
Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases



Abstract
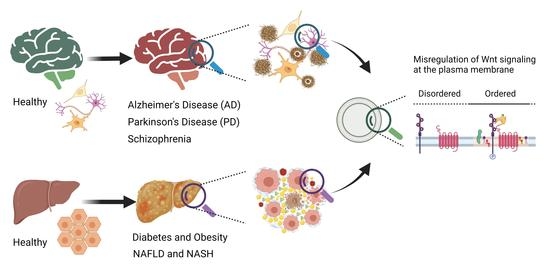
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
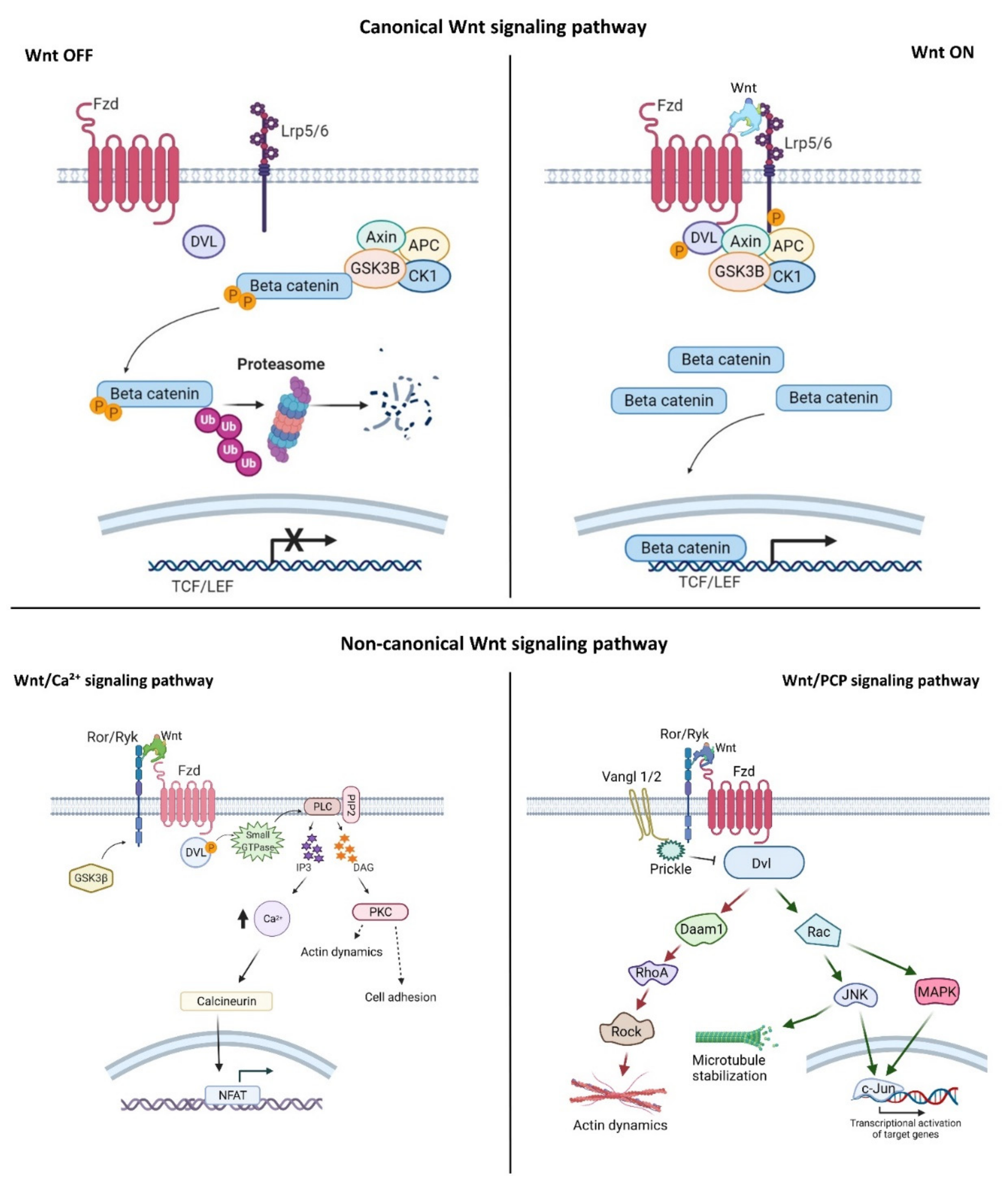
2. Wnt Signaling Pathways
3. Wnt Signaling at the Plasma Membrane in Aging and Brain Disorders
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Schizophrenia
4. Wnt Signaling Pathway in Metabolic Diseases
4.1. Diabetes and Obesity
4.2. Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aman, A.J.; Fulbright, A.N.; Parichy, D.M. Wnt/β-catenin regulates an ancient signaling network during zebrafish scale development. eLife 2018, 7, e37001. [Google Scholar] [CrossRef]
- Behari, J. The Wnt/β-catenin signaling pathway in liver biology and disease. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 745–756. [Google Scholar] [CrossRef]
- Grainger, S.; Willert, K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1422. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Demirci, Y.; Cucun, G.; Poyraz, Y.K.; Mohammed, S.; Heger, G.; Papatheodorou, I.; Ozhan, G. Comparative Transcriptome Analysis of the Regenerating Zebrafish Telencephalon Unravels a Resource with Key Pathways During Two Early Stages and Activation of Wnt/β-Catenin Signaling at the Early Wound Healing Stage. Front. Cell Dev. Biol. 2020, 8, 584604. [Google Scholar] [CrossRef] [PubMed]
- Ozhan, G.; Weidinger, G. Wnt/β-catenin signaling in heart regeneration. Cell Regen. 2015, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, G.; Weidinger, G. Restoring Tissue Homeostasis: Wnt Signaling in Tissue Regeneration after Acute Injury. In Wnt signaling in Development and Disease: Molecular Mechanisms and Biological Functions; Hoppler, S.P., Moon, R.T., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2014. [Google Scholar]
- Bonnet, C.; Brahmbhatt, A.; Deng, S.X.; Zheng, J.J. Wnt signaling activation: Targets and therapeutic opportunities for stem cell therapy and regenerative medicine. RSC Chem. Biol. 2021, 2, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Chen, J.; Deng, Z.-L.; Luo, X.; Song, W.-X.; A Sharff, K.; Tang, N.; Haydon, R.C.; Luu, H.H.; He, T.-C. Wnt signaling and human diseases: What are the therapeutic implications? Lab. Investig. 2007, 87, 97–103. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Levental, I.; Levental, K.R.; Heberle, F.A. Lipid Rafts: Controversies Resolved, Mysteries Remain. Trends Cell Biol. 2020, 30, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Miller, Y.I. Biology of Lipid Rafts: Introduction to the Thematic Review Series. J. Lipid Res. 2020, 61, 598–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezgin, E.; Azbazdar, Y.; Ng, X.W.; Teh, C.; Simons, K.; Weidinger, G.; Wohland, T.; Eggeling, C.; Ozhan, G. Binding of canonical Wnt ligands to their receptor complexes occurs in ordered plasma membrane environments. FEBS J. 2017, 284, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Gutmann, T.; Buhl, T.; Dirkx, R.; Grzybek, M.; Coskun, Ü.; Solimena, M.; Simons, K.; Levental, I.; Schwille, P. Adaptive Lipid Packing and Bioactivity in Membrane Domains. PLoS ONE 2015, 10, e0123930. [Google Scholar] [CrossRef] [PubMed]
- Staubach, S.; Hanisch, F.-G. Lipid rafts: Signaling and sorting platforms of cells and their roles in cancer. Expert Rev. Proteom. 2011, 8, 263–277. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Turnbull, S.; Mei, N.; Filice, C.; Lee, B.Y.; Robinson, M.; Pavlov, E.; Finot, E.; Leonenko, Z. Changes in Lipid Membrane May Trigger Amyloid Toxicity in Alzheimer’s Disease. Biophys. J. 2019, 116, 427a. [Google Scholar] [CrossRef] [Green Version]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell. Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa-Herrera, F.; Taoro-González, L.; Valdés-Baizabal, C.; Diaz, M.; Marín, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef] [Green Version]
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling Pathways at the Plasma Membrane and Their Misregulation in Cancer. Front. Cell Dev. Biol. 2021, 9, 631623. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Clevers, H. WNT signalling events near the cell membrane and their pharmacological targeting for the treatment of cancer. Br. J. Pharmacol. 2017, 174, 4547–4563. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Takada, K.; Zhu, D. Medicine in Drug Discovery Targeting Wnt/β-Catenin Pathway for Drug Therapy. Med. Drug Discov. 2020, 8, 100066. [Google Scholar] [CrossRef]
