
Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life


Abstract
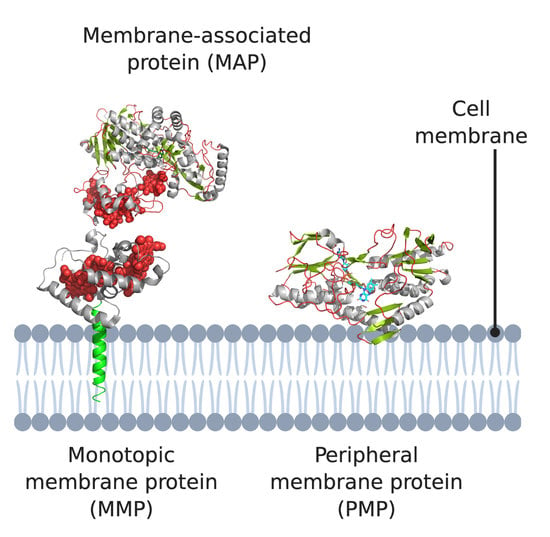
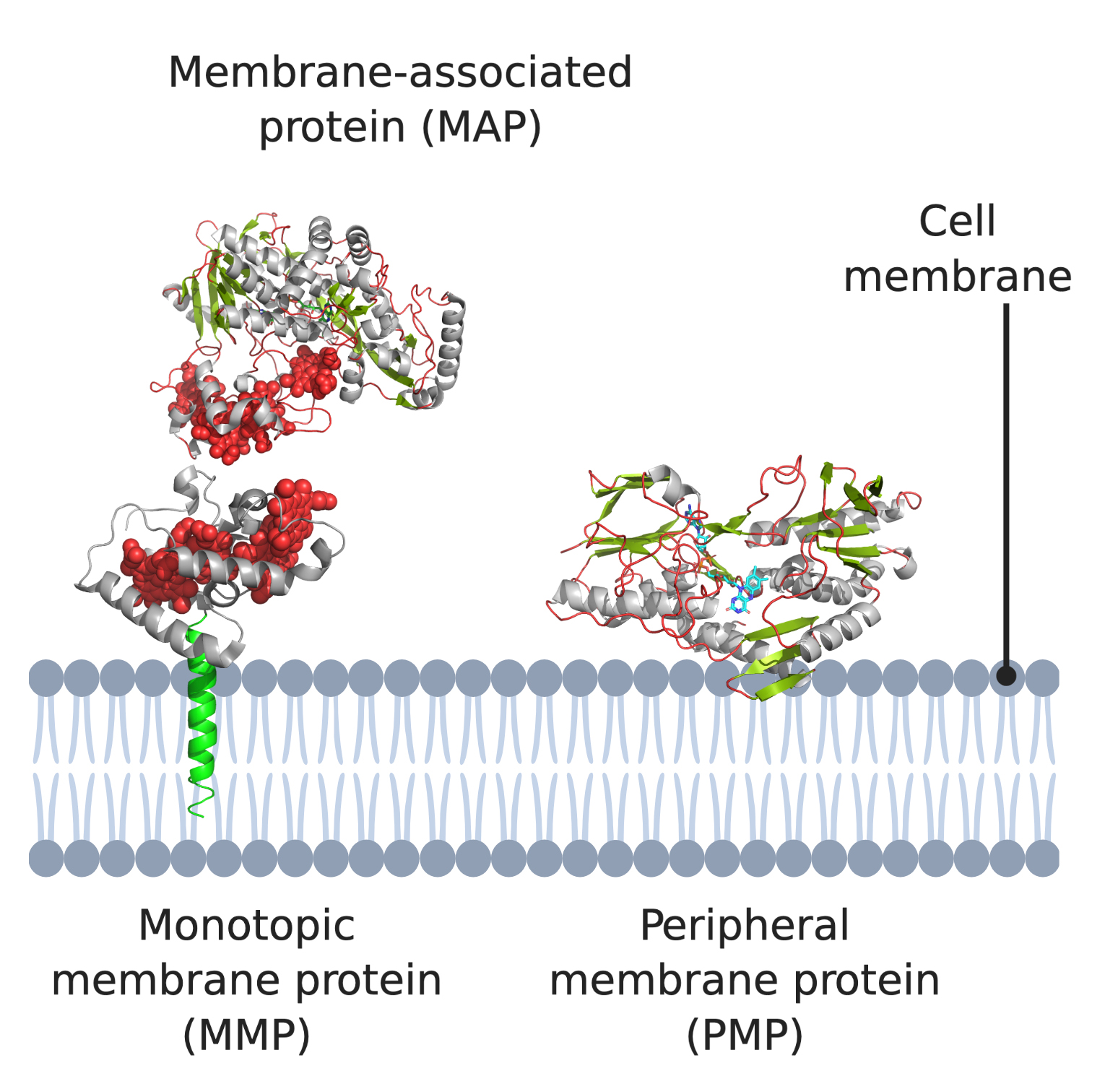
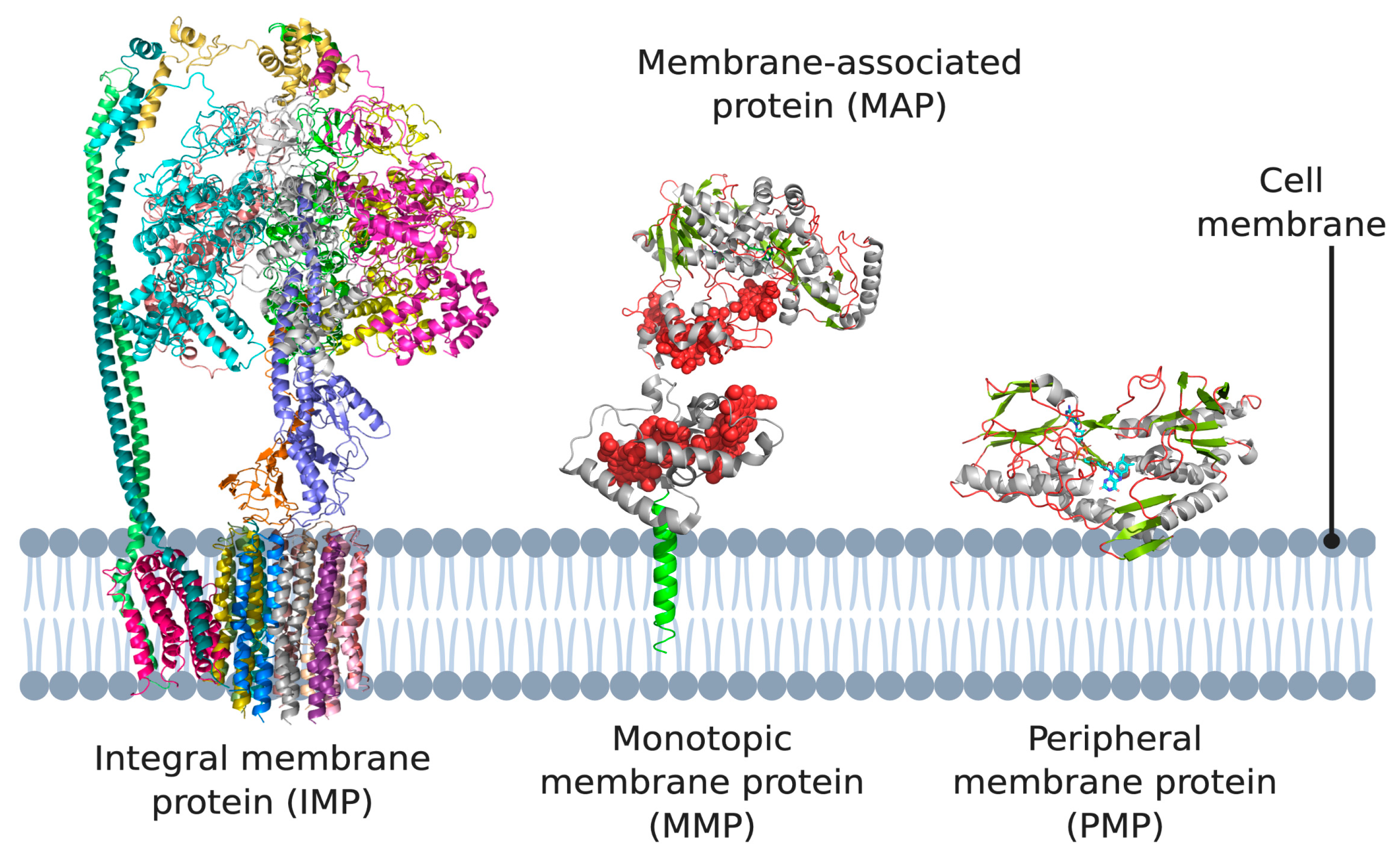
1. Introduction
2. Monotopic Membrane Proteins (MMPs) and Membrane-Associated Proteins (MAPs)
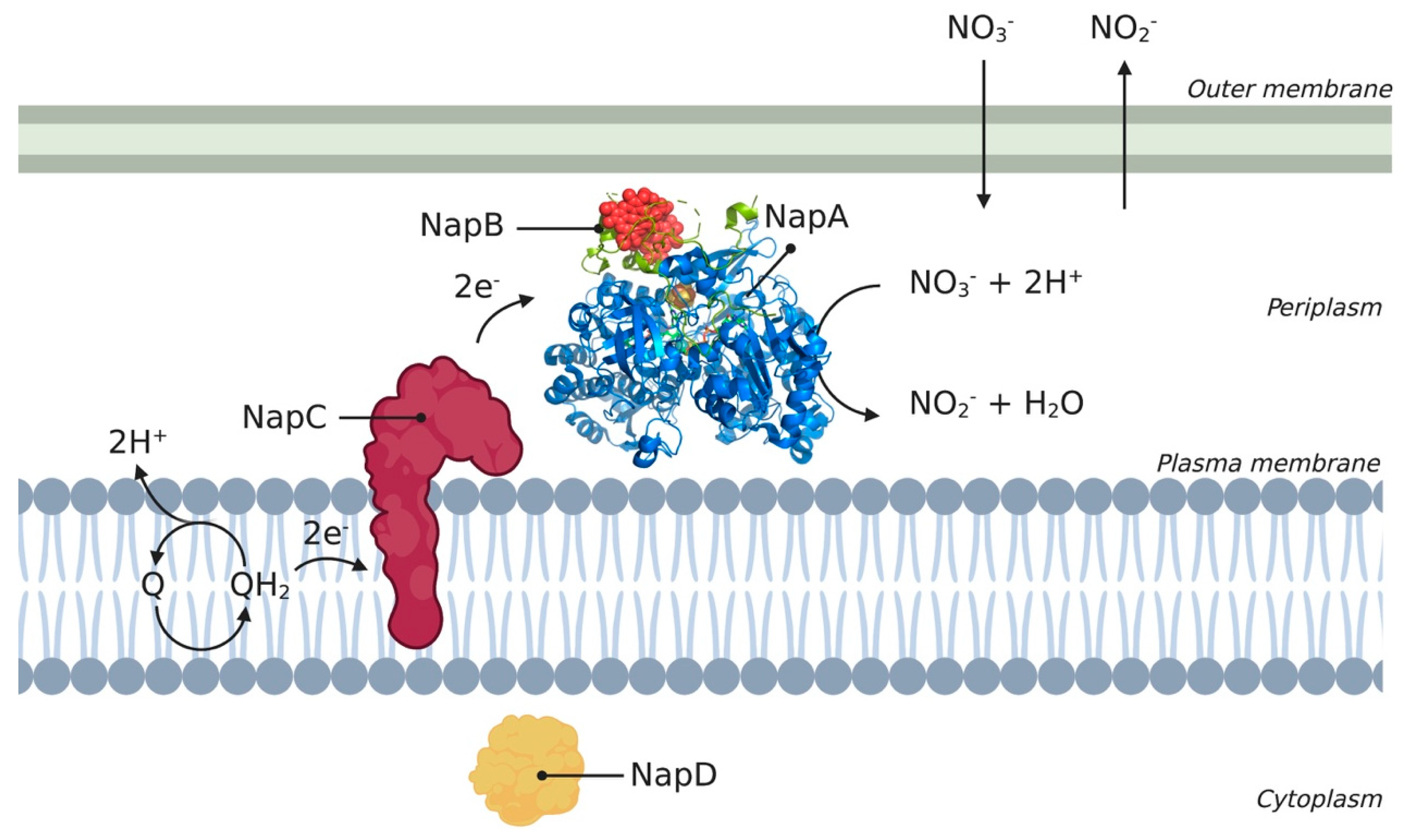
2.1. Periplasmic Nitrate Reductase complex
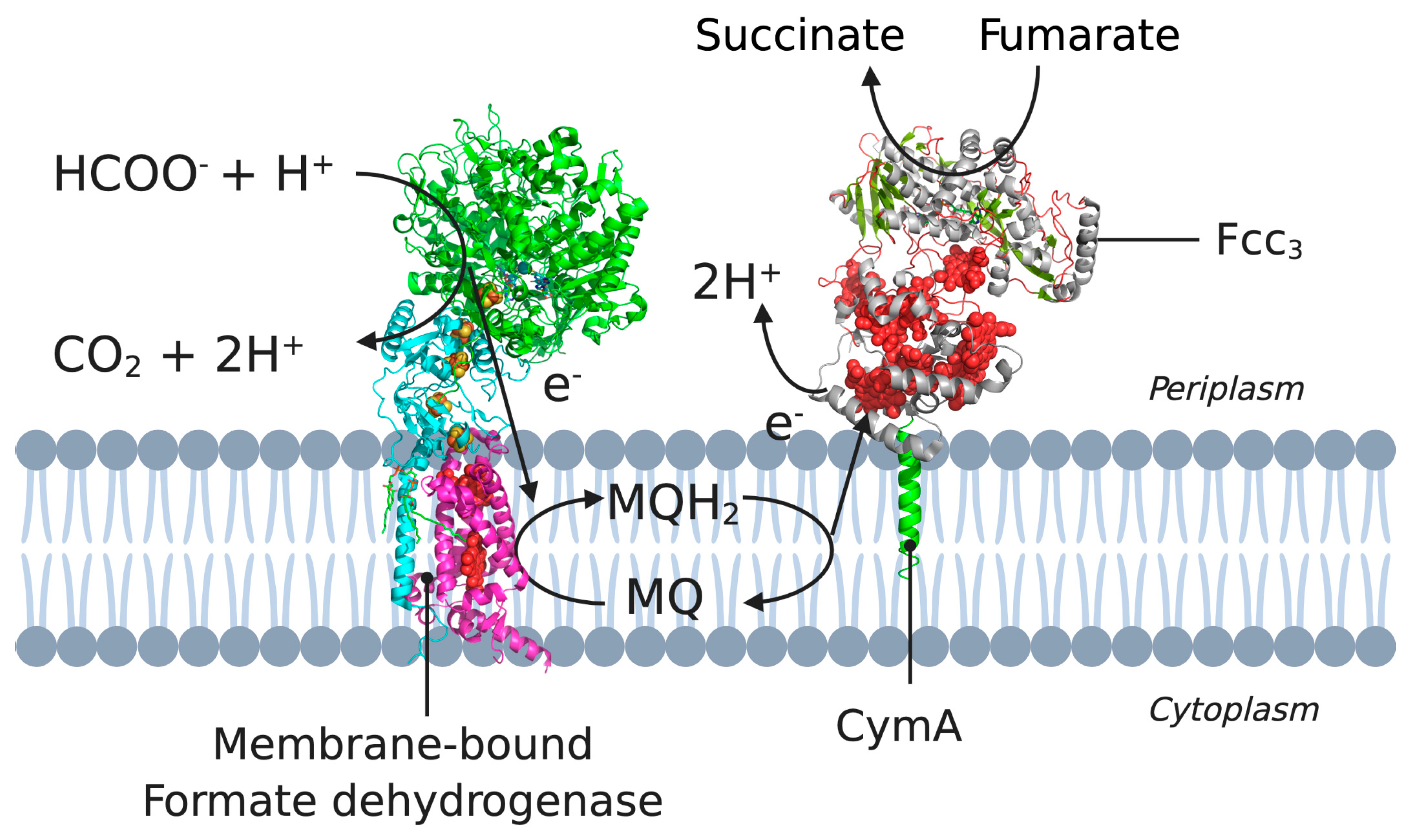
2.2. CymA
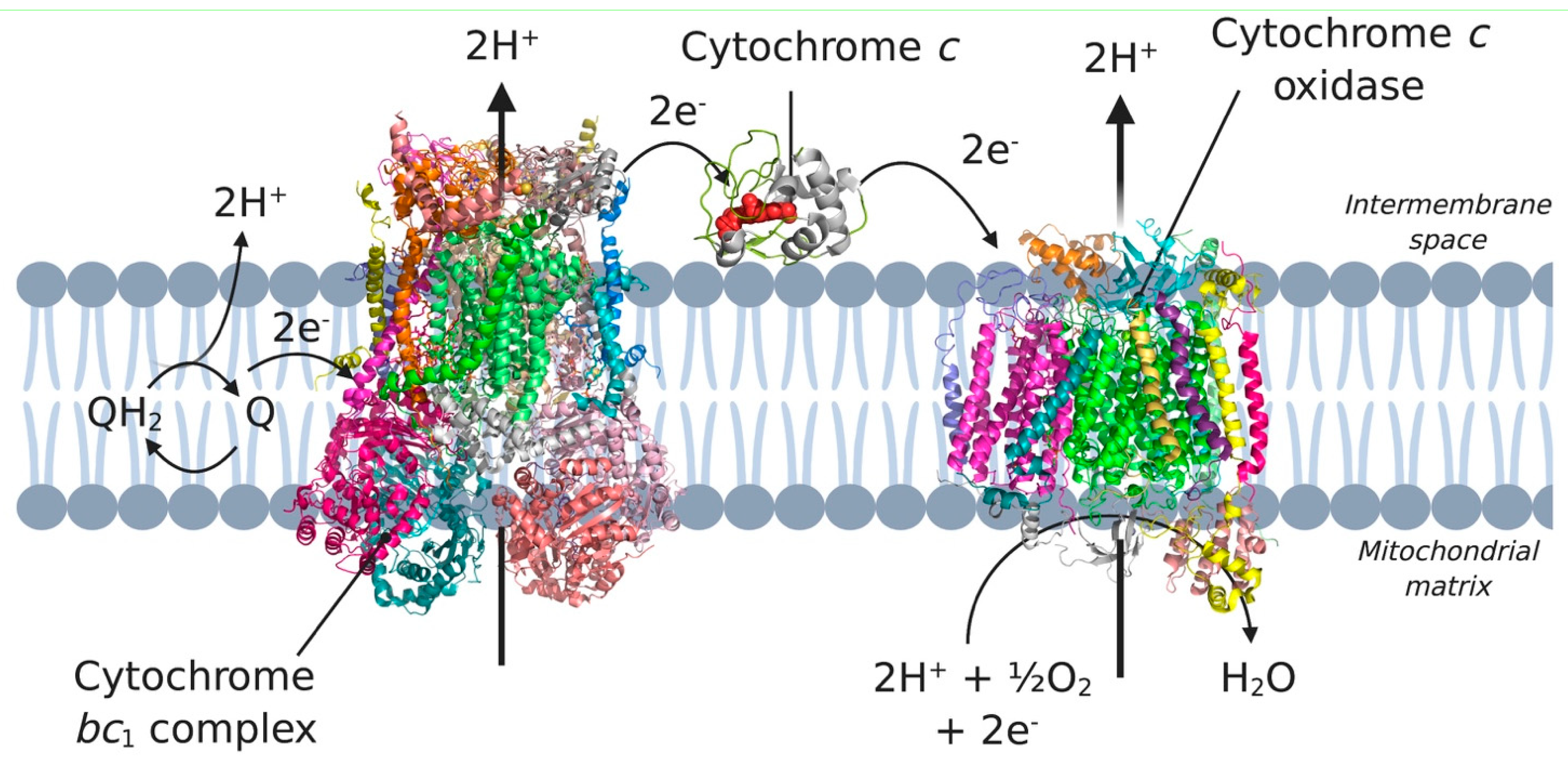
2.3. Membrane-Anchored Cytochrome c and a Paradigm Change
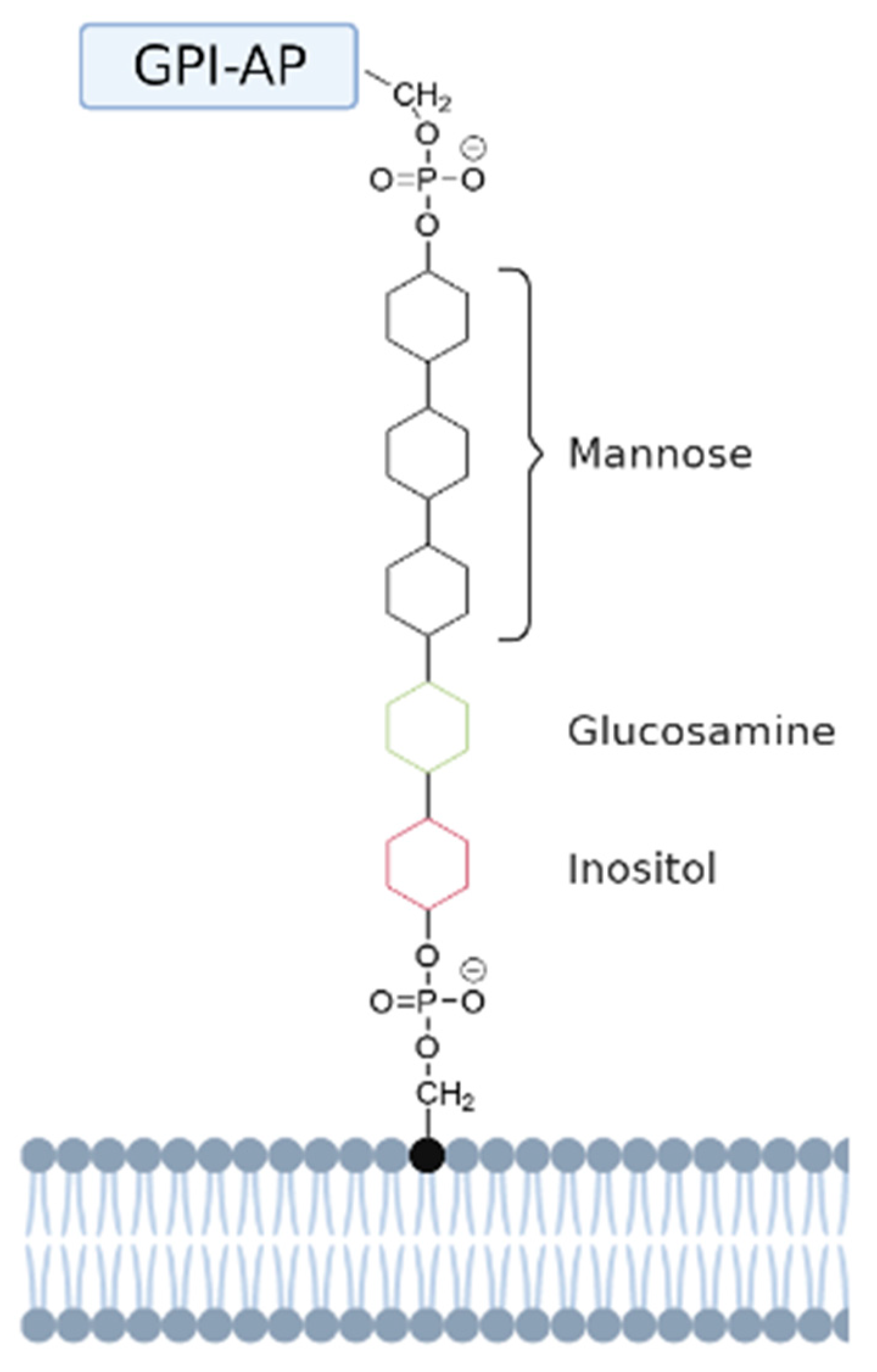
3. Glycosylphosphatidylinositol (GPI)-Anchored PMPs
3.1. Alkaline Phosphatase
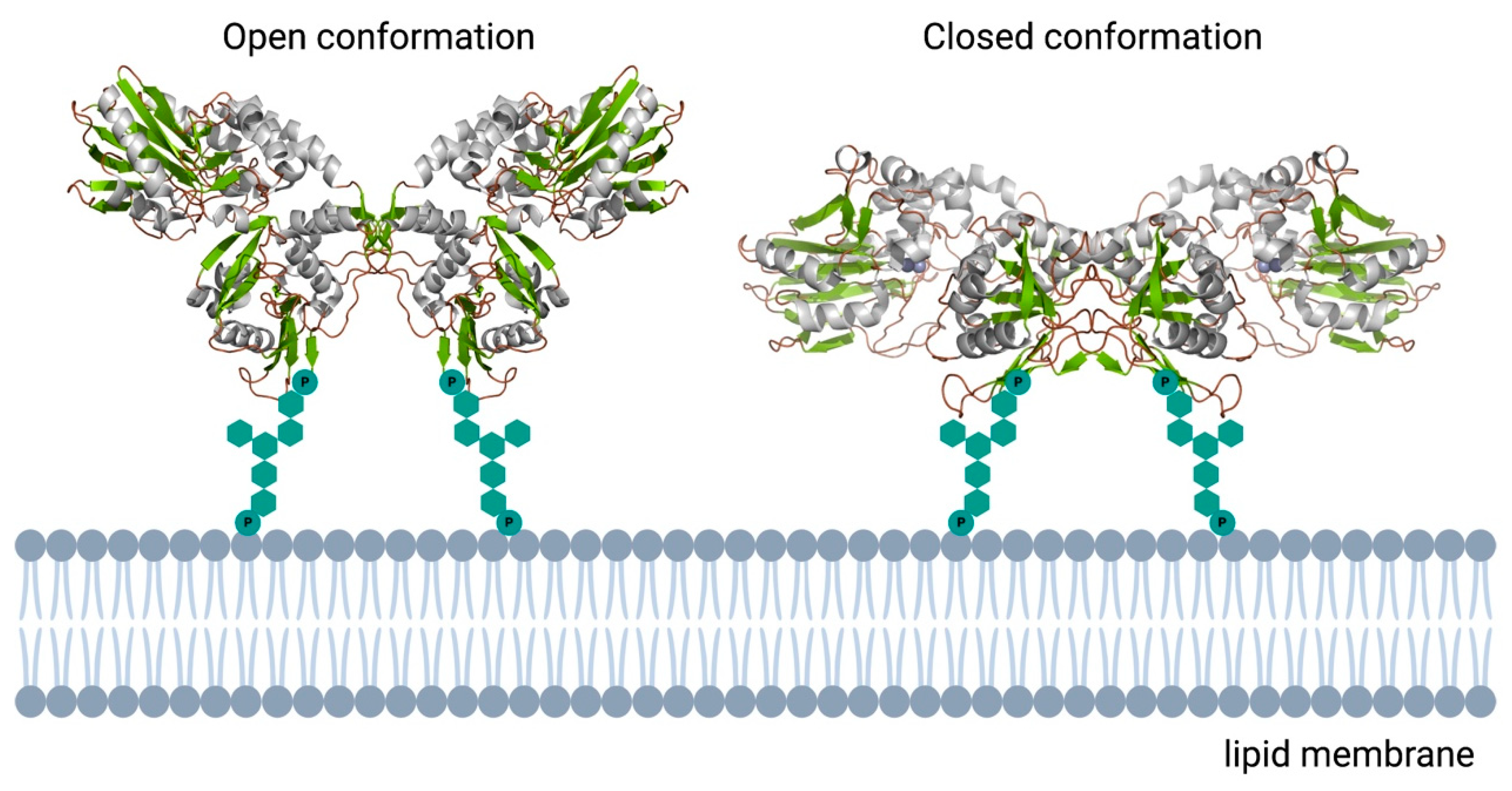
3.2. Ecto-5′-Nucleotidase (CD73)
3.3. Acetylcholinesterase
4. Membrane-Binding PMPs
4.1. Alternative Oxidase
4.2. Cytochrome c
4.3. Type-II NADH Dehydrogenase
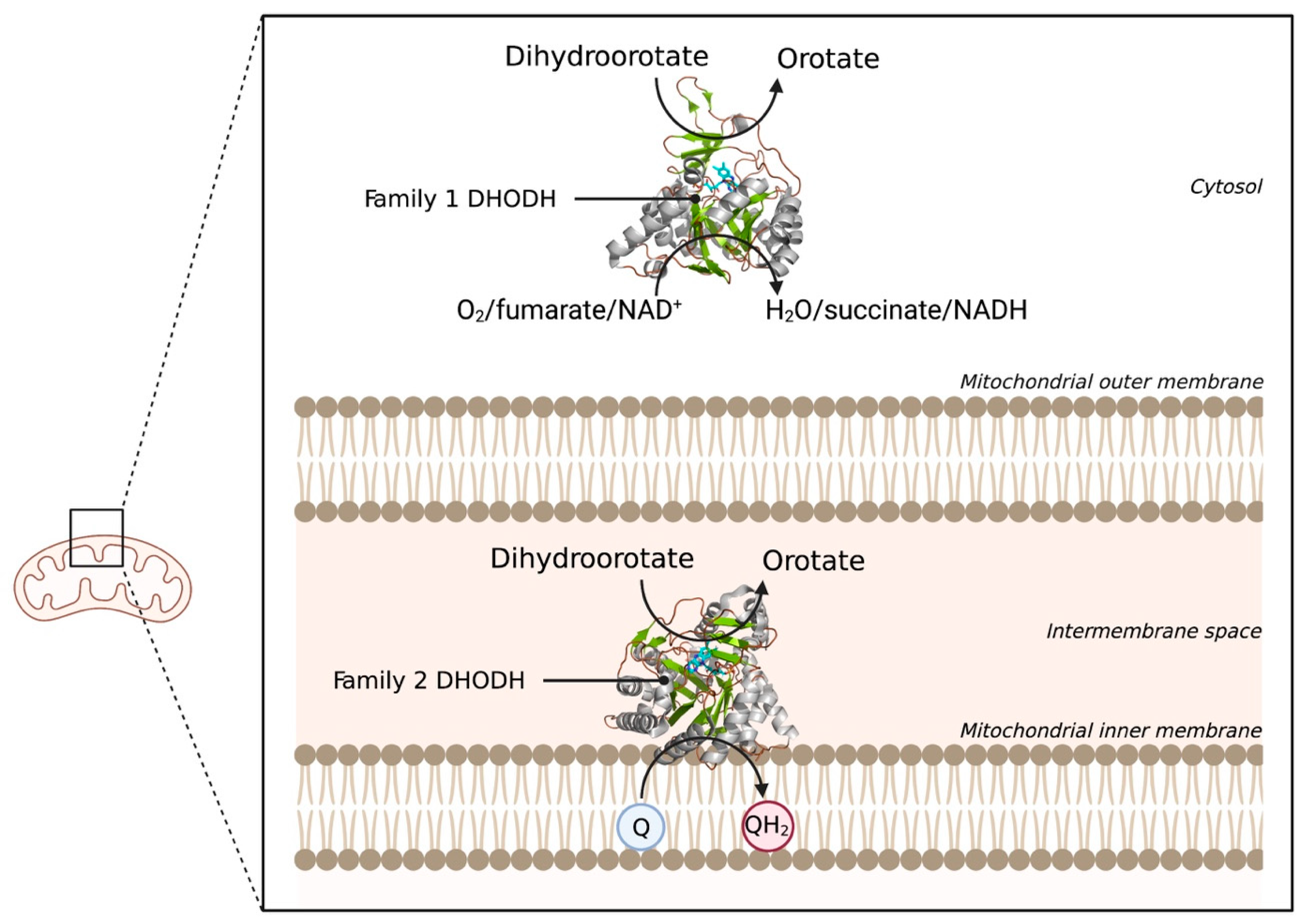
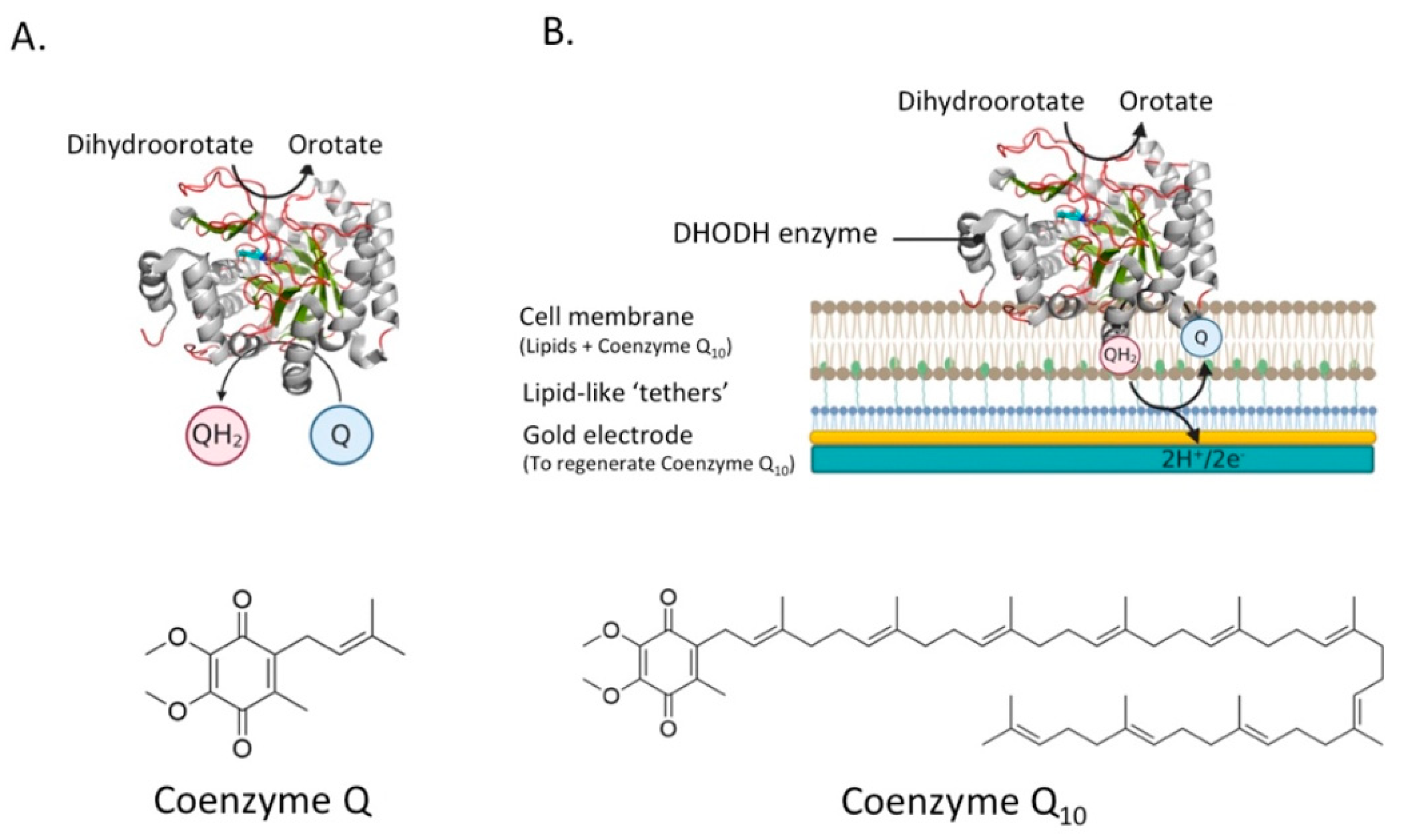
4.4. Dihydroorotate Dehydrogenase
