
Homomeric and Heteromeric α7 Nicotinic Acetylcholine Receptors in Health and Some Central Nervous System Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Dendritic Spines, the Postsynaptic Specialization of Excitatory Synapses in the Brain
3. Fine Structure of the Neuronal nAChR Molecule
4. Functional Pharmacology of Brain nAChR-Mediated Synaptic Plasticity
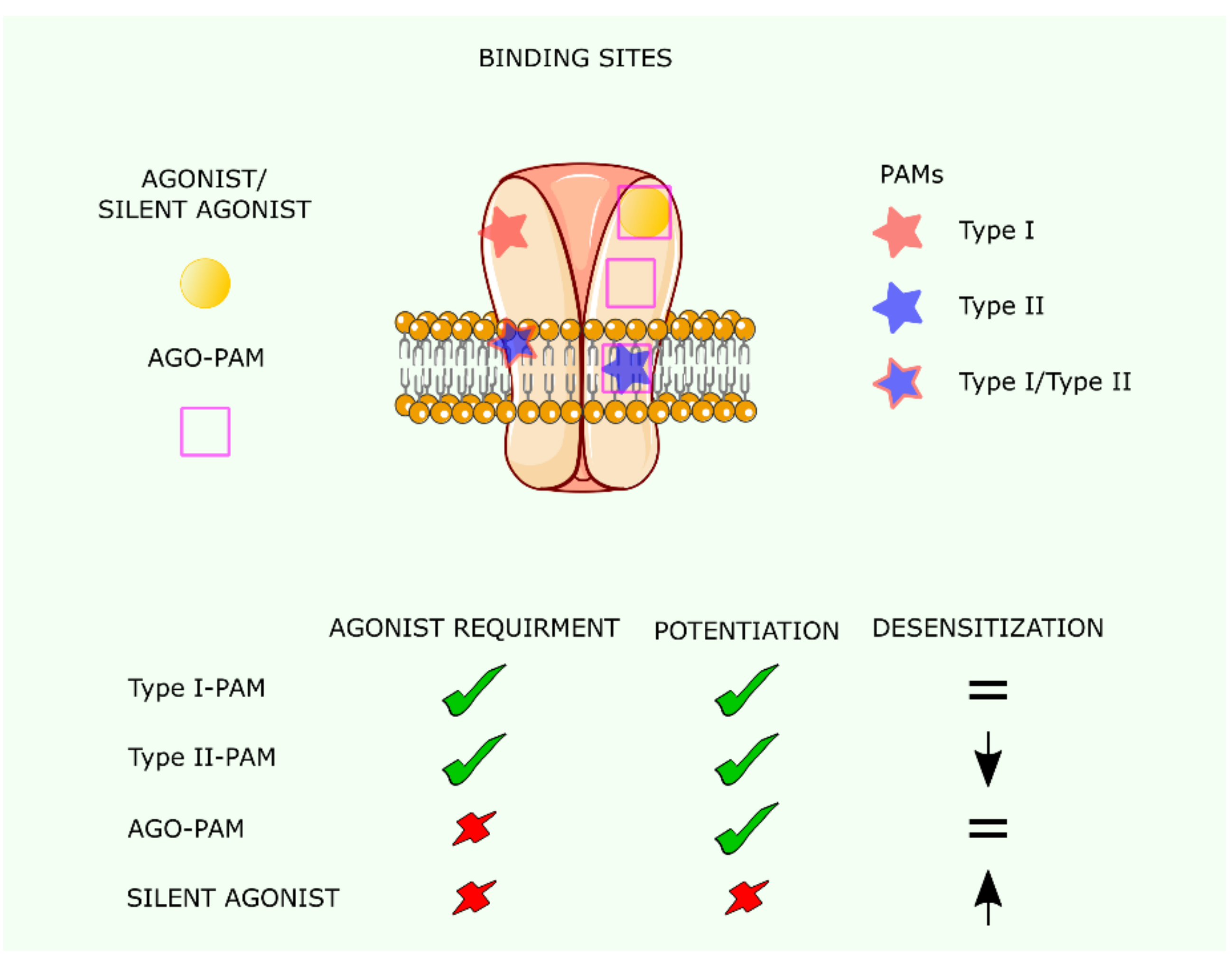
5. Positive Allosteric Modulators (PAMs) and Silent Agonists of the nAChR
6. Dendritic Spines and nAChR-Mediated Synaptic Plasticity
7. α7 nAChR in Disorders of the CNS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Alkondon, M.; Pereira, E.F.; Albuquerque, E.X. α-Bungarotoxin-and Methyllycaconitine-Sensitive Nicotinic Receptors Mediate Fast Synaptic Transmission in Interneurons of Rat Hippocampal Slices. Brain Res. 1998, 810, 257–263. [Google Scholar] [CrossRef]
- Yan, Y.; Peng, C.; Arvin, M.C.; Jin, X.-T.; Kim, V.J.; Ramsey, M.D.; Wang, Y.; Banala, S.; Wokosin, D.L.; McIntosh, J.M. Nicotinic Cholinergic Receptors in VTA Glutamate Neurons Modulate Excitatory Transmission. Cell Rep. 2018, 23, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Hellström-Lindahl, E.; Gorbounova, O.; Seiger, Å.; Mousavi, M.; Nordberg, A. Regional Distribution of Nicotinic Receptors during Prenatal Development of Human Brain and Spinal Cord. Dev. Brain Res. 1998, 108, 147–160. [Google Scholar] [CrossRef]
- Murakami, K.; Ishikawa, Y.; Sato, F. Localization of A7 Nicotinic Acetylcholine Receptor Immunoreactivity on GABAergic Interneurons in Layers I–III of the Rat Retrosplenial Granular Cortex. Neuroscience 2013, 252, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Caruncho, H.J.; Guidotti, A.; Lindstrom, J.; Costa, E.; Pesold, C. Subcellular Localization of the A7 Nicotinic Receptor in Rat Cerebellar Granule Cell Layer. Neuroreport 1997, 8, 1431–1433. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Bizri, H.; Clarke, P.B.S. Blockade of Nicotinic Receptor-Mediated Release of Dopamine from Striatal Synaptosomes by Chlorisondamine Administered in Vivo. Br. J. Pharmacol. 1994, 111, 414–418. [Google Scholar] [CrossRef] [Green Version]
- Lena, C.; Changeux, J.-P.; Mulle, C. Evidence for “Preterminal” Nicotinic Receptors on GABAergic Axons in the Rat Interpeduncular Nucleus. J. Neurosci. 1993, 13, 2680–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bali, Z.K.; Nagy, L.V.; Hernádi, I. Alpha7 Nicotinic Acetylcholine Receptors Play a Predominant Role in the Cholinergic Potentiation of N-Methyl-D-Aspartate Evoked Firing Responses of Hippocampal CA1 Pyramidal Cells. Front. Cell. Neurosci. 2017, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- El-Bizri, H.; Clarke, P.B.S. Blockade of Nicotinic Receptor-Mediated Release of Dopamine from Striatal Synaptosomes by Chlorisondamine and Other Nicotinic Antagonists Administered in Vitro. Br. J. Pharmacol. 1994, 111, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Koukouli, F.; Maskos, U. The Multiple Roles of the A7 Nicotinic Acetylcholine Receptor in Modulating Glutamatergic Systems in the Normal and Diseased Nervous System. Biochem. Pharmacol. 2015, 97, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J.S.; Yin, J.; Long, C.; Spillman, E.; Sheng, C.; Yuan, Q. Temporal Regulation of Nicotinic Acetylcholine Receptor Subunits Supports Central Cholinergic Synapse Development. bioRxiv 2019, 790659. [Google Scholar] [CrossRef] [Green Version]
- Voytenko, L.P.; Lushnikova, I.V.; Savotchenko, A.V.; Isaeva, E.V.; Skok, M.V.; Lykhmus, O.Y.; Patseva, M.A.; Skibo, G.G. Hippocampal GABAergic Interneurons Coexpressing Alpha7-Nicotinic Receptors and Connexin-36 Are Able to Improve Neuronal Viability under Oxygen–Glucose Deprivation. Brain Res. 2015, 1616, 134–145. [Google Scholar] [CrossRef]
- De Jaco, A.; Bernardini, L.; Rosati, J.; Maria Tata, A. Alpha-7 Nicotinic Receptors in Nervous System Disorders: From Function to Therapeutic Perspectives. Cent. Nerv. Syst. Agents Med. Chem. 2017, 17, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Stojakovic, A.; Espinosa, E.P.; Farhad, O.T.; Lutfy, K. Effects of Nicotine on Homeostatic and Hedonic Components of Food Intake. J. Endocrinol. 2017, 235, R13–R31. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, C.; Shichkova, P.; Keller, D.; Markram, H.; Ramaswamy, S. Cellular, Synaptic and Network Effects of Acetylcholine in the Neocortex. Front. Neural Circuits 2019, 13, 24. [Google Scholar] [CrossRef]
