
Secretases Related to Amyloid Precursor Protein Processing

{kind=link}
Abstract
1. Introduction
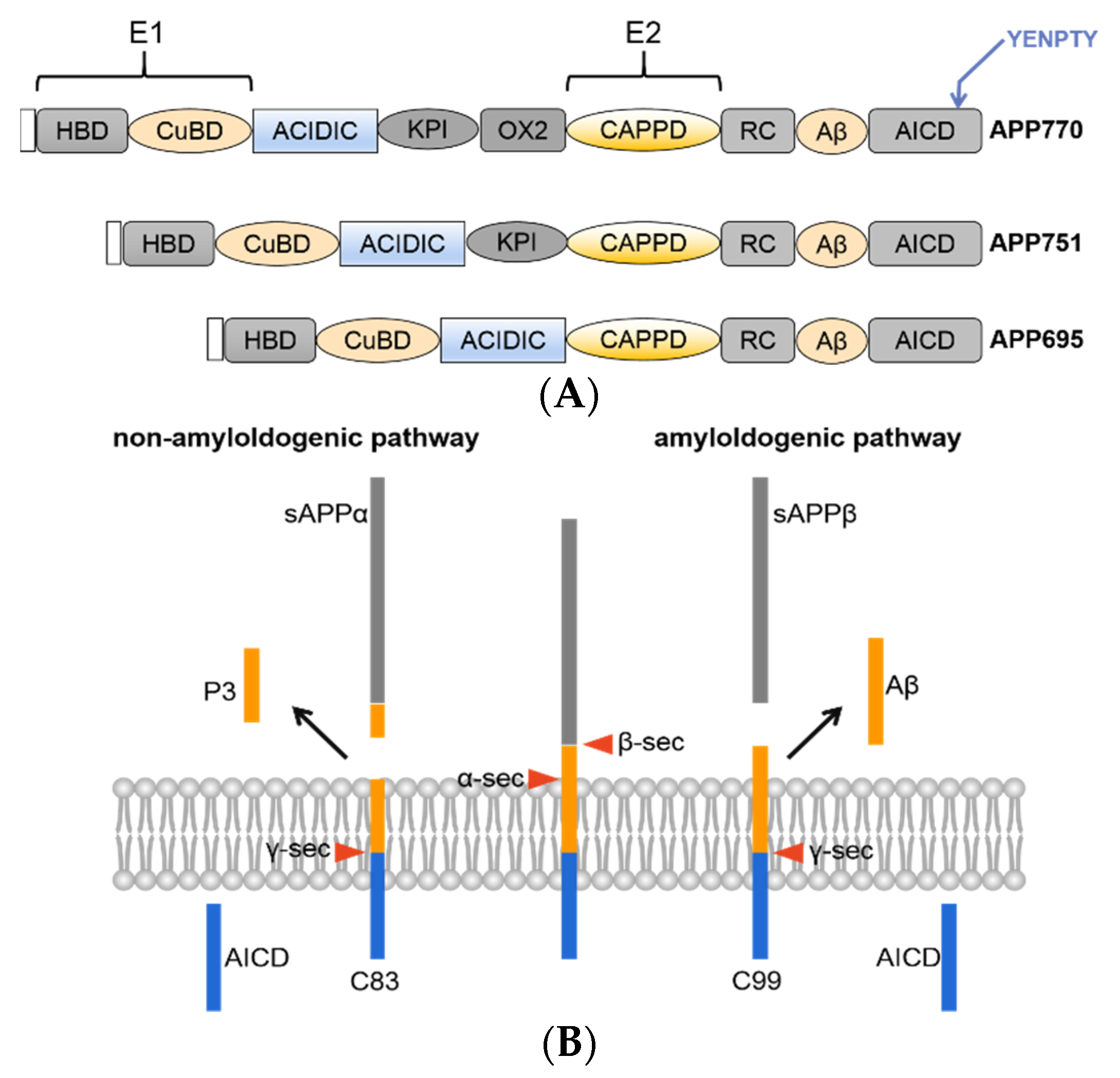
2. APP Structure and Metabolic Processes
3. α-Secretase
4. β-Secretase
5. γ-Secretase
6. Alternative Secretases: Matriptase-2 and MT5-MMP
7. Shedding of the Extracellular Domain of the Membrane Protein APP
8. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Viana, R.J.; Nunes, A.F.; Rodrigues, C.M. Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 522–534. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Reitz, C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimers Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E. Alzheimer disease: Host immune defence, amyloid-beta peptide and Alzheimer disease. Nat. Rev. Neurol. 2016, 12, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Gao, Y.; Ren, R.J.; Zhong, Z.L.; Dammer, E.; Zhao, Q.H.; Shan, S.; Zhou, Z.; Li, X.; Zhang, Y.Q.; Cui, H.L.; et al. Mutation profile of APP, PSEN1, and PSEN2 in Chinese familial Alzheimer’s disease. Neurobiol. Aging 2019, 77, 154–157. [Google Scholar] [CrossRef]
- Wu, M.; Fang, K.; Wang, W.; Lin, W.; Guo, L.; Wang, J. Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus. Biophys. Rep. 2019, 5, 98–109. [Google Scholar] [CrossRef]
- Sisodia, S.S.; Koo, E.H.; Hoffman, P.N.; Perry, G.; Price, D.L. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J. Neurosci. 1993, 13, 3136–3142. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Wilquet, V.; De Strooper, B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588. [Google Scholar] [CrossRef]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.P.; Downer, E.J.; Campbell, V. A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the beta-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem. J. 2003, 371, 789–798. [Google Scholar] [CrossRef]
- Sondag, C.M.; Combs, C.K. Amyloid precursor protein cross-linking stimulates beta amyloid production and pro-inflammatory cytokine release in monocytic lineage cells. J. Neurochem. 2006, 97, 449–461. [Google Scholar] [CrossRef]
- Chen, J.; Luo, B.; Zhong, B.R.; Li, K.Y.; Wen, Q.X.; Song, L.; Xiang, X.J.; Zhou, G.F.; Hu, L.T.; Deng, X.J.; et al. Sulfuretin exerts diversified functions in the processing of amyloid precursor protein. Genes Dis. 2021, 8, 867–881. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 2006, 66, S69–S73. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Soriano, S.; Hayes, J.D.; Ostaszewski, B.; Xia, W.; Selkoe, D.J.; Chen, X.; Stokin, G.B.; Koo, E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 1999, 274, 18851–18856. [Google Scholar] [CrossRef]