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Vinothkumar, K.R.; Henderson, R. Structures of membrane proteins. Q. Rev. Biophys. 2010, 43, 65–158. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Sawyer, A. Elucidating the structure of membrane proteins. Biotechniques 2019, 66, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Membrane Proteins. In Molecular Cell Biology; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Protein Data Bank. RCSB PDB-5T4Q: Autoinhibited E. coli ATP Synthase State 3. 2016. Available online: https://www.rcsb.org/structure/5t4q (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-2J7A: Crystal Structure of Cytochrome C Nitrite Reductase NrfHA Complex from Desulfovibrio vulgaris. 2006. Available online: https://www.rcsb.org/structure/2j7a (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-1Y0P: Flavocytochrome C3 with Mesaconate Bound. 2004. Available online: https://www.rcsb.org/structure/1Y0P (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-6BDO: Structure of Bacterial Type II NADH Dehydrogenase from Caldalkalibacillus thermarum Complexed with a Quinone Inhibitor HQNO at 2.8A Resolution. 2017. Available online: https://www.rcsb.org/structure/6bdo (accessed on 1 April 2021).
- BioRender. Available online: https://biorender.com/ (accessed on 1 April 2021).
- Monje-Galvan, V.; Klauda, J.B. Peripheral membrane proteins: Tying the knot between experiment and computation. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 1584–1593. [Google Scholar] [CrossRef]
- le Maire, M.; Champeil, P.; Møller, J.V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1508, 86–111. [Google Scholar] [CrossRef]
- Ohlendieck, K. Extraction of Membrane Proteins. In Protein Purification Protocols; Humana Press: Totowa, NJ, USA, 2004; pp. 283–294. [Google Scholar] [CrossRef]
- Thomas, T.C.; McNamee, M.G. Purification of membrane proteins. Methods Enzymol. 1990, 182, 499–520. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Rahimian, M.; Aneja, K.K.; Schechter, N.M.; Rubin, H.; Scott, C.P. Mycobacterium tuberculosis Type II NADH-Menaquinone Oxidoreductase Catalyzes Electron Transfer through a Two-Site Ping-Pong Mechanism and Has Two Quinone-Binding Sites. Biochemistry 2014, 53, 1179–1190. [Google Scholar] [CrossRef]