- Gandelman, J.A.; Newhouse, P.; Taylor, W.D. Nicotine and Networks: Potential for Enhancement of Mood and Cognition in Late-Life Depression. Neurosci. Biobehav. Rev. 2018, 84, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lykhmus, O.; Kalashnyk, O.; Uspenska, K.; Skok, M. Positive Allosteric Modulation of Alpha7 Nicotinic Acetylcholine Receptors Transiently Improves Memory but Aggravates Inflammation in LPS-Treated Mice. Front. Aging Neurosci. 2020, 11, 359. [Google Scholar] [CrossRef]
- Sabec, M.H.; Wonnacott, S.; Warburton, E.C.; Bashir, Z.I. Nicotinic Acetylcholine Receptors Control Encoding and Retrieval of Associative Recognition Memory through Plasticity in the Medial Prefrontal Cortex. Cell Rep. 2018, 22, 3409–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukouli, F.; Rooy, M.; Changeux, J.-P.; Maskos, U. Nicotinic Receptors in Mouse Prefrontal Cortex Modulate Ultraslow Fluctuations Related to Conscious Processing. Proc. Natl. Acad. Sci. USA 2016, 113, 14823–14828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caton, M.; Ochoa, E.L.; Barrantes, F.J. The Role of Nicotinic Cholinergic Neurotransmission in Delusional Thinking. NPJ Schizophr. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pucci, S.; Fasoli, F.; Moretti, M.; Benfante, R.; Di Lascio, S.; Viani, P.; Daga, A.; Gordon, T.J.; McIntosh, M.; Zoli, M. Choline and Nicotine Increase Glioblastoma Cell Proliferation by Binding and Activating A7-and A9-Containing Nicotinic Receptors. Pharmacol. Res. 2021, 163, 105336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wilson, K.; Uteshev, V.; He, J.J. Activation of A7 Nicotinic Acetylcholine Receptor Ameliorates HIV-Associated Neurology and Neuropathology. Brain 2021. on-line ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P. The Nicotinic Acetylcholine Receptor: A Typical ‘Allosteric Machine’. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170174. [Google Scholar] [CrossRef] [PubMed]
- Girod, R.; Crabtree, G.; Ernstrom, G.; Ramirez-Latorre, J.; McGehee, D.; Turner, J.; Role, L. Heteromeric Complexes of A5 and/or A7 Subunits: Effects of Calcium and Potential Role in Nicotine-Induced Presynaptic Facilitation. Ann. N. Y. Acad. Sci. 1999, 868, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Khiroug, S.S.; Harkness, P.C.; Lamb, P.W.; Sudweeks, S.N.; Khiroug, L.; Millar, N.S.; Yakel, J.L. Rat Nicotinic ACh Receptor A7 and Β2 Subunits Co-Assemble to Form Functional Heteromeric Nicotinic Receptor Channels. J. Physiol. 2002, 540, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Maggi, L.; Barabino, B.; Eusebi, F.; Ballivet, M. Nicotinic Acetylcholine Receptors Assembled from the A7 and Β3 Subunits. J. Biol. Chem. 1999, 274, 18335–18340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado, M.; Valor, L.M.; Mulet, J.; Gerber, S.; Sala, S.; Sala, F. Expression and Functional Properties of A7 Acetylcholine Nicotinic Receptors Are Modified in the Presence of Other Receptor Subunits. J. Neurochem. 2012, 123, 504–514. [Google Scholar] [CrossRef]
- Cui, C.; Booker, T.K.; Allen, R.S.; Grady, S.R.; Whiteaker, P.; Marks, M.J.; Salminen, O.; Tritto, T.; Butt, C.M.; Allen, W.R. The Β3 Nicotinic Receptor Subunit: A Component of α-Conotoxin MII-Binding Nicotinic Acetylcholine Receptors That Modulate Dopamine Release and Related Behaviors. J. Neurosci. 2003, 23, 11045–11053. [Google Scholar] [CrossRef] [PubMed]
- Drisdel, R.C.; Green, W.N. Neuronal α-Bungarotoxin Receptors Are A7 Subunit Homomers. J. Neurosci. 2000, 20, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotti, C.; Hanke, W.; Maury, K.; Moretti, M.; Ballivet, M.; Clementi, F.; Bertrand, D. Pharmacology and Biophysical Properties of A7 and A7-A8 α-Bungarotoxin Receptor Subtypes Immunopurified from the Chick Optic Lobe. Eur. J. Neurosci. 1994, 6, 1281–1291. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, Y.; Xue, F.; Simard, A.; DeChon, J.; Li, G.; Zhang, J.; Lucero, L.; Wang, M.; Sierks, M. A Novel Nicotinic Acetylcholine Receptor Subtype in Basal Forebrain Cholinergic Neurons with High Sensitivity to Amyloid Peptides. J. Neurosci. 2009, 29, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Dajas-Bailador, F.A.; Mogg, A.J.; Wonnacott, S. Intracellular Ca2+ Signals Evoked by Stimulation of Nicotinic Acetylcholine Receptors in SH-SY5Y Cells: Contribution of Voltage-Operated Ca2+ Channels and Ca2+ Stores. J. Neurochem. 2002, 81, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, N.; Nichols, R.A. Beyond the Channel: Metabotropic Signaling by Nicotinic Receptors. Trends Pharmacol. Sci. 2018, 39, 354–366. [Google Scholar] [CrossRef]