- Karabicici, M.; Azbazdar, Y.; Ozhan, G.; Senturk, S.; Karagonlar, Z.F.; Erdal, E. Changes in Wnt and TGF-β Signaling Mediate the Development of Regorafenib Resistance in Hepatocellular Carcinoma Cell Line HuH. Front. Cell Dev. Biol. 2021, 9, 639779. [Google Scholar] [CrossRef] [PubMed]
- Kimelman, D.; Xu, W. β-Catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- LeCarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Maung, S.M.T.W.; Jenny, A. Planar cell polarity in Drosophila. Organogenesis 2011, 7, 165–179. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.; Roszko, I.; Solnica-Krezel, L. Planar Cell Polarity: Coordinating Morphogenetic Cell Behaviors with Embryonic Polarity. Dev. Cell 2011, 21, 120–133. [Google Scholar] [CrossRef] [Green Version]
- Flores-Hernández, E.; Velázquez, D.M.; Castañeda-Patlán, M.C.; Fuentes-García, G.; Fonseca-Camarillo, G.; Yamamoto-Furusho, J.K.; Romero-Avila, M.T.; García-Sáinz, J.A.; Robles-Flores, M. Canonical and non-canonical Wnt signaling are simultaneously activated by Wnts in colon cancer cells. Cell. Signal. 2020, 72, 109636. [Google Scholar] [CrossRef]
- Wook-Jin, C.; Bothwell, A.L.M. Canonical and Non-Canonical Wnt Signaling in Immune Cells Wook-Jin. Physiol. Behav. 2016, 176, 100–106. [Google Scholar] [CrossRef]
- Xiao, Q.; Chen, Z.; Jin, X.; Mao, R.; Chen, Z. The many postures of noncanonical Wnt signaling in development and diseases. Biomed. Pharmacother. 2017, 93, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2019, 6, 170. [Google Scholar] [CrossRef]
- Cruciat, C.-M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2012, 5, a015081. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.I.; Godoy, J.A.; Inestrosa, N.C. Modulating Wnt signaling at the root: Porcupine and Wnt acylation. Pharmacol. Ther. 2019, 198, 34–45. [Google Scholar] [CrossRef]
- Zhang, X.; MacDonald, B.T.; Gao, H.; Shamashkin, M.; Coyle, A.J.; Martinez, R.V.; He, X. Characterization of Tiki, a New Family of Wnt-specific Metalloproteases. J. Biol. Chem. 2016, 291, 2435–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnerts, M.E.; Kim, K.-A.; Bright, J.M.; Patel, S.M.; Tran, K.; Zhou, M.; Leung, J.M.; Liu, Y.; Lomas, W.E.; Dixon, M.; et al. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP. Proc. Natl. Acad. Sci. USA 2007, 104, 14700–14705. [Google Scholar] [CrossRef] [Green Version]
- Vonica, A.; Bhat, N.; Phan, K.; Guo, J.; Iancu, L.; Weber, J.A.; Karger, A.; Cain, J.W.; Wang, E.C.; DeStefano, G.M.; et al. Apcdd1 is a dual BMP/Wnt inhibitor in the developing nervous system and skin. Dev. Biol. 2020, 464, 71–87. [Google Scholar] [CrossRef]
- Kagermeier-Schenk, B.; Wehner, D.; Ozhan-Kizil, G.; Yamamoto, H.; Li, J.; Kirchner, K.; Hoffmann, C.; Stern, P.; Kikuchi, A.; Schambony, A.; et al. Waif1/5T4 inhibits Wnt/beta-catenin signaling and activates noncanonical Wnt pathways by modifying LRP6 subcellular localization. Dev. Cell 2011, 21, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Özhan, G.; Sezgin, E.; Wehner, D.; Pfister, A.S.; Kühl, S.J.; Kagermeier-Schenk, B.; Kühl, M.; Schwille, P.; Weidinger, G. Lypd6 Enhances Wnt/β-Catenin Signaling by Promoting Lrp6 Phosphorylation in Raft Plasma Membrane Domains. Dev. Cell 2013, 26, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Ozalp, O.; Cark, O.; Azbazdar, Y.; Haykir, B.; Cucun, G.; Kucukaylak, I.; Alkan-Yesilyurt, G.; Sezgin, E.; Ozhan, G. Nradd Acts as a Negative Feedback Regulator of Wnt/β-Catenin Signaling and Promotes Apoptosis. Biomolecules 2021, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Sakane, H.; Yamamoto, H.; Kikuchi, A. LRP6 is internalized by Dkk1 to suppress its phosphorylation in the lipid raft and is recycled for reuse. J. Cell Sci. 2010, 123, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Sakane, H.; Michiue, T.; Kikuchi, A. Wnt3a and Dkk1 Regulate Distinct Internalization Pathways of LRP6 to Tune the Activation of β-Catenin Signaling. Dev. Cell 2008, 15, 37–48. [Google Scholar] [CrossRef]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Mattson, M.P.; Yao, P.J. Communication breakdown: The impact of ageing on synapse structure. Ageing Res. Rev. 2014, 14, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Mcleod, F.; Salinas, P.C. Wnt proteins as modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt Signaling in the Prefrontal Cortex of Alzheimer’s Disease. Mol. Neurobiol. 