- Moreno-Vivián, C.; Cabello, P.; Martínez-Luque, M.; Blasco, R.; Castillo, F. Prokaryotic Nitrate Reduction: Molecular Properties and Functional Distinction among Bacterial Nitrate Reductases. J. Bacteriol. 1999, 181, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Sparacino-Watkins, C.; Stolz, J.F.; Basu, P. Nitrate and periplasmic nitrate reductases. Chem. Soc. Rev. 2014, 43, 676–706. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Mierzejewska, P.; Slominska, E.M.; Smolenski, R.T. The metabolism of ecto-5′-nucleotidase (CD73) inhibitor-α,β-methylene adenosine diphosphate in BALB/c mice. Nucleosides Nucleotides Nucleic Acids 2018, 37, 709–716. [Google Scholar] [CrossRef]
- Saxena, M.; Delgado, Y.; Sharma, R.K.; Sharma, S.; Guzmán, S.L.P.D.L.; Tinoco, A.D.; Griebenow, K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS ONE 2018, 13, e0195542. [Google Scholar] [CrossRef]
- Khutornenko, A.A.; Roudko, V.V.; Chernyak, B.V.; Vartapetian, A.B.; Chumakov, P.M.; Evstafieva, A.G. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12828–12833. [Google Scholar] [CrossRef]
