having low ion channel opening probability, and the so-called second (M2) having a high channel opening probability were applied, and thus an average of the approximately 3 channels open at a time was obtained (
Figure 12b). Examples of the data, with the ground truth and the Deep-Channel idealization, have been shown in
Figure 13 [
64]. All of the Deep-Channel results described here have been achieved without requiring any human intervention beyond providing the script with the correct name of the file or the path.
Illustrates their overall model designing and the testing workflow. The provided Supplementary Information [
64] includes training metrics from the initial validation point and the main text shows the performance metrics that were acquired from the 17 experiments having novel datasets. In training datasets, there were typically contained millions of sample points, and the 17 benchmarking experiments were sequences of the 100,000 samples each.
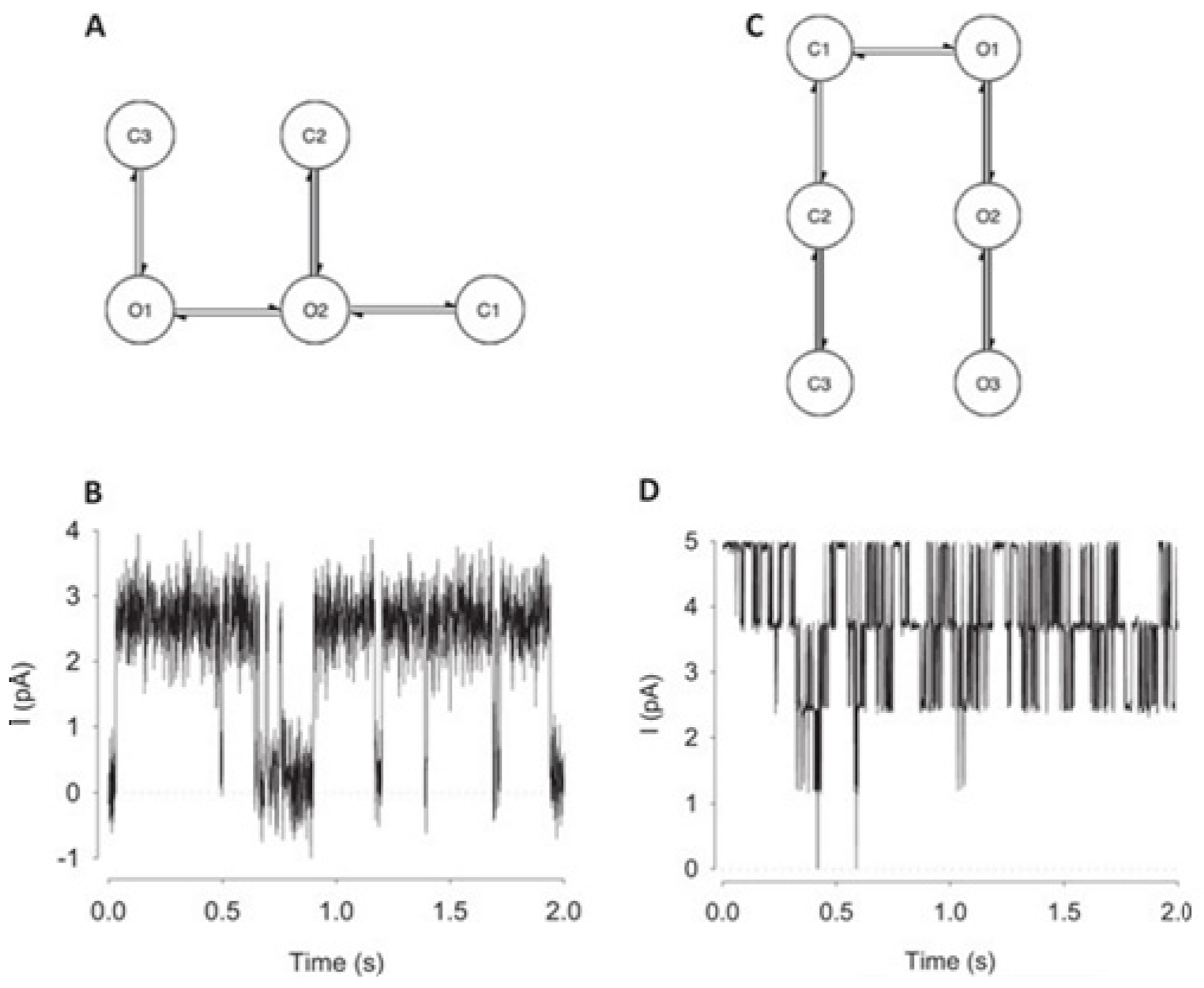
For channels having a relatively low channel-opening probability (see stochastic gating model M1,
Figure 12a), the data idealization process is found to get close to a binary-detection problem (see
Figure 13a), having the channel events’ type closed/open, labels “0”/“1”, respectively. Here, the so-called receiver operating characteristic (ROC) curve, applied for the classification of the channel events for open and closed state detections exceeds a high level of 96%. In low channel open probability case of experiments, the Deep-Channel was found to return a macro-F1 of 97.1 ± 0.02%, but the segmented-k means (SKM) method in the software package QuB was found to result in a macro-F1 of 95.5 ± 0.025%, and 50% threshold method in QuB gave a macro-F1 of 84.7 ± 0.05%, n = 10.