- Moretti, M.; Zoli, M.; George, A.A.; Lukas, R.J.; Pistillo, F.; Maskos, U.; Whiteaker, P.; Gotti, C. The Novel A7β2-Nicotinic Acetylcholine Receptor Subtype Is Expressed in Mouse and Human Basal Forebrain: Biochemical and Pharmacological Characterization. Mol. Pharmacol. 2014, 86, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.R.; Kabbani, N. Alpha 7 Nicotinic Receptor Coupling to Heterotrimeric G Proteins Modulates RhoA Activation, Cytoskeletal Motility, and Structural Growth. J. Neurochem. 2016, 138, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.R.; Nordman, J.C.; Bridges, S.P.; Lin, M.-K.; Kabbani, N. Identification and Characterization of a G Protein-Binding Cluster in A7 Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2015, 290, 20060–20070. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Zwart, R.; Ursu, D.; Jensen, M.M.; Pinborg, L.H.; Gilmour, G.; Wu, J.; Sher, E.; Mikkelsen, J.D. α7 and β2 Nicotinic Acetylcholine Receptor Subunits Form Heteromeric Receptor Complexes that Are Expressed in the Human Cortex and Display Distinct Pharmacological Properties. PLoS ONE 2015, 10, e0130572. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.E.; Minguez, T.; Bermudez, I.; Bouzat, C. Molecular function of the novel α7β2 nicotinic receptor. Cell Mol. Life Sci. 2018, 75, 2457–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.A.; Vieira, J.M.; Xavier-Jackson, C.; Gee, M.T.; Cirrito, J.R.; Bimonte-Nelson, H.A.; Picciotto, M.R.; Lukas, R.J.; Whiteaker, P. Implications of Oligomeric Amyloid-Beta (oAβ42) Signaling through α7β2-Nicotinic Acetylcholine Receptors (nAChRs) on Basal Forebrain Cholinergic Neuronal Intrinsic Excitability and Cognitive Decline. J. Neurosci. 2021, 41, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Hansen, H.H.; Hansen, H.D.; Mease, R.C.; Horti, A.G.; Pomper, M.G.; L’Estrade, E.T.; Herth, M.M.; Peters, D.; Knudsen, G.M. In Vitro and In Vivo Characterization of Dibenzothiophene Derivatives [125I]Iodo-ASEM and [18F]ASEM as Radiotracers of Homo- and Heteromeric α7 Nicotinic Acetylcholine Receptors. Molecules 2020, 25, 1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine—An agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef]
- Sarter, M.; Parikh, V. Choline Transporters, Cholinergic Transmission and Cognition. Nat. Rev. Neurosci. 2005, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Hecker, A.; Küllmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Lerner, S.; Klein, J.; et al. Phosphocholine-Modified Macromolecules and Canonical Nicotinic Agonists Inhibit ATP-Induced IL-1β Release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L.; Bencherif, M.; Lippiello, P. An Evaluation of Neuronal Nicotinic Acetylcholine Receptor Activation by Quaternary Nitrogen Compounds Indicates That Choline Is Selective for the A7 Subtype. Neurosci. Lett. 1996, 213, 201–204. [Google Scholar] [CrossRef]
- Parikh, V.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Sarter, M.; Bruno, J.P. Rapid Assessment of in Vivo Cholinergic Transmission by Amperometric Detection of Changes in Extracellular Choline Levels. Eur. J. Neurosci. 2004, 20, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Mucchietto, V.; Fasoli, F.; Pucci, S.; Moretti, M.; Benfante, R.; Maroli, A.; Di Lascio, S.; Bolchi, C.; Pallavicini, M.; Dowell, C. A9-and A7-Containing Receptors Mediate the pro-Proliferative Effects of Nicotine in the A549 Adenocarcinoma Cell Line. Br. J. Pharmacol. 2018, 175, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Bavo, F.; Pucci, S.; Fasoli, F.; Lammi, C.; Moretti, M.; Mucchietto, V.; Lattuada, D.; Viani, P.; De Palma, C.; Budriesi, R. Potent Antiglioblastoma Agents by Hybridizing the Onium-Alkyloxy-Stilbene Based Structures of an A7-NAChR, A9-NAChR Antagonist and of a pro-Oxidant Mitocan. J. Med. Chem. 2018, 61, 10531–10544. [Google Scholar] [CrossRef]