- Ebiloma, G.U.; Balogun, E.O.; Cueto-Díaz, E.J.; De Koning, H.P.; Dardonville, C. Alternative oxidase inhibitors: Mitochondrion-targeting as a strategy for new drugs against pathogenic parasites and fungi. Med. Res. Rev. 2019, 39, 1553–1602. [Google Scholar] [CrossRef] [PubMed]
- Costeira-Paulo, J.; Gault, J.; Popova, G.; Ladds, M.J.; Van Leeuwen, I.M.; Sarr, M.; Olsson, A.; Lane, D.P.; Laín, S.; Marklund, E.G.; et al. Lipids Shape the Electron Acceptor-Binding Site of the Peripheral Membrane Protein Dihydroorotate Dehydrogenase. Cell Chem. Biol. 2018, 25, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Hernandez, A.; Tate, D.J.; McMillan, D.G.G. Revealing the Membrane-Bound Catalytic Oxidation of NADH by the Drug Target Type-II NADH Dehydrogenase. Biochemistry 2019, 58, 4272–4275. [Google Scholar] [CrossRef] [PubMed]
- Hards, K.; McMillan, D.G.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Shimaki, Y.; Dutta, D.; Muench, S.P.; Ireton, K.; Cook, G.M.; Jeuken, L.J.C. Unprecedented Properties of Phenothiazines Unraveled by a NDH-2 Bioelectrochemical Assay Platform. J. Am. Chem. Soc. 2019, 142, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 24 March 2020).