For datasets including the highly active ion channels (from the model M2), we get it to becoming a multi-class problem of comparison, hence the Deep-Channel was found to outperform both 50% threshold-crossing and the SKM methods in QuB quite considerably. The Deep-Channel macro-F1 was 0.87 ± 0.07, however SKM macro-F1 in QuB, without the manual-baseline corrections, was found to drop sharply to a value 0.57 ± 0.15, and the 50% threshold-crossing macro-F1 was found to fall to a value of 0.47 ± 0.37 (the student’s paired t-test between methods, p = 0.0052).
6.2. Deep Learning to Classify the Ion Transporters and the Channels from the Membrane Proteins
Recently, an article was published proposing a DL method for automatic classification of the ion transporters or pumps and the ion channels from the membrane proteins [
69]. This technique is proposed through training the deep neural networks and by using the position-specific scoring matrix profile used as the input.
From structural and behavioral perspectives ion channels are found to differ significantly from ion transporters, see ref. [
69] (reproduction of the figure is not granted). The DL method of Taju and Ou is dedicated to distinguishably classifying these two structural events. Three-stage approaches have been adopted, where 5 techniques of data normalization have been used; the next 3 imbalanced data techniques have been applied for the minority classes, then 6 classifiers have been compared to the method proposed here, for details see original article [
69]. We shall present here a brief of the results and interpretations.
The goal here is to find a method that will be able to automatically classify the ion transporters and the ion channels from a set of membrane proteins through training the deep neural networks (DNNs) that uses a convolutional neural network (CNN) as its selected algorithm capturing the hidden pattern of information in the set of data. The hidden feature that is extracted in the position-specific scoring matrix (PSSM) from the data set of proteins is thus expected to be the best feature producing the relevant evolution information related to the sequences of the proteins. More importantly, the feature obtained here should be applicable to versatile problems in the fields of bioinformatics and ML, with considerable and promising outcomes or results, when compared to other available feature extraction methods. Firstly, all protein data representation in the format of FASTA (stands for fast-all) is changed into another PSSM profiles’ format. Secondly, DL, demonstrated by the use of such representation will be able for accurately classifying some proteins separated from the data for training. Lastly, for validating this approach, 5 cross-validations are used and test the proposed method’s modeling.
The guidelines of the 5-step rule [
70] are followed for making following 5 steps clearly:
- -
the method of constructing or selecting a valid benchmark data set for training and testing the predictor;
- -
the method of formulating biological-sequence samples with the help of any effective mathematical expression which can accurately reflect their intrinsic correlation to the to-be-predicted target;
- -
the way on introducing or developing a powerful algorithm or engine for operating the prediction;
- -
the way on properly performing the tests for cross-validations for objective evaluation of the anticipated predictor accuracy;
- -
the way on establishing a friendly to the users’ web server for predictor which will be accessible to the mass public.
For details on all these five steps, readers may see ref. [
69]. In ref. [
69] (reproduction of the figure is not granted), a schematic representation of the membrane protein classification prediction steps has been provided. The dataset used is collected from the database of the Universal Protein Knowledgebase (UniProtKB) (accessed on 14 April 2018) (UniProt, 2016) (see
Table 9).
We avoid elaborating on the detailed techniques. To represent the input data, a PSSM-based feature extractor was applied here and a 20 × 20 matrix was produced. Initially, the Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) [
28] against (
ftp://ftp.ncbi.nih.gov/blast/db/FASTA/) (31 August 1997) was used for generating the PSSM profiles. The PSI-BLAST is a method for searching the protein sequence profile and the PSSM is the matrix generated utilizing a protein query that can perform PSI-BLAST search for finding its similarity from biological databases, and thus creates the (position-specific) matrix. For every query of protein, PSSM can produce N × 20 matrix having a component of the profile, where N denotes the protein sequence length and columns represent the scores for the substitution of the amino acids in the protein. For details, see ref. [
69].
The 20 amino acids composition analysis, the n-gram analysis, the sequence motif visualization with the use of the word cloud technique have been shown. The 20 amino acid residues variance has been computed considering the 3 protein data set classes. The experiments compared the DL model performance against 5 different techniques of data normalization and 3 techniques for oversampling. The model was evaluated using the k-fold cross-validation. The best performance of the model was then compared with a few classifiers, such as the Perceptron Gaussian Naïve Bayes, the Random Forest, the Nearest Neighbors, the SVM, and the Nearest Centroid classifiers with the use of independent test data for examining different algorithms’ effects. The analysis of the sequence got performed on the platform of the training data for finding a little information on the amino acids and base pair of the residue patterns at the important motifs in the sets of the data. In ref. [
69] (reproduction of the figure is not granted), we see the amino acids having letter Ala (A), Gly (G), Leu (L), Ser (S), and Val (V) have been dominant and also important, as we see in amino acid composition figure or the amino acids occurrence frequency in all of the proteins. The 20 amino acid residues variance across ion channels, ion transporters, membrane proteins have been computed. The variance analysis got used for measuring the data spreading distance away from the overall average value. Amino acids Leu (L), Ser (S), Ala (A), Val (V), Gly (G), Glu (E), Ile (I), Arg (R), and Thr (T) with high frequencies in analysis top frequent motifs showed a variance value below 0.005, and Cys (C), Lys (K), and Trp (W) showed a variance value above 0.005 of 0.013, 0.012, and 0.032, respectively.