- De Robertis, E.D. Histophysiology of Synapses and Neurosecretion: International Series of Monographs on Pure and Applied Biology: Modern Trends in Physiological Sciences; Elsevier: Amsterdam, The Netherlands, 2013; Volume 20. [Google Scholar]
- Südhof, T.C. The Cell Biology of Synapse Formation. J. Cell Biol. 2021, 220, e202103052. [Google Scholar] [CrossRef] [PubMed]
- Fabian-Fine, R.; Skehel, P.; Errington, M.L.; Davies, H.A.; Sher, E.; Stewart, M.G.; Fine, A. Ultrastructural Distribution of the A7 Nicotinic Acetylcholine Receptor Subunit in Rat Hippocampus. J. Neurosci. 2001, 21, 7993–8003. [Google Scholar] [CrossRef] [PubMed]
- Hazan, L.; Ziv, N.E. Activity Dependent and Independent Determinants of Synaptic Size Diversity. J. Neurosci. 2020, 40, 2828–2848. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Bourne, J.; Harris, K.M. Do Thin Spines Learn to Be Mushroom Spines That Remember? Curr. Opin. Neurobiol. 2007, 17, 381–386. [Google Scholar] [CrossRef]
- Kasai, H.; Ziv, N.E.; Okazaki, H.; Yagishita, S.; Toyoizumi, T. Spine Dynamics in the Brain, Mental Disorders and Artificial Neural Networks. Nat. Rev. Neurosci. 2021, 22, 407–422. [Google Scholar] [CrossRef]
- Ziv, N.E.; Brenner, N. Synaptic Tenacity or Lack Thereof: Spontaneous Remodeling of Synapses. Trends Neurosci. 2018, 41, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, A.; Yamagata, K.; Nakagomi, S.; Uejima, H.; Wiriyasermkul, P.; Ohgaki, R.; Nagamori, S.; Kanai, Y.; Tanaka, H. Nicotine Induces Dendritic Spine Remodeling in Cultured Hippocampal Neurons. J. Neurochem. 2014, 128, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvez, B.; Gross, N.; Sumikawa, K. Activation of A7 Nicotinic Acetylcholine Receptors Protects Potentiated Synapses from Depotentiation during Theta Pattern Stimulation in the Hippocampal CA1 Region of Rats. Neuropharmacology 2016, 105, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Inagaki, R.; Yi, L.; Shibata, M.; Sakagami, H.; Fukunaga, K. Nicotine Rescues Depressive-like Behaviors via A7-Type Nicotinic Acetylcholine Receptor Activation in CaMKIV Null Mice. Mol. Neurobiol. 2020, 57, 4929–4940. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yakel, J.L. Nicotinic Acetylcholine Receptor-Mediated Calcium Signaling in the Nervous System. Acta Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unwin, N. Nicotinic Acetylcholine Receptor and the Structural Basis of Neuromuscular Transmission: Insights from Torpedo Postsynaptic Membranes. Q. Rev. Biophys. 2013, 46, 283–322. [Google Scholar] [CrossRef] [Green Version]
- Fasoli, F.; Gotti, C. Structure of Neuronal Nicotinic Receptors. Neurobiol. Genet. Nicotine Tob. 2015, 23, 1–17. [Google Scholar]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-Ray Structure of the Human A4β2 Nicotinic Receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and Gating Mechanism of the A7 Nicotinic Acetylcholine Receptor. Cell 2021, 184, 2121–2134. [Google Scholar] [CrossRef]
- Walsh, R.M.; Roh, S.-H.; Gharpure, A.; Morales-Perez, C.L.; Teng, J.; Hibbs, R.E. Structural Principles of Distinct Assemblies of the Human A4β2 Nicotinic Receptor. Nature 2018, 557, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Son, C.D.; Moss, F.J.; Cohen, B.N.; Lester, H.A. Nicotine Normalizes Intracellular Subunit Stoichiometry of Nicotinic Receptors Carrying Mutations Linked to Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. Mol. Pharmacol. 2009, 75, 1137–1148. [Google Scholar] [CrossRef]
- Weltzin, M.M.; Lindstrom, J.M.; Lukas, R.J.; Whiteaker, P. Distinctive Effects of Nicotinic Receptor Intracellular-Loop Mutations Associated with Nocturnal Frontal Lobe Epilepsy. Neuropharmacology 2016, 102, 158–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouvatsos, N.; Giastas, P.; Chroni-Tzartou, D.; Poulopoulou, C.; Tzartos, S.J. Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: Ligand-bound α2 homopentamer. Proc. Natl. Acad. Sci. USA 2016, 113, 9635–9640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, N.; Corradi, J.; Sine, S.M.; Bouzat, C. Stoichiometry for Activation of Neuronal α7 Nicotinic Receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 20819–20824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, T.; Döhring, J.; Rohr, A.; Jansen, O.; Deuschl, G. CA1 Neurons in the Human Hippocampus Are Critical for Autobiographical Memory, Mental Time Travel, and Autonoetic Consciousness. Proc. Natl. Acad. Sci. USA 2011, 108, 17562–17567. [Google Scholar] [CrossRef] [Green Version]