- Harbut, M.B.; Yang, B.; Liu, R.; Yano, T.; Vilchèze, C.; Cheng, B.; Lockner, J.; Guo, H.; Yu, C.; Franzblau, S.G.; et al. Small Molecules Targeting Mycobacterium tuberculosis Type II NADH Dehydrogenase Exhibit Antimycobacterial Activity. Angew. Chem. Int. Ed. 2018, 57, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, L.M.; Peters, B.M.; Krom, B.P.; Freiberg, J.A.; Hänsch, G.M.; Filler, S.G.; Jabra-Rizk, M.A.; Shirtliff, M.E. Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology 2015, 161, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Croxen, M.A.; Law, R.J.; Scholz, R.; Keeney, K.M.; Wlodarska, M.; Finlay, B.B. Recent Advances in Understanding Enteric Pathogenic Escherichia coli. Clin. Microbiol. Rev. 2013, 26, 822–880. [Google Scholar] [CrossRef]
- Justice, S.S.; Hunstad, D.A.; Seed, P.C.; Hultgren, S.J. Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc. Natl. Acad. Sci. USA 2006, 103, 19884–19889. [Google Scholar] [CrossRef]
- Salyers, A.A.; Gupta, A.; Wang, Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol. 2004, 12, 412–416. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Disease Burden | Haemophilus influenzae Type B | Health Topics. Available online: http://www.emro.who.int/health-topics/haemophilus-influenzae-type-b/disease-burden.html (accessed on 27 April 2021).
- Su, P.-Y.; Huang, A.-H.; Lai, C.-H.; Lin, H.-F.; Lin, T.-M.; Ho, C.-H. Extensively drug-resistant Haemophilus influenzae—Emergence, epidemiology, risk factors, and regimen. BMC Microbiol. 2020, 20, 102. [Google Scholar] [CrossRef]
- Nguyen, L.; Garcia, J.; Gruenberg, K.; MacDougall, C. Multidrug-Resistant Pseudomonas Infections: Hard to Treat, But Hope on the Horizon? Curr. Infect. Dis. Rep. 2018, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Biggest Threats and Data: Antibiotic/Antimicrobial Resistance. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html (accessed on 27 April 2021).
- Al-Rashida, M.; Iqbal, M.A.-R.A.J. Inhibition of Alkaline Phosphatase: An Emerging New Drug Target. Mini-Rev. Med. Chem. 2015, 15, 41–51. [Google Scholar] [CrossRef]
- Klontzas, M.E.; Vassalou, E.E.; Zibis, A.H.; Karantanas, A.H. Hydroxyapatite deposition disease around the hip: Outcomes of CT-guided treatment. Diagn. Interv. Radiol. 2016, 22, 466–470. [Google Scholar] [CrossRef]
- Chen, Q.; Pu, N.; Yin, H.; Zhang, J.; Zhao, G.; Lou, W.; Wu, W. CD73 acts as a prognostic biomarker and promotes progression and immune escape in pancreatic cancer. J. Cell. Mol. Med. 2020, 24, 8674–8686. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F. Potential of CD73 as a target for cancer immunotherapy. Immunotherapy 2019, 11, 1353–1355. [Google Scholar] [CrossRef]
- Alzheimer’s Association Report. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- World Health Organization. WHO | Human African Trypanosomiasis, WHO. 2020. Available online: http://www.who.int/gho/neglected_diseases/human_african_trypanosomiasis/en/ (accessed on 27 April 2021).
- Menzies, S.K.; Tulloch, L.B.; Florence, G.J.; Smith, T.K. The trypanosome alternative oxidase: A potential drug target? Parasitology 2016, 145, 175–183. [Google Scholar] [CrossRef]
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 27 April 2021).
- Zhu, X.; Zhang, K.; Wang, Q.; Chen, S.; Gou, Y.; Cui, Y.; Li, Q. Cisplatin-mediated c-myc overexpression and cytochrome c (cyt c) release result in the up-regulation of the death receptors DR4 and DR5 and the activation of caspase 3 and caspase 9, likely responsible for the TRAIL-sensitizing effect of cisplatin. Med. Oncol. 2015, 32, 133. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.; Merz, C.; Evans, L.; Gradl, S.; Seidel, H.; Friberg, A.; Eheim, A.; Lejeune, P.; Brzezinka, K.; Zimmermann, K.; et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 2019, 33, 2403–2415. [Google Scholar] [CrossRef]
- Brondijk, T.H.C.; Nilavongse, A.; Filenko, N.; Richardson, D.J.; Cole, J.A. NapGH Components of the Periplasmic Nitrate Reductase of Escherichia Coli K-12: Location, Topology and Physiological Roles in Quinol Oxidation and Redox Balancing. Biochem. J. 2004, 379, 47–55. Available online: https://www.academia.edu/29265246/NapGH_components_of_the_periplasmic_nitrate_reductase_of_Escherichia_coli_K_12_location_topology_and_physiological_roles_in_quinol_oxidation_and_redox_balancing (accessed on 16 October 2020). [CrossRef]
- Sohaskey, C.D.; Wayne, L.G. Role of narK2X and narGHJI inHypoxic Upregulation of Nitrate Reduction byMycobacteriumtuberculosis. J. Bacteriol. 2003, 185, 7247–7256. [Google Scholar] [CrossRef]
- Bedzyk, L.; Wang, T.; Ye, R.W. The Periplasmic Nitrate Reductase in Pseudomonas sp. Strain G-179 Catalyzes the First Step of Denitrification. J. Bacteriol. 1999, 181, 2802–2806. [Google Scholar] [CrossRef]
- Protein Data Bank. RCSB PDB-3ML1: Crystal Structure of the Periplasmic Nitrate Reductase from Cupriavidus Necator. 2010. Available online: https://www.rcsb.org/structure/3ML1 (accessed on 1 April 2021).
- Myers, C.R.; Myers, J.M. Outer membrane cytochromes of Shewanella putrefaciens MR-1: Spectral analysis, and purification of the 83-kDa c-type cytochrome. Biochim. Biophys. Acta (BBA) Biomembr. 1997, 1326, 307–318. [Google Scholar] [CrossRef]
- Schwalb, C.; Chapman, A.S.K.; Reid, G.A. The Tetraheme Cytochrome CymA Is Required for Anaerobic Respiration with Dimethyl Sulfoxide and Nitrite inShewanella oneidensis. Biochemistry 2003, 42, 9491–9497. [Google Scholar] [CrossRef]
- Schwalb, C.; Chapman, S.K.; Reid, G.A. The membrane-bound tetrahaem c-type cytochrome CymA interacts directly with the soluble fumarate reductase in Shewanella. Biochem. Soc. Trans. 2002, 30, 658–662. Available online: www.tigr.org (accessed on 15 October 2020). [CrossRef]
- Marritt, S.J.; McMillan, D.G.G.; Shi, L.; Fredrickson, J.K.; Zachara, J.M.; Richardson, D.J.; Jeuken, L.J.; Butt, J.N. The roles of CymA in support of the respiratory flexibility of Shewanella oneidensis MR-1. Biochem. Soc. Trans. 2012, 40, 1217–1221. [Google Scholar] [CrossRef]
- Marritt, S.J.; Lowe, T.G.; Bye, J.; McMillan, D.G.G.; Shi, L.; Fredrickson, J.; Zachara, J.; Richardson, D.J.; Cheesman, M.R.; Jeuken, L.J.C.; et al. A functional description of CymA, an electron-transfer hub supporting anaerobic respiratory flexibility in Shewanellae. Biochem. J. 2012, 444, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Habte, M.L.; Beyene, E.A. Biological Application and Disease of Oxidoreductase Enzymes. In Oxidoreductase [Working Title]; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar]
- McMillan, D.G.G.; Marritt, S.J.; Butt, J.N.; Jeuken, L.J.C. Menaquinone-7 is specific cofactor in tetraheme quinol dehydrogenase CymA. J. Biol. Chem. 2012, 287, 14215–14225. [Google Scholar] [CrossRef]
- Protein Data Bank. RCSB PDB-1KQG: Formate Dehydrogenase N from E. coli. 2002. Available online: https://www.rcsb.org/structure/1KQG (accessed on 1 April 2021).