- Groot, A.J.; Habets, R.; Yahyanejad, S.; Hodin, C.M.; Reiss, K.; Saftig, P.; Theys, J.; Vooijs, M. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol. Cell. Biol. 2014, 34, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Zolkiewska, A. ADAM proteases: Ligand processing and modulation of the Notch pathway. Cell. Mol. Life Sci. 2008, 65, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Skovronsky, D.M.; Moore, D.B.; Milla, M.E.; Doms, R.W.; Lee, V.M. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J. Biol. Chem. 2000, 275, 2568–2575. [Google Scholar] [CrossRef]
- Sisodia, S.S. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA 1992, 89, 6075–6079. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Bennett, B.D.; Denis, P.; Haniu, M.; Teplow, D.B.; Kahn, S.; Louis, J.C.; Citron, M.; Vassar, R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta -secretase. J. Biol. Chem. 2000, 275, 37712–37717. [Google Scholar] [CrossRef]
- Schneider, A.; Rajendran, L.; Honsho, M.; Gralle, M.; Donnert, G.; Wouters, F.; Hell, S.W.; Simons, M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 2008, 28, 2874–2882. [Google Scholar] [CrossRef]
- Prabhu, Y.; Burgos, P.V.; Schindler, C.; Farias, G.G.; Magadan, J.G.; Bonifacino, J.S. Adaptor protein 2-mediated endocytosis of the beta-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol. Biol. Cell 2012, 23, 2339–2351. [Google Scholar] [CrossRef]
- Sannerud, R.; Declerck, I.; Peric, A.; Raemaekers, T.; Menendez, G.; Zhou, L.; Veerle, B.; Coen, K.; Munck, S.; De Strooper, B.; et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. USA 2011, 108, e559–e568. [Google Scholar] [CrossRef]
- Obregon, D.; Hou, H.; Deng, J.; Giunta, B.; Tian, J.; Darlington, D.; Shahaduzzaman, M.; Zhu, Y.; Mori, T.; Mattson, M.P.; et al. Soluble amyloid precursor protein-alpha modulates beta-secretase activity and amyloid-beta generation. Nat. Commun. 2012, 3, 777. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Evin, G.; Barakat, A.; Masters, C.L. BACE: Therapeutic target and potential biomarker for Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1923–1926. [Google Scholar] [CrossRef]
- Tan, J.; Evin, G. Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 2012, 120, 869–880. [Google Scholar] [CrossRef]
- Buggia-Prevot, V.; Fernandez, C.G.; Riordan, S.; Vetrivel, K.S.; Roseman, J.; Waters, J.; Bindokas, V.P.; Vassar, R.; Thinakaran, G. Axonal BACE1 dynamics and targeting in hippocampal neurons: A role for Rab11 GTPase. Mol. Neurodegener. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and gamma-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, Y.; Zhang, X.; Bu, G.; Xu, H.; Zhang, Y.W. Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 6. [Google Scholar] [CrossRef]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef]
- Dries, D.R.; Yu, G. Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. Gamma-secretase: Proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef]