The datasets were then classified to distinguish three classes, namely ion channels (class A), ion transporters (class B), and other proteins (class C) using the following techniques, for details (not presented here) see ref. [
69]:
- -
Comparative results were extracted using different techniques for feature normalization
- -
Comparative results on different techniques for the imbalanced data set
To evaluate the predictor model performance, 5-fold cross validations have been applied in training data sets.
Table 10 reports the results of the fivefold cross-validation technique that was applied in the training data, which is a challenging step to find the best model prediction of independent test sets. The performance is seen to reach the highest Sen (89.20%), Spec (84.89%), Overall Acc (87.05%), and MCC (0.75) for class A. Class B achieves Sen (86.76%), Spec (88.23%), Overall Acc (87.49%), and MCC (0.75), and performance of Sen (92.50%), Spec (96.19%), Overall Acc (94.35%), and MCC (0.89) are seen for class C.
The application of all these Deep-Channel algorithms and models has been found possible, though with limitations, for the case of biological data on ion channels. We have presented here basically two example studies where DL algorithms have been utilized to demonstrate various ion channel features. The effectiveness of Deep-Channel to detect events in the single-molecule datasets has been mainly demonstrated. The method is exclusively applicable not only for patch-clamp experimental data, but it has potential for the deep learning convolution or LSTM networks for tackling other related biological data analysis problems. The ion channel, ion pump, and other membrane protein classification using DL algorithms and modeling has been found quite impressive and time and resource saving initiative. Over the next decade, we may see an exponential increase in use of AI, ML, and DL in understanding natural status and mutated conditions of ion channels of biological cells.
7. ML in Ion Channel Engineering
AI techniques are found helpful in ion channel engineering. Recently, ML is reported to help in designing the membrane hosted channelrhodopsins (ChRs) for the eukaryotic expression and the plasma membrane localization efficiently [
14]. Here a predictive ML approach has been used that can capture the complexity and facilitates successfully the MP design and engineering. The application of ML on the training sequences that are made by the structure-guided SCHEMA recombination enables to predict accurately the rare sequences in a library of pretty diverse members of channelrhodopsins (ChRs), expressed and localized to the mammalian cells’ plasma membranes.
In protein engineering, membrane protein (MP) sequence changes, influencing expression and membrane localization, are highly context-dependent. That means what changes are found to eliminate localization in one sequence context may have almost no effect in another. Subtle amino acid changes may have dramatic effects [
14,
71]. It is conclusive that the MP sequence determinants of expression and membrane localization are not necessarily captured by ordinary rules as applied in studies [
72] providing information on signal peptide sequence having positive charge at the membrane–cytoplasm interface “positive-inside” rule [
73], and an enhanced hydrophobicity in transmembrane domains.
Considering all available knowledge, it is clear that any accurate atomistic models considering physics principles relating a sequence to its expression and plasma membrane localization levels aren’t available due to advanced level stochasticity and large scale complexity of the biological process. Statistical models, offering an alternative, are useful to predict the outcomes of any complex processes, because they do rely on energetics of the system (e.g., for ion channels see ref. [
17,
18]) and not require any prior knowledge of underlying mechanisms. Empirical data such as expression or localization values of MPs’ known sequences can be used to train statistical models. While training, this model infers input/output relationships between the sequence as input and expression or localization as output. These relationships are then used to make predictions on the properties of the unmeasured sequence variants. The process of the use of empirical data for training and selecting optimal statistical models is referred to as ML [
14].
In predicting protein properties such as solubility [
74], crystallization propensity, periplasm trafficking [
75], and general functions [
76] ML has been found useful. Although these models can identify protein sequence elements that are predicted to contribute to specific property of interest in the respective studies, they are not generally useful to identify subtle sequences and features, such as amino acids or their interactions. Specific condition of expression and pinpointed localization for any certain class of any related sequences (e.g., of ChRs) aren’t identifiable with confidence. The common reason behind this is that the ML models utilized here are trained using many protein classes and utilizing large data sets that are composed of literature/published data from various sources having almost no standardization on the versatile experimental conditions, and trained using many protein classes. Bedbrook and colleagues focused on a model building on ChRs, utilizing training data that are collected from a considerable range of ChR sequences and under standardized conditions [
14]. Here the Gaussian process (GP) classification has been applied and the regression followed from ref. [
77] to build ML models that are able to predict expression and localization of ChR directly from the data. Sitting on a background of their earlier work where GP models were found to successfully predict a few biophysical conditions such as the thermal stability, binding affinity, enzyme kinetics, etc. [
78]. In this work Arnold group asked whether GP models could successfully predict mammalian expression and heterologous integral membrane localization of ChRs, and to do so the amount of experimental data might be required. To generate a training set, SCHEMA recombination chimeras were used, which are useful for producing large scale libraries of quite diverse, functional chimeric sequences from parent protein homologues [
79]. Synthesis and measurements of expression and localization were made for a small subset (0.18%) of the sequences from the recombination library of ChR. These data were used to train GP classification and regression models and predict the expression and localization properties of ChR sequences.