2018, 56, 873–891. [Google Scholar] [CrossRef]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The effects of aging on the expression of Wnt pathway genes in mouse tissues. AGE 2014, 36, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.L.; Brann, D.W. Estrogen regulation of Dkk1 and Wnt/β-Catenin signaling in neurodegenerative disease. Brain Res. 2012, 1514, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Makdissy, N.; Haddad, K.; AlBacha, J.D.; Chaker, D.; Ismail, B.; Azar, A.; Oreibi, G.; Ayoub, D.; Achkar, I.; Quilliot, D.; et al. Essential role of ATP6AP2 enrichment in caveolae/lipid raft microdomains for the induction of neuronal differentiation of stem cells. Stem Cell Res. Ther. 2018, 9, 132. [Google Scholar] [CrossRef]
- Honda, A.; Ito, Y.; Takahashi-Niki, K.; Matsushita, N.; Nozumi, M.; Tabata, H.; Takeuchi, K.; Igarashi, M. Extracellular Signals Induce Glycoprotein M6a Clustering of Lipid Rafts and Associated Signaling Molecules. J. Neurosci. 2017, 37, 4046–4064. [Google Scholar] [CrossRef] [PubMed]
- Azbazdar, Y.; Özalp, O.; Sezgin, E.; Veerapathiran, S.; Duncan, A.; Sansom, M.S.P.; Eggeling, C.; Wohland, T.; Karaca, E.; Ozhan, G. More Favorable Palmitic Acid Over Palmitoleic Acid Modification of Wnt3 Ensures Its Localization and Activity in Plasma Membrane Domains. Front. Cell Dev. Biol. 2019, 7, 281. [Google Scholar] [CrossRef]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Levental, I.; Lingwood, D.; Grzybek, M.; Coskun, Ü.; Simons, K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 22050–22054. [Google Scholar] [CrossRef] [Green Version]
- Young, F.B.; Butland, S.L.; Sanders, S.S.; Sutton, L.M.; Hayden, M.R. Putting proteins in their place: Palmitoylation in Huntington disease and other neuropsychiatric diseases. Prog. Neurobiol. 2012, 97, 220–238. [Google Scholar] [CrossRef]
- Abeysinghe, A.; Deshapriya, R.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 425. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Colom-Cadena, M.; the Synaptic Health Endpoints Working Group; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [Green Version]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a Negative Modulator of the Wnt Pathway, Is Associated with Neuronal Degeneration in Alzheimer’s Brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef] [PubMed]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Alarcon, M.; Medina, M.; Hu, Q.; Avila, M.E.; Bustos, B.; Pérez-Palma, E.; Peralta, A.; Salazar, P.; Ugarte, G.D.; Reyes, A.E.; et al. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1709.e9–1709.e18. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, W.F.; Avila, M.E.; Major, M.B.; Myers, A.; Saez, K.; Henriquez, J.P.; Zhao, A.; et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.-J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of Active Glycogen Synthase Kinase 3β (GSK-3β) in Brains Staged for Alzheimer Disease Neurofibrillary Changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hartmann, H.; Do, V.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Van De Wetering, M.; Clevers, H.; Saftig, P.; et al. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395, 698–702. [Google Scholar] [CrossRef]
- Kawamura, Y.; Kikuchi, A.; Takada, R.; Takada, S.; Sudoh, S.; Shibamoto, S.; Yanagisawa, K.; Komano, H. Inhibitory effect of a presenilin 1 mutation on the Wnt signalling pathway by enhancement of β-catenin phosphorylation. J. Biol. Inorg. Chem. 2001, 268, 3036–3041. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.-C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e7. [Google Scholar] [CrossRef]
- Elliott, C.; I Rojo, A.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R.; et al. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry 2018, 8, 179. [Google Scholar] [CrossRef]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 43. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The Secreted Wnt Antagonist Dickkopf-1 Is Required for Amyloid-Mediated Synaptic Loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Sellers, K.J.; Elliott, C.; Jackson, J.; Ghosh, A.; Ribe, E.; Rojo, A.I.; Jarosz-Griffiths, H.H.; Watson, I.A.; Xia, W.; Semenov, M.; et al. Amyloid β synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimer’s Dement. 2017, 14, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.; Lopes, D.; Ammari, R.; Kopra, J.; Millar, S.; Gibb, A.; Salinas, P.C. Deficient Wnt signalling triggers striatal synaptic degeneration and impaired motor behaviour in adult mice. Nat. Commun. 2014, 5, 4992. [Google Scholar] [CrossRef] [Green Version]