- Nakauchi, S.; Sumikawa, K. Endogenously Released ACh and Exogenous Nicotine Differentially Facilitate Long-Term Potentiation Induction in the Hippocampal CA1 Region of Mice. Eur. J. Neurosci. 2012, 35, 1381–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakauchi, S.; Sumikawa, K. Endogenous ACh Suppresses LTD Induction and Nicotine Relieves the Suppression via Different Nicotinic ACh Receptor Subtypes in the Mouse Hippocampus. Life Sci. 2014, 111, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Horenstein, N.A.; Papke, R.L.; Kulkarni, A.R.; Chaturbhuj, G.U.; Stokes, C.; Manther, K.; Thakur, G.A. Critical Molecular Determinants of A7 Nicotinic Acetylcholine Receptor Allosteric Activation: Separation of Direct Allosteric Activation and Positive Allosteric Modulation. J. Biol. Chem. 2016, 291, 5049–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papke, R.L.; Lindstrom, J.M. Nicotinic Acetylcholine Receptors: Conventional and Unconventional Ligands and Signaling. Neuropharmacology 2020, 168, 108021. [Google Scholar] [CrossRef] [PubMed]
- Grønlien, J.H.; Håkerud, M.; Ween, H.; Thorin-Hagene, K.; Briggs, C.A.; Gopalakrishnan, M.; Malysz, J. Distinct Profiles of A7 NAChR Positive Allosteric Modulation Revealed by Structurally Diverse Chemotypes. Mol. Pharmacol. 2007, 72, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targowska-Duda, K.M.; Kaczor, A.A.; Jozwiak, K.; Arias, H.R. Molecular Interactions of Type I and Type II Positive Allosteric Modulators with the Human A7 Nicotinic Acetylcholine Receptor: An in silico Study. J. Biomol. Struct. Dyn. 2019, 37, 411–439. [Google Scholar] [CrossRef] [PubMed]
- Corrie, J.B.; Free, C.R.; Corradi, J.; Bouzat, C.; Sine, S.M. Single-Channel and Structural Foundations of Neuronal A7 Acetylcholine Receptor Potentiation. J. Neurosci. 2011, 31, 13870–13879. [Google Scholar]
- Sitzia, F.; Brown, J.T.; Randall, A.; Dunlop, J. Voltage-and Temperature-Dependent Allosteric Modulation of A7 Nicotinic Receptors by PNU120596. Front. Pharmacol. 2011, 2, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Kimbrell, M.R.; Tian, C.; Pack, T.F.; Crooks, P.A.; Fifer, E.K.; Papke, R.L. Multiple Modes of A7 NAChR Noncompetitive Antagonism of Control Agonist-Evoked and Allosterically Enhanced Currents. Mol. Pharmacol. 2013, 84, 459–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, M.; Garai, S.; Thakur, G.A.; Stokes, C.; Gulsevin, A.; Horenstein, N.A.; Papke, R.L. Macroscopic and Microscopic Activation of A7 Nicotinic Acetylcholine Receptors by the Structurally Unrelated Allosteric Agonist-Positive Allosteric Modulators (Ago-PAMs) B-973B and GAT107. Mol. Pharmacol. 2019, 95, 43–61. [Google Scholar] [CrossRef] [Green Version]
- Vallés, A.S.; Borroni, M.V.; Barrantes, F.J. Targeting Brain A7 Nicotinic Acetylcholine Receptors in Alzheimer’s Disease: Rationale and Current Status. CNS Drugs 2014, 28, 975–987. [Google Scholar] [CrossRef]
- Gill, J.K.; Savolainen, M.; Young, G.T.; Zwart, R.; Sher, E.; Millar, N.S. Agonist Activation of A7 Nicotinic Acetylcholine Receptors via an Allosteric Transmembrane Site. Proc. Natl. Acad. Sci. USA 2011, 108, 5867–5872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrachi, T.; Marsha, O.; Brusin, K.; Ben-David, Y.; Thakur, G.A.; Vaknin-Dembinsky, A.; Treinin, M.; Brenner, T. Suppression of Neuroinflammation by an Allosteric Agonist and Positive Allosteric Modulator of the A7 Nicotinic Acetylcholine Receptor GAT107. J. Neuroinflamm. 2021, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bagdas, D.; Wilkerson, J.L.; Kulkarni, A.; Toma, W.; AlSharari, S.; Gul, Z.; Lichtman, A.H.; Papke, R.L.; Thakur, G.A.; Damaj, M.I. The A7 Nicotinic Receptor Dual Allosteric Agonist and Positive Allosteric Modulator GAT107 Reverses Nociception in Mouse Models of Inflammatory and Neuropathic Pain. Br. J. Pharmacol. 2016, 173, 2506–2520. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Mikkelsen, J.D. The A7 Nicotinic Acetylcholine Receptor Ligands Methyllycaconitine, NS6740 and GTS-21 Reduce Lipopolysaccharide-Induced TNF-α Release from Microglia. J. Neuroimmunol. 2012, 251, 65–72. [Google Scholar] [CrossRef]