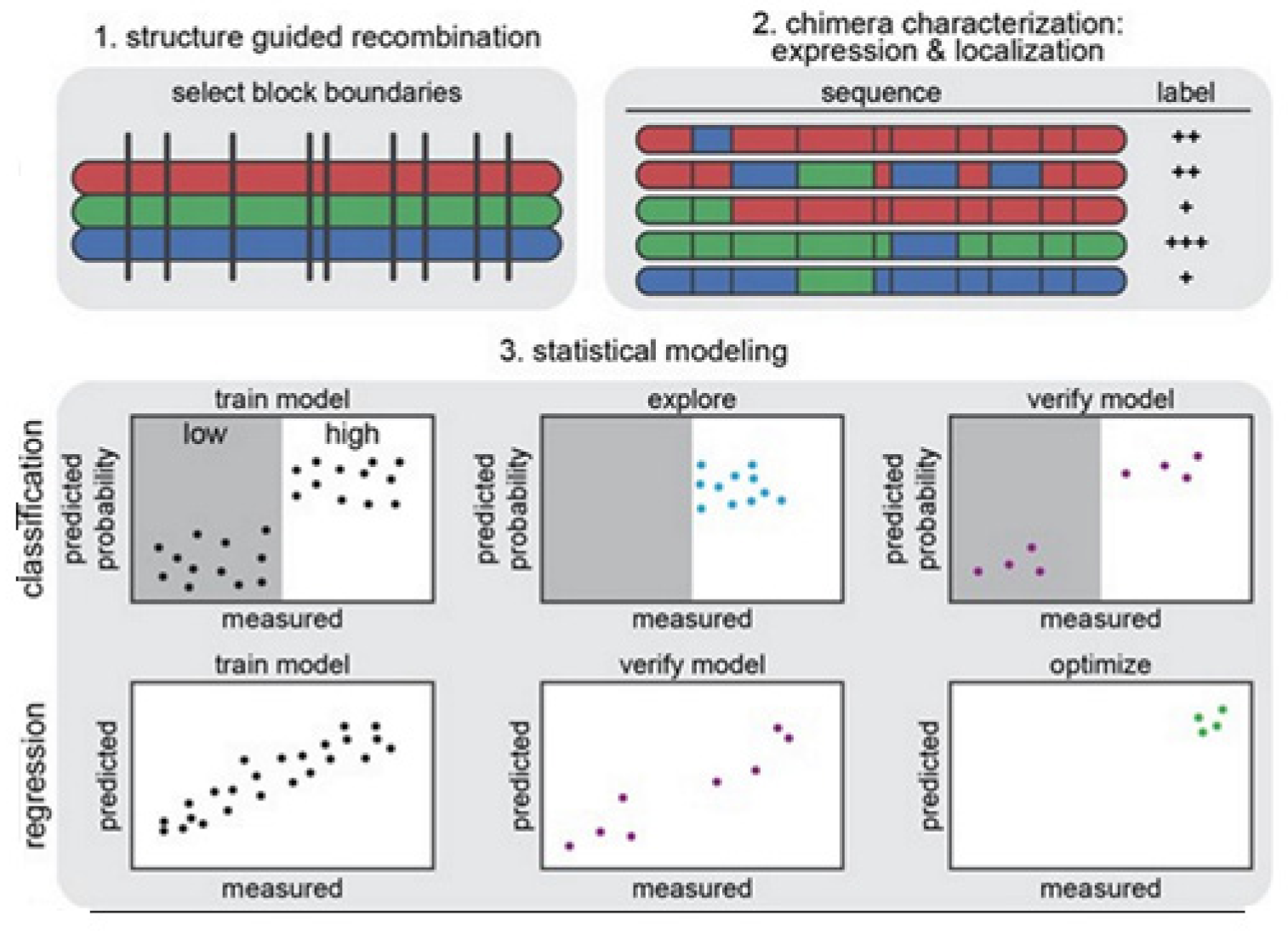
The strategy on development of the predictive ML models is illustrated in
Figure 14 [
14]. In
Figure 14 (1) we see that the structure-guided recombination SCHEMA is for selecting the block boundaries. This is done for the purpose of shuffling the sequences of the protein and generate any library for sequence-diverse ChRs by starting with 3 ChRs parents, referring to 3 colors (red, green, and blue). In
Figure 14 (2), we present a library subset which will serve as the set for the training. The genes of the chimeras have been synthesized and cloned into the mammalian expression vector, transfected cells being assayed for ChR expressions and localizations. In
Figure 14 (3), 2 models, classification and regression, are trained with the utilization of the training data and then verified. The model for classification here is used for exploring the diverse sequences that are predicted to show ‘high’ level of localizations. The model for regression here is used for designing the ChRs having the optimal level of localizations to plasma membranes.