- Marzo, A.; Galli, S.; Lopes, D.; Mcleod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of Synapse Degeneration by Restoring Wnt Signaling in the Adult Hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-C.; Tsai, C.-W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-Mediated Wnt Signaling Contributes to Synaptic Abnormalities and Amyloid Pathology in Alzheimer’s Disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.-J.; Xu, H.-L. Silencing of Long Noncoding RNA SOX21-AS1 Relieves Neuronal Oxidative Stress Injury in Mice with Alzheimer’s Disease by Upregulating FZD3/5 via the Wnt Signaling Pathway. Mol. Neurobiol. 2018, 56, 3522–3537. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; A Chacon, M.; I Barría, M.; Garrido, J.L.; A Godoy, J.; Olivares, G.; E Reyes, A.; Alvarez, A.R.; Bronfman, M.; Inestrosa, N.C. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol. Psychiatry 2003, 8, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- Zampagni, M.; Evangelisti, E.; Cascella, R.; Liguri, G.; Becatti, M.; Pensalfini, A.; Uberti, D.; Cenini, G.; Memo, M.; Bagnoli, S.; et al. Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J. Mol. Med. 2010, 88, 597–608. [Google Scholar] [CrossRef]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef]
- Ji, S.-R.; Wu, Y.; Sui, S.-F. Cholesterol Is an Important Factor Affecting the Membrane Insertion of β-Amyloid Peptide (Aβ1–40), Which May Potentially Inhibit the Fibril Formation. J. Biol. Chem. 2002, 277, 6273–6279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, A.M.; Ferreira, A. Increased Membrane Cholesterol Might Render Mature Hippocampal Neurons More Susceptible to -Amyloid-Induced Calpain Activation and Tau Toxicity. J. Neurosci. 2009, 29, 4640–4651. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Okada, T.; Kozutsumi, Y.; Matsuzaki, K. GM1 ganglioside-mediated accumulation of amyloid β-protein on cell membranes. Biochem. Biophys. Res. Commun. 2005, 328, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Odaka, A.; Suzuki, N.; Ihara, Y. GM1 ganglioside–bound amyloid β–protein (Aβ): A possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1995, 1, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Kakio, A.; Nishimoto, S.-I.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Cholesterol-dependent Formation of GM1 Ganglioside-bound Amyloid β-Protein, an Endogenous Seed for Alzheimer Amyloid. J. Biol. Chem. 2001, 276, 24985–24990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Matsubara, T.; Sato, T.; Yanagisawa, K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochim. Biophys. Acta (BBA)—Biomembr. 2008, 1778, 2717–2726. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, K.; Yanagisawa, K. Sphingomyelin accumulation provides a favorable milieu for GM1 ganglioside-induced assembly of amyloid β-protein. Neurosci. Lett. 2010, 481, 168–172. [Google Scholar] [CrossRef]
- Amaro, M.; Šachl, R.; Aydogan, G.; Mikhalyov, I.I.; Vácha, R.; Hof, M. GM 1 Ganglioside Inhibits β-Amyloid Oligomerization Induced by Sphingomyelin. Angew. Chem. Int. Ed. 2016, 55, 9411–9415. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Xiong, W.C.; Mei, L. Lipid Rafts Serve as a Signaling Platform for Nicotinic Acetylcholine Receptor Clustering. J. Neurosci. 2006, 26, 4841–4851. [Google Scholar] [CrossRef] [Green Version]