- Papke, R.L.; Bagdas, D.; Kulkarni, A.R.; Gould, T.; AlSharari, S.D.; Thakur, G.A.; Damaj, M.I. The Analgesic-like Properties of the Alpha7 NAChR Silent Agonist NS6740 Is Associated with Non-Conducting Conformations of the Receptor. Neuropharmacology 2015, 91, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Pieschl, R.L.; Miller, R.; Jones, K.M.; Post-Munson, D.J.; Chen, P.; Newberry, K.; Benitex, Y.; Molski, T.; Morgan, D.; McDonald, I.M. Effects of BMS-902483, an A7 Nicotinic Acetylcholine Receptor Partial Agonist, on Cognition and Sensory Gating in Relation to Receptor Occupancy in Rodents. Eur. J. Pharmacol. 2017, 807, 1–11. [Google Scholar] [CrossRef]
- Papke, R.L.; Peng, C.; Kumar, A.; Stokes, C. NS6740, an A7 Nicotinic Acetylcholine Receptor Silent Agonist, Disrupts Hippocampal Synaptic Plasticity. Neurosci. Lett. 2018, 677, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Álvarez, M.; Moreno-Ortega, A.J.; Navarro, E.; Fernández-Morales, J.C.; Egea, J.; López, M.G.; Cano-Abad, M.F. Positive Allosteric Modulation of Alpha-7 Nicotinic Receptors Promotes Cell Death by Inducing Ca2+ Release from the Endoplasmic Reticulum. J. Neurochem. 2015, 133, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Khoshbouei, H.; Garai, S.; Cantwell, L.N.; Stokes, C.; Thakur, G.; Papke, R.L. Allosterically Potentiated A7 Nicotinic Acetylcholine Receptors: Reduced Calcium Permeability and Current-Independent Control of Intracellular Calcium. Mol. Pharmacol. 2020, 98, 695–709. [Google Scholar] [CrossRef]
- Pismataro, M.C.; Horenstein, N.A.; Stokes, C.; Quadri, M.; De Amici, M.; Papke, R.L.; Dallanoce, C. Design, Synthesis, and Electrophysiological Evaluation of NS6740 Derivatives: Exploration of the Structure-Activity Relationship for Alpha7 Nicotinic Acetylcholine Receptor Silent Activation. Eur. J. Med. Chem. 2020, 205, 112669. [Google Scholar] [CrossRef]
- Campbell, N.R.; Fernandes, C.C.; Halff, A.W.; Berg, D.K. Endogenous Signaling through A7-Containing Nicotinic Receptors Promotes Maturation and Integration of Adult-Born Neurons in the Hippocampus. J. Neurosci. 2010, 30, 8734–8744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, B.J.; Mervis, R.F. Dendritic Spine Alterations in the Hippocampus and Parietal Cortex of Alpha7 Nicotinic Acetylcholine Receptor Knockout Mice. Neuroscience 2013, 233, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Hsu, F.C.; Baumann, B.H.; Coulter, D.A.; Lynch, D.R. Cortical Synaptic NMDA Receptor Deficits in A7 Nicotinic Acetylcholine Receptor Gene Deletion Models: Implications for Neuropsychiatric Diseases. Neurobiol. Dis. 2014, 63, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros-Yáñez, I.; Benavides-Piccione, R.; Bourgeois, J.P.; Changeux, J.P.; DeFelipe, J. Alterations of Cortical Pyramidal Neurons in Mice Lacking High-Affinity Nicotinic Receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 11567–11572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozada, A.F.; Wang, X.; Gounko, N.V.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Induction of Dendritic Spines by Β2-Containing Nicotinic Receptors. J. Neurosci. 2012, 32, 8391–8400. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.C.; Alves, N.C.; Nashmi, R.; De Biasi, M.; Lambe, E.K. Nicotinic A5 Subunits Drive Developmental Changes in the Activation and Morphology of Prefrontal Cortex Layer VI Neurons. Biol. Psychiatry 2012, 71, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Tian, M.K.; Bailey, C.D.C.; Lambe, E.K. Dendritic Spine Density of Prefrontal Layer 6 Pyramidal Neurons in Relation to Apical Dendrite Sculpting by Nicotinic Acetylcholine Receptors. Front. Cell. Neurosci. 2015, 9, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-L.; Chen, X.-T.; Luo, L.; Lou, Z.-Y.; Wang, S.; Chen, J.-T.; Wang, M.; Sun, L.-G.; Ruan, D.-Y. Reparatory Effects of Nicotine on NMDA Receptor-Mediated Synaptic Plasticity in the Hippocampal CA1 Region of Chronically Lead-Exposed Rats. Eur. J. Neurosci. 2006, 23, 1111–1119. [Google Scholar] [CrossRef]
- Welsby, P.; Rowan, M.; Anwyl, R. Nicotinic Receptor-Mediated Enhancement of Long-Term Potentiation Involves Activation of Metabotropic Glutamate Receptors and Ryanodine-Sensitive Calcium Stores in the Dentate Gyrus. Eur. J. Neurosci. 2006, 24, 3109–3118. [Google Scholar] [CrossRef]