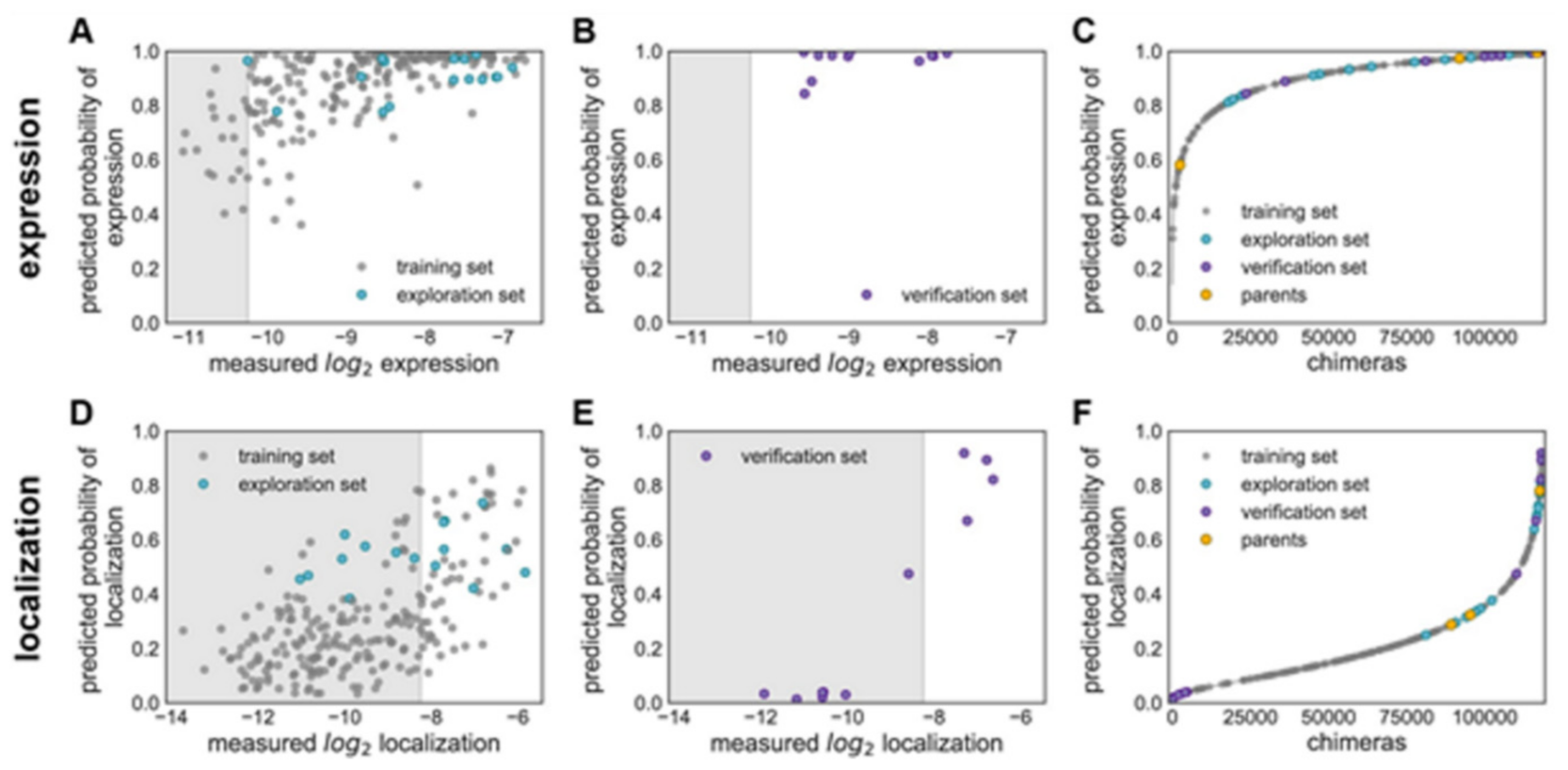
Figure 15 explains various features on building GP classification models of ChR properties, for details including data sets used see ref. [
14]. In
Figure 15, the plots of the predicted probability versus the measured properties have been divided into many sections, the ‘high’ performer represented by the white background, and the ‘low’ performer represented by the gray background, for each property: expression and localization. In
Figure 15A,D, the predicted probability vs. measured properties for the training set (see gray points) and the exploration set (see cyan points). LOO cross-validation was utilized for predictions for training and exploration sets. In
Figure 15B,E, the predicted probabilities vs. measured properties for the verification set are presented. A model trained on the training and exploration sets was utilized for predictions. In
Figure 15C,F, the predicted probability of the ‘high’ expression, and the localization for all chimeras in the recombination library (having 118,098 chimeras) that is made by the use of the models, trained on data taken from the training and the exploration sets. All the library contained chimeras are shown by the gray lines; the gray, cyan, purple, and yellow points indicating the sets for training, exploration, verification, and the parents, respectively.
Figure 15A–C Show the expression and D–F the localization.