- Isbert, S.; Wagner, K.; Eggert, S.; Schweitzer, A.; Multhaup, G.; Weggen, S.; Kins, S.; Pietrzik, C.U. APP dimer formation is initiated in the endoplasmic reticulum and differs between APP isoforms. Cell. Mol. Life Sci. 2012, 69, 1353–1375. [Google Scholar] [CrossRef][Green Version]
- Antalis, T.M.; Bugge, T.H.; Wu, Q. Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci. 2011, 99, 1–50. [Google Scholar] [CrossRef]
- Ramsay, A.J.; Reid, J.C.; Velasco, G.; Quigley, J.P.; Hooper, J.D. The type II transmembrane serine protease matriptase-2--identification, structural features, enzymology, expression pattern and potential roles. Front. Biosci. 2008, 13, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Cal, S.; Quesada, V.; Sanchez, L.M.; Lopez-Otin, C. Matriptase-2, a membrane-bound mosaic serine proteinase predominantly expressed in human liver and showing degrading activity against extracellular matrix proteins. J. Biol. Chem. 2002, 277, 37637–37646. [Google Scholar] [CrossRef]
- Hooper, J.D.; Campagnolo, L.; Goodarzi, G.; Truong, T.N.; Stuhlmann, H.; Quigley, J.P. Mouse matriptase-2: Identification, characterization and comparative mRNA expression analysis with mouse hepsin in adult and embryonic tissues. Biochem. J. 2003, 373, 689–702. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef]
- Beliveau, F.; Brule, C.; Desilets, A.; Zimmerman, B.; Laporte, S.A.; Lavoie, C.L.; Leduc, R. Essential role of endocytosis of the type II transmembrane serine protease TMPRSS6 in regulating its functionality. J. Biol. Chem. 2011, 286, 29035–29043. [Google Scholar] [CrossRef]
- Jackle, F.; Schmidt, F.; Wichert, R.; Arnold, P.; Prox, J.; Mangold, M.; Ohler, A.; Pietrzik, C.U.; Koudelka, T.; Tholey, A.; et al. Metalloprotease meprin beta is activated by transmembrane serine protease matriptase-2 at the cell surface thereby enhancing APP shedding. Biochem. J. 2015, 470, 91–103. [Google Scholar] [CrossRef]
- Baranger, K.; Rivera, S.; Liechti, F.D.; Grandgirard, D.; Bigas, J.; Seco, J.; Tarrago, T.; Leib, S.L.; Khrestchatisky, M. Endogenous and synthetic MMP inhibitors in CNS physiopathology. Prog. Brain Res. 2014, 214, 313–351. [Google Scholar] [CrossRef]
- Lafleur, M.A.; Handsley, M.M.; Knauper, V.; Murphy, G.; Edwards, D.R. Endothelial tubulogenesis within fibrin gels specifically requires the activity of membrane-type-matrix metalloproteinases (MT-MMPs). J. Cell Sci. 2002, 115, 3427–3438. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.M.; Reeves, T.M.; Phillips, L.L. MT5-MMP, ADAM-10, and N-cadherin act in concert to facilitate synapse reorganization after traumatic brain injury. J. Neurotrauma 2012, 29, 1922–1940. [Google Scholar] [CrossRef]