- McMillan, D.G.G.; Marritt, S.J.; Firer-Sherwood, M.A.; Shi, L.; Richardson, D.J.; Evans, S.D.; Elliott, S.J.; Butt, J.N.; Jeuken, L.J.C. Protein—Protein Interaction Regulates the Direction of Catalysis and Electron Transfer in a Redox Enzyme Complex. J. Am. Chem. Soc. 2013, 135, 10550–10556. [Google Scholar] [CrossRef]
- Gates, A.J.; Marritt, S.J.; Bradley, J.M.; Shi, L.; McMillan, D.G.G.; Jeuken, L.J.; Richardson, D.J.; Butt, J.N. Electrode assemblies composed of redox cascades from microbial respiratory electron transfer chains. Biochem. Soc. Trans. 2013, 41, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Daldal, F.; Deshmukh, M.; Prince, R.C. Membrane-anchored cytochrome c as an electron carrier in photosynthesis and respiration: Past, present and future of an unexpected discovery. Photosynth. Res. 2003, 76. [Google Scholar] [CrossRef]
- McMillan, D.G.G.; Marritt, S.J.; Kemp, G.L.; Gordon-Brown, P.; Butt, J.N.; Jeuken, L.J.C. The impact of enzyme orientation and electrode topology on the catalytic activity of adsorbed redox enzymes. Electrochim. Acta 2013, 110, 79–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Myllykallio, H.; Drepper, F.; Mathis, P.; Daldal, F. Membrane-Anchored CytochromecyMediated Microsecond Time Range Electron Transfer from the Cytochrome bc1 Complex to the Reaction Center in Rhodobacter capsulatus. Biochemistry 1998, 37, 5501–5510. [Google Scholar] [CrossRef]
- Lee, D.-W.; Ozturk, Y.; Mamedova, A.; Osyczka, A.; Cooley, J.W.; Daldal, F. A functional hybrid between the cytochrome bc1 complex and its physiological membrane-anchored electron acceptor cytochrome cy in Rhodobacter capsulatus. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 346–352. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) Anchors: Biochemistry and Cell Biology: Introduction to a Thematic Review Series. J. Lipid Res. 2016, 57, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.J.; Hart, G.W.; Kinoshita, T. Glycosylphosphatidylinositol Anchors. In Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2017; Volume 3. [Google Scholar]
- Zurzolo, C.; Simons, K. Glycosylphosphatidylinositol-anchored proteins: Membrane organization and transport. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 632–639. [Google Scholar] [CrossRef]
- Montenegro, M.F.; Moral-Naranjo, M.T.; Campoy, F.J.; Muñoz-Delgado, E.; Vidal, C.J. The lipid raft-bound alkaline phosphatase activity increases and the level of transcripts remains unaffected in liver of merosin-deficient LAMA2dy mouse. Chem. Biol. Interact. 2014, 216, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug Discov. 2008, 7, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.; Sanvictores, T.; John, S. Alkaline Phosphatase; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Giocondi, M.-C.; Seantier, B.; Dosset, P.; Milhiet, P.-E.; Le Grimellec, C. Characterizing the interactions between GPI-anchored alkaline phosphatases and membrane domains by AFM. Pflügers Arch. Eur. J. Physiol. 2008, 456, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Sträter, N. Ecto-5′-nucleotidase: Structure function relationships. Purinergic Signal. 2006, 2, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Zukowska, P.; Kutryb-Zajac, B.; Toczek, M.; Smolenski, R.T.; Slominska, E.M. The role of ecto-5′-nucleotidase in endothelial dysfunction and vascular pathologies. Pharmacol. Rep. 2015, 67, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Knapp, K.; Zebisch, M.; Pippel, J.; El-Tayeb, A.; Müller, C.E.; Sträter, N. Crystal Structure of the Human Ecto-5′-Nucleotidase (CD73): Insights into the Regulation of Purinergic Signaling. Structure 2012, 20, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. RCSB PDB-4H2I: Human Ecto-5′-Nucleotidase (CD73): Crystal form III (Closed) in Complex with AMPCP. 2012. Available online: https://www.rcsb.org/structure/4H2I (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-4H2B: Human Ecto-5′-Nucleotidase (CD73): Crystal form II (Open) in Complex with Baicalin. 2012. Available online: https://www.rcsb.org/structure/4H2B (accessed on 1 April 2021).
- Adzic, M.; Nedeljkovic, N. Unveiling the role of Ecto-5′-nucleotidase/CD73 in astrocyte migration by using pharmacological tools. Front. Pharmacol. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, S.; Liu, G.; Kong, F.; Chen, S.; Yan, H. Multiple steps determine CD73 shedding from RPE: Lipid raft localization, ARA1 interaction, and MMP-9 up-regulation. Purinergic Signal. 2018, 14, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Perrier, A.L.; Massoulié, J.; Krejci, E. PRiMA: The membrane anchor of acetylcholinesterase in the brain. Neuron 2002, 33, 275–285. [Google Scholar] [CrossRef]
- Haas, R.; Jackson, B.C.; Reinhold, B.; Foster, J.D.; Rosenberry, T.L. Glycoinositol phospholipid anchor and protein C-terminus of bovine erythrocyte acetylcholinesterase: Analysis by mass spectrometry and by protein and DNA sequencing. Biochem. J. 1996, 314, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Paulick, M.G.; Bertozzi, C.R. The glycosylphosphatidylinositol anchor: A complex membrane-anchoring structure for proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef]
- McDonald, A.E. Alternative oxidase: What information can protein sequence comparisons give us? Physiol. Plant. 2009, 137, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.A.; Andersson, M.E.; Nordlund, P. New insight into the structure and function of the alternative oxidase. Biochim. Biophys. Acta (BBA) Bioenerg. 2000, 1460, 241–254. [Google Scholar] [CrossRef]
- Minagawa, N.; Sakajo, S.; Komiyama, T.; Yoshimoto, A. Essential role of ferrous iron in cyanide-resistant respiration in Hansenula anomala. FEBS Lett. 1990, 267, 114–116. [Google Scholar] [CrossRef]
- Siedow, J.N.; Umbach, A.L.; Moore, A.L. The active site of the cyanide-resistant oxidase from plant mitochondria contains a binuclear iron center. FEBS Lett. 1995, 362, 10–14. [Google Scholar] [CrossRef]
- Andersson, M.E.; Nordlund, P. A revised model of the active site of alternative oxidase. FEBS Lett. 1999, 449, 17–22. [Google Scholar] [CrossRef]
- Trusova, V.M.; Gorbenko, G.P.; Molotkovsky, J.G.; Kinnunen, P.K. Cytochrome c-Lipid Interactions: New Insights from Resonance Energy Transfer. Biophys. J. 2010, 99, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Spaar, A.; Flöck, D.; Helms, V. Association of cytochrome c with membrane-bound cytochrome c oxidase proceeds parallel to the membrane rather than in bulk solution. Biophys. J. 2009, 96, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. RCSB PDB-5XTE: Cryo-EM Structure of Human Respiratory Complex III (Cytochrome bc1 Complex). 2017. Available online: https://www.rcsb.org/structure/5XTE (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-5Z62: Structure of Human Cytochrome C Oxidase. 2019. Available online: https://www.rcsb.org/structure/5Z62 (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-4Q5P: Lysine-Ligated Yeast Iso-1 Cytochrome C. 2014. Available online: https://www.rcsb.org/structure/4Q5P (accessed on 1 April 2021).