Multi-block-swap sequences (from the training set) mostly did not localize to the membrane. The model for the localization classification was used for identifying the multi-block-swap chimeras of the library, having a pretty high probability of prediction, >0.4, falling into a ‘high’ localizer category (see previous
Figure 15D). Among the multi-block-swap chimeras having the predicted ‘high’ localization, a 16 diverse chimeras set (having average of 69 mutations in amino acids) of the closest parent have been chosen and then named as ‘exploration’, see ref. [
14]. Bedbrook and colleagues synthesized and tested these chimeras, and found that the model had accurately predicted chimeras showing good localization (
Figure 15 and
Figure 16). 50% of the exploration set was found to show ‘high’ localization compared to just 12% of the multi-block-swap sequences from the original training set, although both have similar levels of mutation, data shown in [
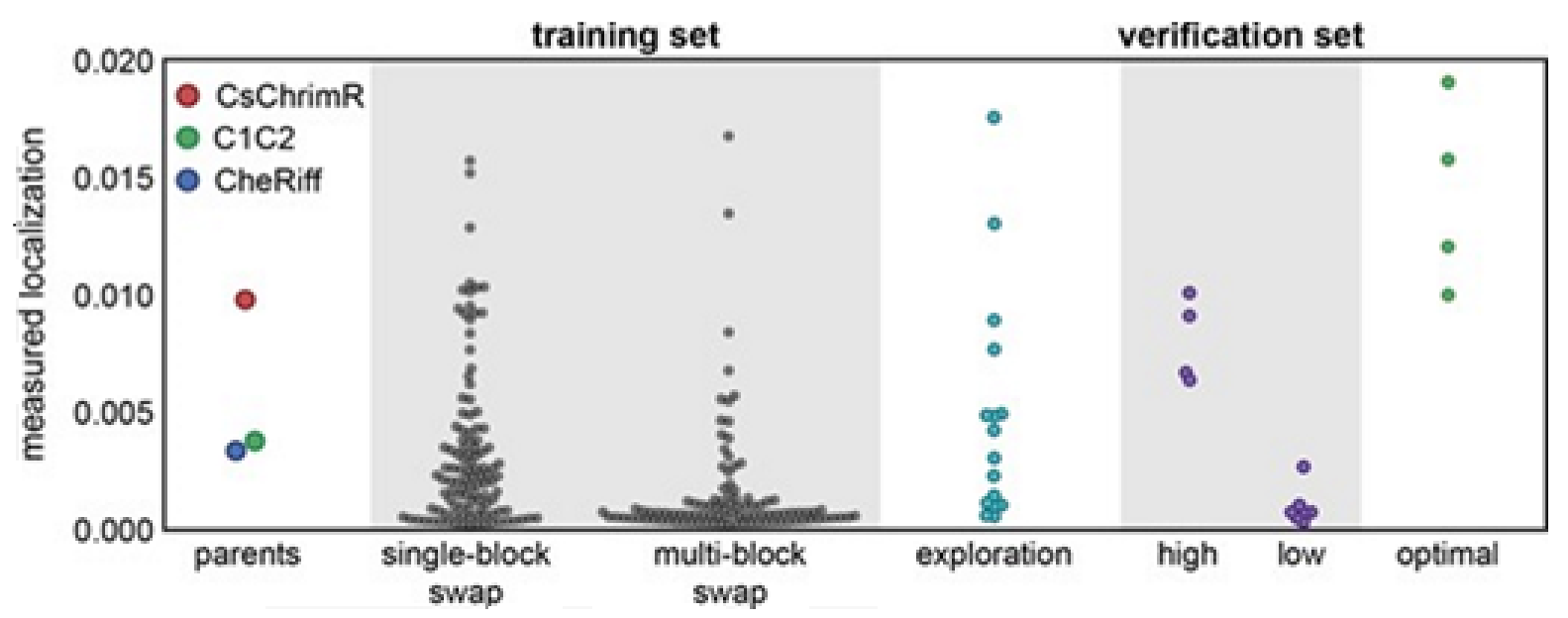
14]. Exploration set chimeras to have on average 69 ± 12 amino acid mutations from the closest parent versus 73 ± 21 for multi-block-swap chimeras in the training set.
Although the model for the classification predicts the probability of any sequence that falls into the ‘high’ localizer category, it may lack in giving a necessary quantitative prediction. The designing of the chimera sequences having the optimal localization has then been made [
14]. For optimal localization, one has to be at or above the CsChrimR level, which is the best localizing parent [
80]. A regression model for the localization of ChR in the plasma membrane is required to help predict sequences with optimal localization. A GP regression model is presented which utilized the localization data from training and exploration sets [
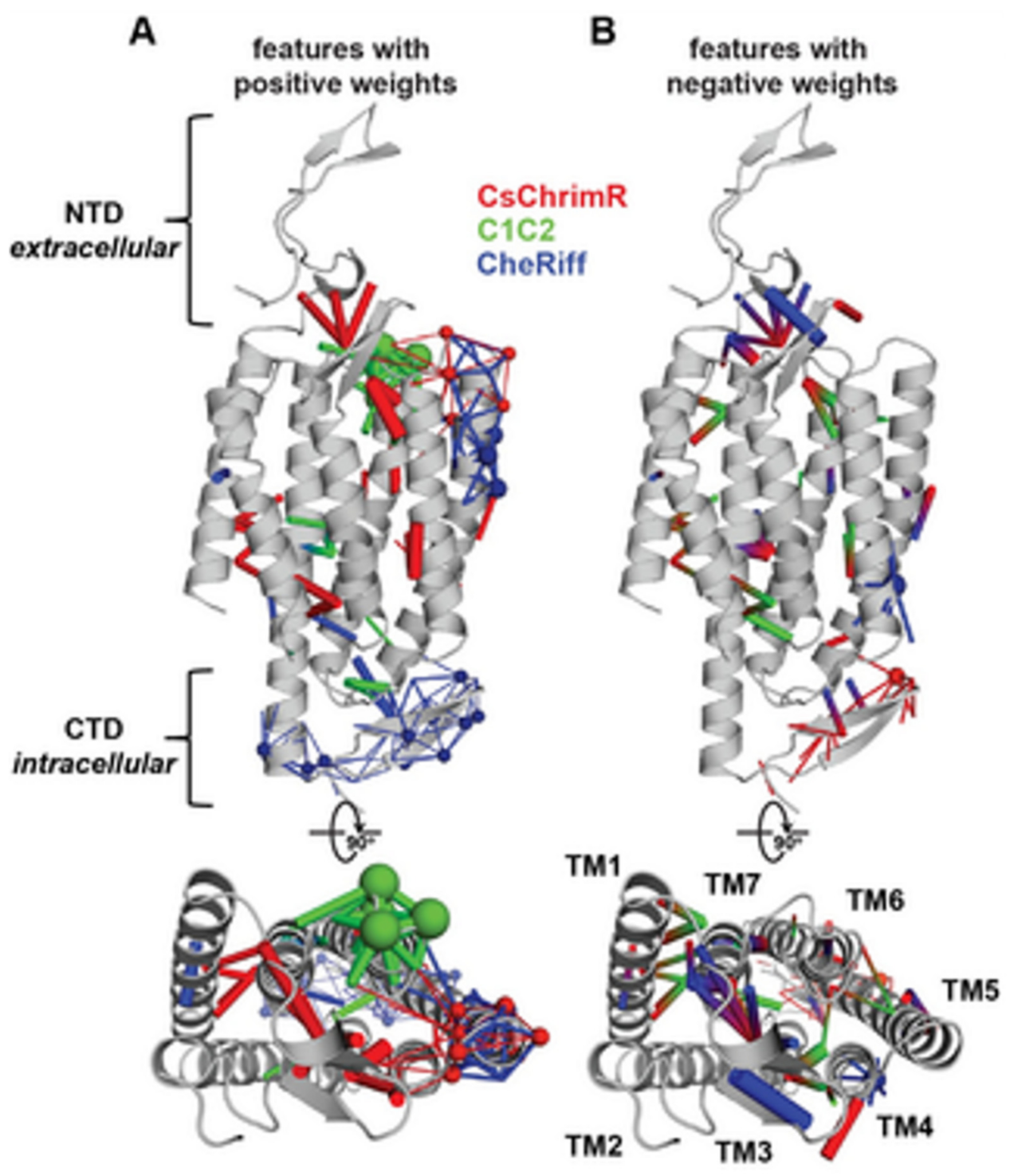