- Colón-Sáez, J.O.; Yakel, J.L. The α7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 2011, 589, 3163–3174. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Chou, K.L.; Stacy, M.; Simuni, T.; Miyasaki, J.; Oertel, W.H.; Sethi, K.; Fernandez, H.H.; Stocchi, F. The spectrum of “off” in Parkinson’s disease: What have we learned over 40 years? Park. Relat. Disord. 2018, 51, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, J.; Ceravolo, R.; Qamhawi, Z.; Lee, J.-Y.; Deuschl, G.; Brooks, D.; Bonuccelli, U.; Pavese, N. Progression of tremor in early stages of Parkinson’s disease: A clinical and neuroimaging study. Brain 2018, 141, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, D.; Torch, S.; Nissou, M.-F.; Benabid, A.-L.; Verna, J.-M. Extracellular toxicity of 6-hydroxydopamine on PC12 cells. Neurosci. Lett. 2000, 283, 193–196. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Rocha, E.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, F.; Xu, P.; Qu, S. Recent Advance in the Relationship between Excitatory Amino Acid Transporters and Parkinson’s Disease. Neural Plast. 2016, 2016, 8941327. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Chen, C.; Ding, L.; Mo, M.; Zou, J.; Lu, Z.; Li, H.; Wu, H.; Dai, Y.; Xu, P.; et al. Wnt1 Promotes EAAT2 Expression and Mediates the Protective Effects of Astrocytes on Dopaminergic Cells in Parkinson’s Disease. Neural Plast. 2019, 2019, 1247276. [Google Scholar] [CrossRef] [Green Version]
- Castello, P.R.; Drechsel, D.A.; Patel, M. Mitochondria Are a Major Source of Paraquat-induced Reactive Oxygen Species Production in the Brain. J. Biol. Chem. 2007, 282, 14186–14193. [Google Scholar] [CrossRef] [Green Version]
- Colle, D.; Santos, D.B.; Naime, A.A.; Gonçalves, C.L.; Ghizoni, H.; Hort, M.A.; Farina, M. Early Postnatal Exposure to Paraquat and Maneb in Mice Increases Nigrostriatal Dopaminergic Susceptibility to a Re-challenge with the Same Pesticides at Adulthood: Implications for Parkinson’s Disease. Neurotox. Res. 2019, 37, 210–226. [Google Scholar] [CrossRef]
- Ma, J.; Huang, C.; Ma, K.; Wu, Y.-P.; Li, B.-X.; Sun, Y. Effect of Wnt1 and Wnt5a on the development of dopaminergic neurons, and toxicity induced by combined exposure to paraquat and maneb during gestation and lactation. Mol. Med. Rep. 2017, 16, 9721–9728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola-Guzmán, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- De Gregorio, R.; Pulcrano, S.; De Sanctis, C.; Volpicelli, F.; Guatteo, E.; von Oerthel, L.; Latagliata, E.C.; Esposito, R.; Piscitelli, R.M.; Perrone-Capano, C.; et al. miR-34b/c Regulates Wnt1 and Enhances Mesencephalic Dopaminergic Neuron Differentiation. Stem Cell Rep. 2018, 10, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Han, Y.; Fan, X.; Li, Q.; Sun, L.; Qinghu, L. Protective mechanism of Wnt4 gene on Parkinson’s disease (PD) transgenic Drosophila. Int. J. Neurosci. 2019, 129, 703–714. [Google Scholar] [CrossRef]
- Marinko, J.T.; Huang, H.; Penn, W.D.; Capra, J.A.; Schlebach, J.P.; Sanders, C.R. Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis. Chem. Rev. 2019, 119, 5537–5606. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.-I. Lipid rafts involvement in the pathogenesis of Parkinson s disease. Front. Biosci. 2015, 20, 263–279. [Google Scholar] [CrossRef] [Green Version]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.; Kwak, H.B.; Le, S.W.; Kim, H.-H.; Lee, Z.H. Lipid rafts are important for the association of RANK and TRAF. Exp. Mol. Med. 2003, 35, 279–284. [Google Scholar] [CrossRef]
- Gaspar, R.; Pallbo, J.; Weininger, U.; Linse, S.; Sparr, E. Ganglioside lipids accelerate α-synuclein amyloid formation. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 1062–1072. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, A.S.; Fink, A.L. GM1 Specifically Interacts with α-Synuclein and Inhibits Fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Peer Rev. J. Formul. Manag. 2014, 39, 638–645. [Google Scholar]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Doren, V.V.; et al. Schizophrenia risk from complex variation of complement component 4 Schizophrenia Working Group of the Psychiatric Genomics Consortium HHS Public Access. Nature 2016, 11, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E. Neurodevelopmental abnormalities in Schizophrenia: Insights from neuropathology. Dev. Psychopathol. 1999, 11, 439–456. [Google Scholar] [CrossRef]
- Brzózka, M.M.; Radyushkin, K.; Wichert, S.P.; Ehrenreich, H.; Rossner, M. Cognitive and Sensorimotor Gating Impairments in Transgenic Mice Overexpressing the Schizophrenia Susceptibility Gene Tcf4 in the Brain. Biol. Psychiatry 2010, 68, 33–40. [Google Scholar] [CrossRef]