- Sekine-Aizawa, Y.; Hama, E.; Watanabe, K.; Tsubuki, S.; Kanai-Azuma, M.; Kanai, Y.; Arai, H.; Aizawa, H.; Iwata, N.; Saido, T.C. Matrix metalloproteinase (MMP) system in brain: Identification and characterization of brain-specific MMP highly expressed in cerebellum. Eur. J. Neurosci. 2001, 13, 935–948. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef]
- Zhu, X.; Li, X.; Zhu, M.; Xu, K.; Yang, L.; Han, B.; Huang, R.; Zhang, A.; Yao, H. Metalloprotease Adam10 suppresses epilepsy through repression of hippocampal neuroinflammation. J. Neuroinflamm. 2018, 15, 221. [Google Scholar] [CrossRef]
- Ehlers, M.R.; Riordan, J.F. Membrane proteins with soluble counterparts: Role of proteolysis in the release of transmembrane proteins. Biochemistry 1991, 30, 10065–10074. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Deciphering Alzheimer’s disease: The amyloid precursor protein yields new clues. Science 1990, 248, 1058–1060. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Choi, S.H.; Romano, D.M.; Gannon, M.A.; Lesinski, A.N.; Kim, D.Y.; Tanzi, R.E. ADAM10 missense mutations potentiate beta-amyloid accumulation by impairing prodomain chaperone function. Neuron 2013, 80, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Prox, J.; Bernreuther, C.; Altmeppen, H.; Grendel, J.; Glatzel, M.; D’Hooge, R.; Stroobants, S.; Ahmed, T.; Balschun, D.; Willem, M.; et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 2013, 33, 12915–12928. [Google Scholar] [CrossRef]
- Picon-Pages, P.; Gutierrez, D.A.; Barranco-Almohalla, A.; Crepin, G.; Tajes, M.; Ill-Raga, G.; Guix, F.X.; Menendez, S.; Arumi-Uria, M.; Vicente, R.; et al. Amyloid Beta-Peptide Increases BACE1 Translation through the Phosphorylation of the Eukaryotic Initiation Factor-2alpha. Oxid. Med. Cell. Longev. 2020, 2020, 2739459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Xiang, X.; Filser, S.; Marinkovic, P.; Dorostkar, M.M.; Crux, S.; Neumann, U.; Shimshek, D.R.; Rammes, G.; Haass, C.; et al. Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibition Impairs Synaptic Plasticity via Seizure Protein 6. Biol. Psychiatry 2018, 83, 428–437. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Liu, Y.; Ji, S. Secretases Related to Amyloid Precursor Protein Processing. Membranes 2021, 11, 983. https://doi.org/10.3390/membranes11120983
Liu X, Liu Y, Ji S. Secretases Related to Amyloid Precursor Protein Processing. Membranes. 2021; 11(12):983. https://doi.org/10.3390/membranes11120983
Chicago/Turabian StyleLiu, Xiaoling, Yan Liu, and Shangrong Ji. 2021. "Secretases Related to Amyloid Precursor Protein Processing" Membranes 11, no. 12: 983. https://doi.org/10.3390/membranes11120983
APA StyleLiu, X., Liu, Y., & Ji, S. (2021). Secretases Related to Amyloid Precursor Protein Processing. Membranes, 11(12), 983. https://doi.org/10.3390/membranes11120983