14]. While developing the GP regression model for the localization, L1-regularized linear regression was used to identify a limited set of sequences and specific structural features that are known to strongly influence the ChR localization. The features include inter-residue contacts and individual residues, and help to offer insight into the structural determinants for the ChR localization. While mapping onto the C1C2 structure, these features can highlight parts of the ChR sequence and the structural contacts which are important for ChR’s plasma membrane localization, see
Figure 17 [
14]. Both beneficial and deleterious features are distributed throughout the protein, with no single feature dictating localization properties (
Figure 17).
In
Figure 17, the features with the positive (A) and the negative (B) weights have been displayed on the crystal structure of C1C2 (in grey color). The features can be the residues (see spheres) or the contacts (sticks) from the ChRs parents. The CsChrimR features have been shown in color ‘red’, the features from the C1C2 are presented in color ‘green’, and the features from the CheRiff are presented in color ‘blue’. For cases where any feature appears in 2 parents, the color priorities have been used differently, as follows: the red above and the green above the blue. Sticks are shown to connect beta carbons of the contacting residues (or specifically the alpha carbon for the glycine). The spheres’ size and the sticks’ thickness have been used as proportional to the weights of the parameter. 2 contact residues can either be from the same or different parents. The Single-color contacts occur as both contributing residues appear from the same parent. The occurrence of multi-color contacts happens when residues from different parents come in contact. N-terminal domain (NTD), C-terminal domain (CTD), seven transmembrane helices (TM1-7) are labeled.