- Okerlund, N.D.; Cheyette, B.N.R. Synaptic Wnt signaling—a contributor to major psychiatric disorders? J. Neurodev. Disord. 2011, 3, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, H.; Genetic Risk and Outcome in Psychosis (GROUP); Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.H.; Mors, O.; et al. Common variants conferring risk of Schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Ebrisch, R.; Esaniotis, A.; Ewolf, R.; Ebielau, H.; Ebernstein, H.-G.; Esteiner, J.; Ebogerts, B.; Braun, A.K.; Jankowski, Z.P.; Ekumaratilake, J.; et al. The Role of Dopamine in Schizophrenia from a Neurobiological and Evolutionary Perspective: Old Fashioned, but Still in Vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef]
- Marchetti, B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3743. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.; Kerwin, R.; Al-Sarraji, S.; Brion, J.P.; Chadwich, A.; Lovestone, S.; Anderton, B.; Everall, I. Abnormalities of Wnt signalling in Schizophrenia—Evidence for neurodevelopmental abnormality. NeuroReport 1998, 9, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Alsabban, A.H.; Morikawa, M.; Tanaka, Y.; Takei, Y.; Hirokawa, N. Kinesin Kif3b mutation reduces NMDAR subunit NR 2A trafficking and causes Schizophrenia-like phenotypes in mice. EMBO J. 2019, 39, e101090. [Google Scholar] [CrossRef]
- Lovestone, S.; Killick, R.; Di Forti, M.; Murray, R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007, 30, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Hoseth, E.Z.; Krull, F.; Dieset, I.; Mørch, R.H.; Hope, S.; Gardsjord, E.S.; Steen, N.E.; Melle, I.; Brattbakk, H.-R.; Steen, V.M.; et al. Exploring the Wnt signaling pathway in Schizophrenia and bipolar disorder. Transl. Psychiatry 2018, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Ftouh, S.; Akbar, M.T.; Hirsch, S.R.; De Belleroche, J.S. Down-regulation of Dickkopf 3, a regulator of the Wnt signalling pathway, in elderly schizophrenic subjects. J. Neurochem. 2005, 94, 520–530. [Google Scholar] [CrossRef]
- Proitsi, P.; Li, T.; Hamilton, G.; Di Forti, M.; Collier, D.; Killick, R.; Chen, R.; Sham, P.; Murray, R.; Powell, J.; et al. Positional Pathway Screen of wnt Signaling Genes in Schizophrenia: Association with DKK. Biol. Psychiatry 2008, 63, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, T.; Seno, H.; Ishino, H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999, 38, 36313823. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Armoskus, C.; Wrobel, B.B.; Spitsyna, V.N.; Souaiaia, T.; Herstein, J.S.; Walker, C.P.; Nguyen, J.D.; Camarena, A.; Weitz, J.R.; et al. Gene Expression in Patient-Derived Neural Progenitors Implicates WNT5A Signaling in the Etiology of Schizophrenia. Biol. Psychiatry 2020, 88, 236–247. [Google Scholar] [CrossRef]
- Liu, X.; Low, S.-K.; Atkins, J.R.; Wu, J.; Reay, W.R.; Cairns, H.M.; Green, M.J.; Schall, U.; Jablensky, A.; Mowry, B.; et al. Wnt receptor gene FZD1 was associated with Schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort. Aust. New Zealand J. Psychiatry 2019, 54, 902–908. [Google Scholar] [CrossRef]
- Katsu, T.; Ujike, H.; Nakano, T.; Tanaka, Y.; Nomura, A.; Nakata, K.; Takaki, M.; Sakai, A.; Uchida, N.; Imamura, T.; et al. The human frizzled-3 (FZD3) gene on chromosome 8p21, a receptor gene for Wnt ligands, is associated with the susceptibility to Schizophrenia. Neurosci. Lett. 2003, 353, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Si, T.; Ling, Y.; Ruan, Y.; Han, Y.; Wang, X.; Zhang, H.; Kong, Q.; Li, X.; Liu, C.; et al. Association study of the human FZD3 locus with Schizophrenia. Biol. Psychiatry 2003, 54, 1298–1301. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, X.; Yuan, Y.; Ling, Y.; Ruan, Y.; Si, T.; Lu, T.; Wu, S.; Gong, X.; Zhu, Z.; et al. Positive association of the human frizzled 3 (FZD3) gene haplotype with Schizophrenia in Chinese Han population. Am. J. Med Genet. 2004, 129B, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Muratake, T.; Yamada, K.; Iwayama-Shigeno, Y.; Iwamoto, K.; Takao, H.; Toyota, T.; Kaneko, N.; Minabe, Y.; Nakamura, K.; et al. Genetic and expression analyses of FZD3 in Schizophrenia. Biol. Psychiatry 2004, 56, 462–465. [Google Scholar] [CrossRef]
- Wei, J.; Hemmings, G.P. Lack of a genetic association between the frizzled-3 gene and Schizophrenia in a British population. Neurosci. Lett. 2004, 366, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-C.; Huang, Y.-S.; Ouyang, W.-C. Beneficial effects of omega-3 fatty acid supplementation in Schizophrenia: Possible mechanisms. Lipids Health Dis. 2020, 19, 159. [Google Scholar] [CrossRef]