- Welsby, P.J.; Rowan, M.J.; Anwyl, R. Intracellular Mechanisms Underlying the Nicotinic Enhancement of LTP in the Rat Dentate Gyrus. Eur. J. Neurosci. 2009, 29, 65–75. [Google Scholar] [CrossRef]
- Nordberg, A. Human Nicotinic Receptors—Their Role in Aging and Dementia. Neurochem. Int. 1994, 25, 93–97. [Google Scholar] [CrossRef]
- Nordberg, A.; Alafuzoff, I.; Winblad, B. Nicotinic and Muscarinic Subtypes in the Human Brain: Changes with Aging and Dementia. J. Neurosci. Res. 1992, 31, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Adams, C.; Breese, C.R.; Adler, L.E.; Bickford, P.; Byerley, W.; Coon, H.; Griffith, J.M.; Miller, C.; Myles-Worsley, M. Nicotinic Receptor Function in Schizophrenia. Schizophr. Bull. 1996, 22, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Gault, J.; Adams, C.; Breese, C.R.; Rollins, Y.; Adler, L.E.; Olincy, A.; Freedman, R. Nicotinic Receptors, Smoking and Schizophrenia. Restor. Neurol. Neurosci. 1998, 12, 195–201. [Google Scholar] [PubMed]
- Mexal, S.; Berger, R.; Logel, J.; Ross, R.G.; Freedman, R.; Leonard, S. Differential Regulation of A7 Nicotinic Receptor Gene (CHRNA7) Expression in Schizophrenic Smokers. J. Mol. Neurosci. 2010, 40, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, A.S.; Picciotto, M.R. High-Affinity Nicotinic Acetylcholine Receptor Expression and Trafficking Abnormalities in Psychiatric Illness. Psychopharmacology 2013, 229, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Weiland, S.; Bertrand, D.; Leonard, S. Neuronal Nicotinic Acetylcholine Receptors: From the Gene to the Disease. Behav. Brain Res. 2000, 113, 43–56. [Google Scholar] [CrossRef]
- Palma, E.; Conti, L.; Roseti, C.; Limatola, C. Novel Approaches to Study the Involvement of A7-NAChR in Human Diseases. Curr. Drug Targets 2012, 13, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Barrantes, F.J. Molecular pathology of the nicotinic acetylcholine receptor. In The Nicotinic Acetylcholine Receptor; Springer: Berlin/Heidelberg, Germany, 1998; pp. 175–212. [Google Scholar]
- Freedman, R.; Hall, M.; Adler, L.E.; Leonard, S. Evidence in Postmortem Brain Tissue for Decreased Numbers of Hippocampal Nicotinic Receptors in Schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [Google Scholar] [CrossRef]
- Olincy, A.; Stevens, K.E. Treating Schizophrenia Symptoms with an A7 Nicotinic Agonist, from Mice to Men. Biochem. Pharmacol. 2007, 74, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.-Z.; Zhang, X.; Blennow, K.; Nordberg, A. Decreased Protein Level of Nicotinic Receptor A7 Subunit in the Frontal Cortex from Schizophrenic Brain. Neuroreport 1999, 10, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Court, J.; Spurden, D.; Lloyd, S.; McKeith, I.; Ballard, C.; Cairns, N.; Kerwin, R.; Perry, R.; Perry, E. Neuronal Nicotinic Receptors in Dementia with Lewy Bodies and Schizophrenia: α-Bungarotoxin and Nicotine Binding in the Thalamus. J. Neurochem. 1999, 73, 1590–1597. [Google Scholar] [CrossRef]
- Bertelsen, B.; Oranje, B.; Melchior, L.; Fagerlund, B.; Werge, T.M.; Mikkelsen, J.D.; Tümer, Z.; Glenthøj, B.Y. Association Study of CHRNA7 Promoter Variants with Sensory and Sensorimotor Gating in Schizophrenia Patients and Healthy Controls: A Danish Case–Control Study. Neuromol. Med. 2015, 17, 423–430. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The Human CHRNA7 and CHRFAM7A Genes: A Review of the Genetics, Regulation, and Function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozada, A.F.; Wang, X.; Gounko, N.V.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Glutamatergic Synapse Formation Is Promoted by A7-Containing Nicotinic Acetylcholine Receptors. J. Neurosci. 2012, 32, 7651–7661. [Google Scholar] [CrossRef] [Green Version]