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef]
- Gorbenko, G.P.; Molotkovsky, J.G.; Kinnunen, P.K. Cytochrome c Interaction with Cardiolipin/Phosphatidylcholine Model Membranes: Effect of Cardiolipin Protonation. Biophys. J. 2006, 90, 4093–4103. [Google Scholar] [CrossRef]
- Schurig-Briccio, L.A.; Yano, T.; Rubin, H.; Gennis, R.B. Characterization of the type 2 NADH: Menaquinone oxidoreductases from Staphylococcus aureus and the bactericidal action of phenothiazines. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.M.; Volentini, S.I.; Rintoul, M.R.; Rapisarda, V.A. Amphipathic C-terminal region of Escherichia coli NADH dehydrogenase-2 mediates membrane localization. Arch. Biochem. Biophys. 2011, 505, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.M.P.; Bandeiras, T.M.; Teixeira, M. New Insights into Type II NAD(P)H: Quinone Oxidoreductases. Microbiol. Mol. Biol. Rev. 2004, 68, 603–616. [Google Scholar] [CrossRef]
- Rapisarda, V.A.; Chehín, R.N.; Rivas, J.D.L.; Rodríguez-Montelongo, L.; Farías, R.N.; Massa, E.M. Evidence for Cu(I)-thiolate ligation and prediction of a putative copper-binding site in the Escherichia coli NADH dehydrogenase-2. Arch. Biochem. Biophys. 2002, 405, 87–94. [Google Scholar] [CrossRef]
- Schmid, R.; Gerloff, D.L. Functional properties of the alternative NADH: Ubiquinone oxidoreductase from E. coli through comparative 3-D modelling. FEBS Lett. 2004, 578, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Blaza, J.N.; Bridges, H.R.; Aragão, D.; Dunn, E.A.; Heikal, A.; Cook, G.M.; Nakatani, Y.; Hirst, J. The mechanism of catalysis by type-II NADH: Quinone oxidoreductases. Sci. Rep. 2017, 7, 40165. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.I.; Broek, M.A.V.D.; Merkel, A.Y.; Cortes, P.D.L.T.; Kalamorz, F.; Cook, G.M.; Van Loosdrecht, M.C.M.; McMillan, D.G.G. Genomic analysis of Caldalkalibacillus thermarum TA2.A1 reveals aerobic alkaliphilic metabolism and evolutionary hallmarks linking alkaliphilic bacteria and plant life. Extremophiles 2020, 24, 923–935. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.G.G.; Keis, S.; Berney, M.; Cook, G.M. Nonfermentative thermoalkaliphilic growth is restricted to alkaline environments. Appl. Environ. Microbiol. 2009, 75, 7649–7654. [Google Scholar] [CrossRef][Green Version]
- Feng, Y.; Li, W.; Li, J.; Wang, J.; Ge, J.; Xu, D.; Liu, Y.; Wu, K.; Zeng, Q.; Wu, J.-W.; et al. Structural insight into the type-II mitochondrial NADH dehydrogenases. Nat. Cell Biol. 2012, 491, 478–482. [Google Scholar] [CrossRef]
- Iwata, M.; Lee, Y.; Yamashita, T.; Yagi, T.; Iwata, S.; Cameron, A.D.; Maher, M.J. The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates. Proc. Natl. Acad. Sci. USA 2012, 109, 15247–15252. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Inaoka, D.K.; Shiba, T.; Saimoto, H.; Sakura, T.; Amalia, E.; Kido, Y.; Sakai, C.; Nakamura, M.; Moore, A.L.; et al. Selective Cytotoxicity of Dihydroorotate Dehydrogenase Inhibitors to Human Cancer Cells Under Hypoxia and Nutrient-Deprived Conditions. Front. Pharmacol. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Madak, J.T.; Bankhead, A.; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. RCSB PDB-5XFV: Crystal Structures of FMN-Bound form of Dihydroorotate Dehydrogenase from Trypanosoma Brucei. 2017. Available online: https://www.rcsb.org/structure/5XFV (accessed on 1 April 2021).
- Protein Data Bank. RCSB PDB-6VTN: Crystal Structure of Plasmodium Falciparum Dihydroorotate Dehydrogenase Bound with Inhibitor DSM557. 2020. Available online: https://www.rcsb.org/structure/6VTN (accessed on 1 April 2021).
- Rossi, C.; Chopineau, J. Biomimetic tethered lipid membranes designed for membrane-protein interaction studies. Eur. Biophys. J. 2007, 36, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. RCSB PDB-2PRH: The Structures of Apo- and Inhibitor Bound Human Dihydroorotate Dehydrogenase Reveal Conformational Flexibility within the Inhibitor Binding Site. 2007. Available online: https://www.rcsb.org/structure/2PRH (accessed on 1 April 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Example Protein (Model Organism) | Examples of Pathogenic Organism(s) | Effect on Human Health | Current Available Drugs/Treatments | Homologues among Domains/Species |
|---|---|---|---|---|
| Type-II NADH dehydrogenase (NDH-2) (Caldalkalibacillus thermarum) | Mycobacterium tuberculosis | Tuberculosis; 1.4M deaths worldwide in 2019 [24]. | Bedaquiline (against M. tuberculosis)NDH-2 targeting thioquinazoline (TQZ)-based and tetrahydroindazole (THI)-based inhibitor candidates [25]. | Not reported in mammalian biology; is in prokaryotes and yeast. |
| Staphylococcus aureus | Opportunistic and nosocomial infections, 50,000 deaths/year in the USA [26]. | |||
| Escherichia coli | Gastrointestinal infections causing an estimated 325,000 deaths in developing countries [27]. Cause of 90% of urinary tract infections, 135M cases/year [28]. Horizontal gene transfer of antibiotic resistance to other species [29]. | |||
| Periplasmic nitrate reductase (Nap) (Cupriavidus necator, Rhodobacter sphaeroides) | Haemophilus influenzae | Respiratory disease; 199,000 deaths of children/year in 2008 [30]. | Not reported targeting Nap. Cefotaxime 80% effective against extensive drug resistant (XDR) strains [31]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
| Pseudomonas aeruginosa | Sixth most common nosocomial pathogen in the USA [32]. Lung infection; 2700 deaths/year in the USA [33]. | Not reported targeting Nap. Against multi-drug-resistant strains, cefiderocol and imipenem-cilastatin/relebactam in phase II clinical trials [32]. | ||
| CymA | Shewanella putrificans Shewanella alga | Food spoilage, necrosis, seafood toxin producing (opportunistic pathogen). | N/A | Reported in prokaryotes (specifically in bacteria). Not reported in mammalian biology. |
| Alkaline phosphatase (AP) (Homo sapiens) | Causes disease in humans | Hydroxyapatite deposition disease (HADD) [34]. | Paracetamol and/or nonsteroidal anti-inflammatory drugs, barbotage, and steroid injections for severe cases [35]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
| Ecto-5′-nucleotidase (CD73) (Homo sapiens) | N/A | Tumor progression; 47,050 deaths/year in the USA in 2020 [36]. | Monoclonal antibodies: CPI-006, CPI-444, oleclumab, TJ004309, NZV930, and BMS-986179 [37]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
| Acetylcholine esterase (Homo sapiens) | Causes disease in humans | Senile plaque formation (Alzheimer’s disease); 122,019 deaths/year in the USA in 2018 [38]. | Donepezil, rivastigmine (Exelon) and galantamine (Razadyne, Nivalin) [39]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
| Alternative oxidase (AO) (Trypanosoma brucei) | Trypanosoma brucei | African trypanosomiasis (sleeping sickness); 116 deaths in 2019 [40]. | Pentamidine (early stage), nifurtimox and eflornithine (late stage) for T. brucei gambiense; Suramin (early stage) and melarsoprol (late stage) for T. brucei rhodesiense [41]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes. Not reported in mammalian biology. |
| Cytochrome c | N/A | Inhibits cancer progression;9,900,000 total cancer deaths/year [42]. | Cisplatin [43]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
| Dihydroorotate dehydrogenase (DHODH) (Homo sapiens) | N/A | Inhibits cancer progression; 9,900,000 total cancer deaths/year [42]. | Brequinar and leflunomide [44]. | Reported in prokaryotes (specifically in bacteria) and eukaryotes; also, in humans. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boes, D.M.; Godoy-Hernandez, A.; McMillan, D.G.G. Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life. Membranes 2021, 11, 346. https://doi.org/10.3390/membranes11050346
Boes DM, Godoy-Hernandez A, McMillan DGG. Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life. Membranes. 2021; 11(5):346. https://doi.org/10.3390/membranes11050346
Chicago/Turabian StyleBoes, Deborah M., Albert Godoy-Hernandez, and Duncan G. G. McMillan. 2021. "Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life" Membranes 11, no. 5: 346. https://doi.org/10.3390/membranes11050346
APA StyleBoes, D. M., Godoy-Hernandez, A., & McMillan, D. G. G. (2021). Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life. Membranes, 11(5), 346. https://doi.org/10.3390/membranes11050346