- Kim, W.; Fan, Y.-Y.; Barhoumi, R.; Smith, R.; McMurray, D.N.; Chapkin, R.S. n-3 polyunsaturated fatty acids suppress the localization and activation of signaling proteins at the immunological synapse in murine CD4+ T cells by affecting lipid raft formation. J. Immunol. 2008, 181, 6236–6243. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.; Wilczek, K.; Blennow, K.; Maras, A.; Jatzko, A.; A Petroianu, G.; Braus, D.F.; Gattaz, W.F. Altered thalamic membrane phospholipids in Schizophrenia: A postmortem study. Biol. Psychiatry 2004, 56, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Asaba, H.; Kang, M.-J.; Ikeda, Y.; Sone, H.; Takada, S.; Kim, D.-H.; Ioka, R.X.; Ono, M.; Tomoyori, H.; et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA 2002, 100, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Habener, J.F. Wnt signaling in pancreatic islets. In The Islets of Langerhans; Islam, M.S., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 391–419. [Google Scholar]
- Rulifson, I.C.; Karnik, S.K.; Heiser, P.; Berge, D.T.; Chen, H.; Gu, X.; Taketo, M.M.; Nusse, R.; Hebrok, M.; Kim, S.K. Wnt signaling regulates pancreatic beta cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 6247–6252. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Geng, H.; Liu, X.; Wang, X.; Li, R.; Lv, Q.; Liu, Y.; Wang, J.; Yang, M.; Jones, P.M.; et al. Wingless-type MMTV integration site family member 5a: A novel biomarker regulated in type 2 diabetes mellitus and diabetic kidney disease. J. Diabetes Metab. Disord. 2019, 18, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Azar, F.A.; Lim, G.E. Metabolic Contributions of Wnt Signaling: More than Controlling Flight. Front. Cell Dev. Biol. 2021, 9, 709823. [Google Scholar] [CrossRef]
- Müller-Wieland, M.D. Definition, Klassifikation und Diagnostik des Diabetes mellitus; Definition, Classification and Diagnosis of Diabetes Mellitus. Der Diabetol. 2019, 15, 128–134. [Google Scholar] [CrossRef]
- Elhourch, S.; Arrouchi, H.; Mekkaoui, N.; Allou, Y.; Ghrifi, F.; Allam, L.; Elhafidi, N.; Belyamani, L.; Ibrahimi, A.; Elomri, N.; et al. Significant Association of Polymorphisms in the TCF7L2 Gene with a Higher Risk of Type 2 Diabetes in a Moroccan Population. J. Pers. Med. 2021, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Muendlein, A.; Saely, C.H.; Geller-Rhomberg, S.; Sonderegger, G.; Rein, P.; Winder, T.; Beer, S.; Vonbank, A.; Drexel, H. Single Nucleotide Polymorphisms of TCF7L2 Are Linked to Diabetic Coronary Atherosclerosis. PLoS ONE 2011, 6, e17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghera, D.K.; Nath, S.K.; Ortega, L.; Gambarelli, M.; Kim-Howard, X.; Singh, J.R.; Ralhan, S.K.; Wander, G.S.; Mehra, N.K.; Mulvihill, J.J.; et al. TCF7L2 Polymorphisms are Associated with Type 2 Diabetes in Khatri Sikhs from North India: Genetic Variation Affects Lipid Levels. Ann. Hum. Genet. 2008, 72, 499–509. [Google Scholar] [CrossRef]
- He, Q.J.; Wang, P.; Liu, Q.Q.; Wu, Q.G.; Li, Y.F.; Wang, J.; Lee, S.C. Secreted Wnt6 mediates diabetes-associated centrosome amplification via its receptor FZD. Am. J. Physiol. Physiol. 2020, 318, C48–C62. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.-X. Activation of the Wnt Pathway Plays a Pathogenic Role in Diabetic Retinopathy in Humans and Animal Models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toomes, C.; Bottomley, H.M.; Jackson, R.M.; Towns, K.V.; Scott, S.; Mackey, D.A.; Craig, J.E.; Jiang, L.; Yang, Z.; Trembath, R.; et al. Mutations in LRP5 or FZD4 Underlie the Common Familial Exudative Vitreoretinopathy Locus on Chromosome 11q. Am. J. Hum. Genet. 2004, 74, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Gaudio, A.; Privitera, F.; Battaglia, K.; Torrisi, V.; Sidoti, M.H.; Pulvirenti, I.; Canzonieri, E.; Tringali, G.; Fiore, C.E. Sclerostin Levels Associated with Inhibition of the Wnt/β-Catenin Signaling and Reduced Bone Turnover in Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 3744–3750. [Google Scholar] [CrossRef]