GP regression model as briefed here can be utilized to engineer novel sequences that localize better [
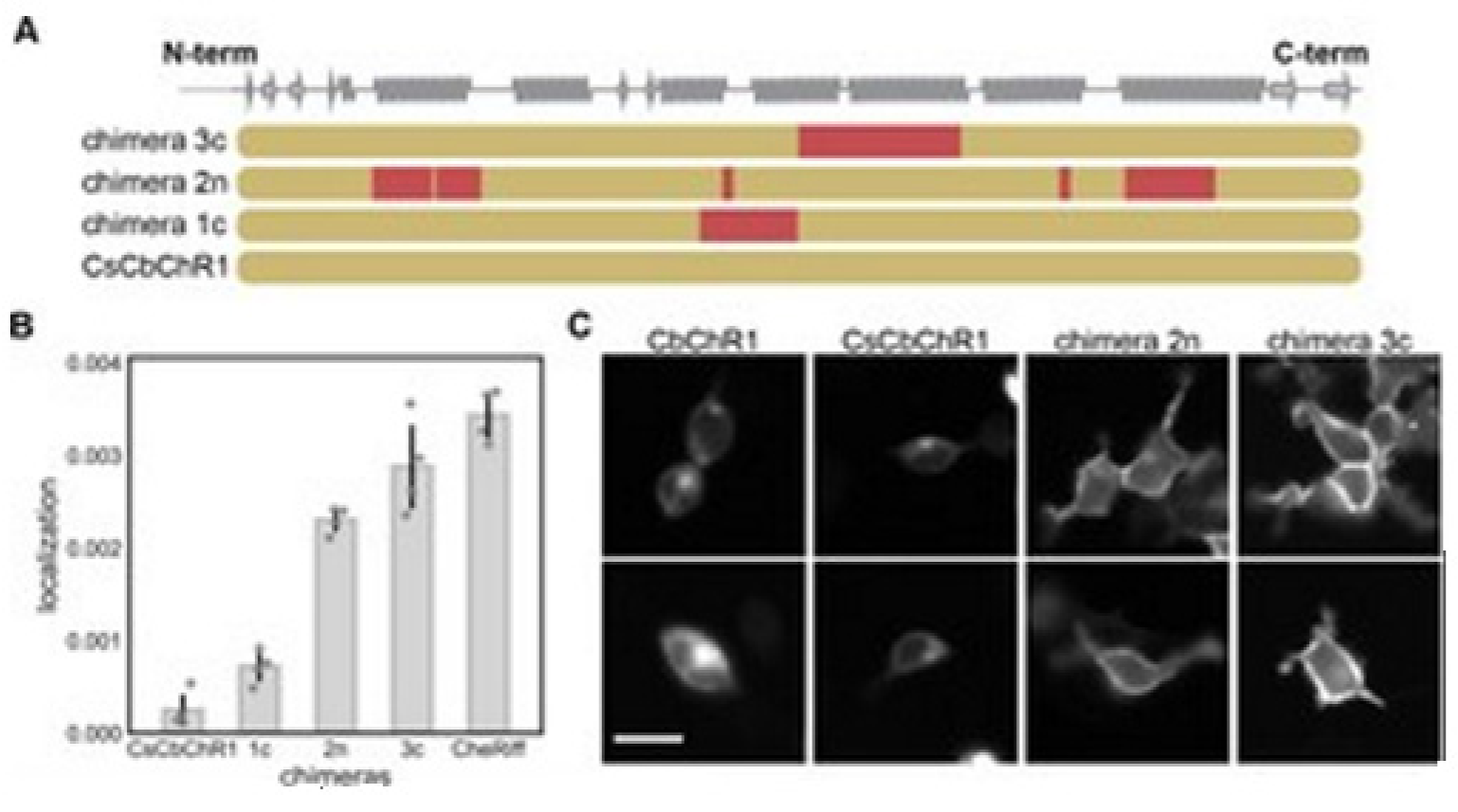
14]. Bedbrook and colleagues chose a nonfunctional natural ChR variant, CbChR1, expressed in HEK cells and neurons, but does not localize to the plasma membrane [
80]. Being distant from three parental sequences CbChR1 is only 60% identical to CsChrimR and 40% to CheRiff and C1C2. CbChR1 was optimized by introducing minor changes in amino acids, predicted by the localization regression model might be beneficial for membrane localization. To enable CbChR1 localization measurements with the SpyTag-based labeling method, the N-terminus of CbChR1 was substituted with the CsChrimR N-terminus that contains the SpyTag sequence downstream of the signal peptide to make the chimera CsCbChR1 [
81]. This block swap was not found to cause any change in CbChR1′s membrane localization properties, see
Figure 18C.
Figure 18A presents the blocks’ identities in CsCbChR1 chimeras-each row is representing a chimera. Yellow color representation for the CbChR1 parent and red color representation for the CsChrimR parent. The Chimeras 1c, 2n, and 3c contain 4, 21, and 17 mutations, respectively with respect to the CsCbChR1. In
Figure 18B, the plot represents the measured CsCbChR1 localization, compared to 3 CsCbChR1 single-block-swap chimeras and CheRiff parent. In
Figure 18C, 2 cell images of mKate expression in CbChR1 and CsCbChR1 compared with top-performing CsCbChR1 single-block-swap chimeras showing the differences in the ChR localization properties–the chimera 2n and the chimera 3c localize to the plasma membrane. The bar of the scale here is 20 μm.
The classification and the models of the regression models of the GP were trained using the expression and the localization data that were collected from the 218 ChR chimeras, which were chosen from the library of 118,098 variants designed using the SCHEMA recombination of three ChRs parents. The GP models were used for identifying the ChRs, which were expressed and well-localized, showing that these models elucidate the sequence and the structure elements important for the processes.
Bedbrook and colleagues have successfully detailed the steps in building ML models and highlighted these artificial techniques’ power in predicting certain protein properties considering a specific ion channel protein ChR [
14]. Combining recombination-based library design with statistical modeling methods, they could scan a highly functional portion of protein sequence space through training on only a few sequences. Model developments have yielded a tool having been used to not only predict optimally performing chimeric proteins but also be applied to improve related ChR proteins that are outside the library. These ML methods may appear as powerful tools for general protein engineering in general. As shown here for ChR these ML models may also be found applicable to regulate any ion channel functions by engineering the channel proteins.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}