- Choueiry, J.; Blais, C.M.; Shah, D.; Smith, D.; Fisher, D.; Illivitsky, V.; Knott, V. Combining CDP-Choline and Galantamine: Effects of a Selective A7 Nicotinic Acetylcholine Receptor Agonist Strategy on P50 Sensory Gating of Speech Sounds in Healthy Volunteers. J. Psychopharmacol. 2019, 33, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Tanaka, H. Activities of Nicotinic Acetylcholine Receptors Modulate Neurotransmission and Synaptic Architecture. Neural Regen. Res. 2014, 9, 2128. [Google Scholar] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh Receptors as Therapeutic Targets in CNS Disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.-Z.; Zhang, X.; Ravid, R.; Nordberg, A. Decreased Protein Levels of Nicotinic Receptor Subunits in the Hippocampus and Temporal Cortex of Patients with Alzheimer’s Disease. J. Neurochem. 2000, 74, 237–243. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Meyer-Luehmann, M.; Osetek, J.D.; Jones, P.B.; Stern, E.A.; Bacskai, B.J.; Hyman, B.T. Impaired Spine Stability Underlies Plaque-Related Spine Loss in an Alzheimer’s Disease Mouse Model. Am. J. Pathol. 2007, 171, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasala, M.; Fabiani, C.; Corradi, J.; Antollini, S.; Bouzat, C. Molecular Modulation of Human A7 Nicotinic Receptor by Amyloid-β Peptides. Front. Cell. Neurosci. 2019, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Das, D.; Karunakaran, S.; Nanguneri, S.; Bapat, D.; Ray, A.; Shaw, E.; Bennett, D.A.; Nair, D.; Ravindranath, V. Aβ Mediates F-Actin Disassembly in Dendritic Spines Leading to Cognitive Deficits in Alzheimer’s Disease. J. Neurosci. 2018, 38, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Hrynchak, M.V.; Rierola, M.; Golovyashkina, N.; Penazzi, L.; Pump, W.C.; David, B.; Sündermann, F.; Brandt, R.; Bakota, L. Chronic Presence of Oligomeric Aβ Differentially Modulates Spine Parameters in the Hippocampus and Cortex of Mice with Low APP Transgene Expression. Front. Synaptic Neurosci. 2020, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Reese, L.C.; Zhang, W.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective Induction of Calcineurin Activity and Signaling by Oligomeric Amyloid Beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Stallings, N.R.; O’Neal, M.A.; Hu, J.; Kavalali, E.T.; Bezprozvanny, I.; Malter, J.S. Pin1 Mediates Aβ42-Induced Dendritic Spine Loss. Sci. Signal. 2018, 11, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.-Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L. Amyloid β Induces the Morphological Neurodegenerative Triad of Spine Loss, Dendritic Simplification, and Neuritic Dystrophies through Calcineurin Activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, T.; Fuhrmann, M.; Burgold, S.; Ochs, S.M.; Hoffmann, N.; Mitteregger, G.; Kretzschmar, H.; LaFerla, F.M.; Herms, J. Multiple Events Lead to Dendritic Spine Loss in Triple Transgenic Alzheimer’s Disease Mice. PLoS ONE 2010, 5, e15477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkwood, C.M.; Ciuchta, J.; Ikonomovic, M.D.; Fish, K.N.; Abrahamson, E.E.; Murray, P.S.; Klunk, W.E.; Sweet, R.A. Dendritic Spine Density, Morphology, and Fibrillar Actin Content Surrounding Amyloid-β Plaques in a Mouse Model of Amyloid-β Deposition. J. Neuropathol. Exp. Neurol. 2013, 72, 791–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Karuppan, M.K.M.; Devadoss, D.; Nair, M.; Chand, H.S.; Lakshmana, M.K. TFEB protein expression is reduced in aged brains and its overexpression mitigates senescence-associated biomarkers and memory deficits in mice. Neurobiol. Aging 2021, 106, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.K.; Herskowitz, J.H. Dendritic Spines: Mediators of Cognitive Resilience in Aging and Alzheimer’s Disease. Neuroscientist 2020. Available online: https://journals.sagepub.com/doi/abs/10.1177/1073858420945964 (accessed on 29 August 2021). [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borroni, V.; Barrantes, F.J. Homomeric and Heteromeric α7 Nicotinic Acetylcholine Receptors in Health and Some Central Nervous System Diseases. Membranes 2021, 11, 664. https://doi.org/10.3390/membranes11090664
Borroni V, Barrantes FJ. Homomeric and Heteromeric α7 Nicotinic Acetylcholine Receptors in Health and Some Central Nervous System Diseases. Membranes. 2021; 11(9):664. https://doi.org/10.3390/membranes11090664
Chicago/Turabian StyleBorroni, Virginia, and Francisco J. Barrantes. 2021. "Homomeric and Heteromeric α7 Nicotinic Acetylcholine Receptors in Health and Some Central Nervous System Diseases" Membranes 11, no. 9: 664. https://doi.org/10.3390/membranes11090664
APA StyleBorroni, V., & Barrantes, F. J. (2021). Homomeric and Heteromeric α7 Nicotinic Acetylcholine Receptors in Health and Some Central Nervous System Diseases. Membranes, 11(9), 664. https://doi.org/10.3390/membranes11090664