- Semënov, M.; Tamai, K.; He, X. SOST Is a Ligand for LRP5/LRP6 and a Wnt Signaling Inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, X.; Li, F.; Ji, Q. MiR-128-3p accelerates cardiovascular calcification and insulin resistance through ISL1-dependent Wnt pathway in type 2 diabetes mellitus rats. J. Cell. Physiol. 2018, 234, 4997–5010. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Tian, P.; Liu, Z.; Zhang, F.; Zhang, Y.; Qu, L.; Liu, X.; Wang, Y.; Zhou, X.; Xiao, Y.; et al. MicroRNA-27a targets Sfrp1 to induce renal fibrosis in diabetic nephropathy by activating Wnt/β-Catenin signalling. Biosci. Rep. 2020, 40, BSR20192794. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.P.; Nishii, A.; Li, Z.; DelProposto, J.B.; Corsa, C.A.; Mori, H.; Hardij, J.; Learman, B.S.; Lumeng, C.N.; MacDougald, O.A. Wnt/β-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Mol. Metab. 2020, 42, 101078. [Google Scholar] [CrossRef]
- Chen, N.; Wang, J. Wnt/β-Catenin Signaling and Obesity. Front. Physiol. 2018, 9, 792. [Google Scholar] [CrossRef]
- Akoumianakis, I.; Sanna, F.; Margaritis, M.; Badi, I.; Akawi, N.; Herdman, L.; Coutinho, P.; Fagan, H.; Antonopoulos, A.S.; Oikonomou, E.K.; et al. Adipose tissue–derived WNT5A regulates vascular redox signaling in obesity via USP17/RAC1-mediated activation of NADPH oxidases. Sci. Transl. Med. 2019, 11, eaav5055. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, R.; Wang, F.; Hong, J.; Li, X.; Chen, M.; Ke, Y.; Zhang, X.; Ma, Q.; Wang, R.; et al. Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nature 2013, 15, 1455–1463. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2011, 8, 1031–1063. [Google Scholar] [CrossRef]
- Giannessi, D.; Maltinti, M.; Del Ry, S. Adiponectin circulating levels: A new emerging biomarker of cardiovascular risk. Pharmacol. Res. 2007, 56, 459–467. [Google Scholar] [CrossRef]
- Salinas, M.L.; Fuentes, N.; Choate, R.; Wright, R.C.; McMurray, D.N.; Chapkin, R.S. AdipoRon Attenuates Wnt Signaling by Reducing Cholesterol-Dependent Plasma Membrane Rigidity. Biophys. J. 2019, 118, 885–897. [Google Scholar] [CrossRef]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Mok, M.T.; Yang, P.; Cheng, A.S. Epigenetic Activation of Wnt/β-Catenin Signaling in NAFLD-Associated Hepatocarcinogenesis. Cancers 2016, 8, 76. [Google Scholar] [CrossRef]
- Bhala, N.; Angulo, P.; Van Der Poorten, D.; Lee, E.; Hui, J.; Saracco, G.M.; Adams, L.A.; Charatcharoenwitthaya, P.; Topping, J.H.; Bugianesi, E.; et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: An international collaborative study. Hepatology 2011, 54, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Song, K.; Srivastava, R.; Dong, C.; Go, G.-W.; Li, N.; Iwakiri, Y.; Mani, A. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 2015, 29, 3436–3445. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-P.; Li, H.-J.; Nie, J.; Chen, X.; Zhou, X. Mutation of miR-21 targets endogenous lipoprotein receptor-related protein 6 and nonalcoholic fatty liver disease. Am. J. Transl. Res. 2017, 9, 715–721. [Google Scholar] [PubMed]
- Bacle, A.; Kadri, L.; Khoury, S.; Ferru-Clément, R.; Faivre, J.-F.; Cognard, C.; Bescond, J.; Krzesiak, A.; Contzler, H.; Delpech, N.; et al. A comprehensive study of phospholipid fatty acid rearrangements in the metabolic syndrome: Correlations to organ dysfunction. Dis. Model. Mech. 2020, 13, dmm043927. [Google Scholar] [CrossRef] [Green Version]
- Imamura, F.; Sharp, S.J.; Koulman, A.; Schulze, M.B.; Kröger, J.; Griffin, J.L.; Huerta, J.M.; Guevara, M.; Sluijs, I.; Agudo, A.; et al. A combination of plasma phospholipid fatty acids and its association with incidence of type 2 diabetes: The EPIC-InterAct case-cohort study. PLoS Med. 2017, 14, e1002409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Arendt, B.M.; Hillyer, L.M.; Fung, S.K.; McGilvray, I.; Guindi, M.; Allard, J.P. Plasma phospholipids and fatty acid composition differ between liver biopsy-proven nonalcoholic fatty liver disease and healthy subjects. Nutr. Diabetes 2016, 6, e220. [Google Scholar] [CrossRef] [Green Version]
- Perona, J.S. Membrane lipid alterations in the metabolic syndrome and the role of dietary oils. Biochim. et Biophys. Acta (BBA) Biomembr. 2017, 1859, 1690–1703. [Google Scholar] [CrossRef]
- Imran, M.; Sergent, O.; Tête, A.; Gallais, I.; Chevanne, M.; Lagadic-Gossmann, D.; Podechard, N. Membrane Remodeling as a Key Player of the Hepatotoxicity Induced by Co-Exposure to Benzo[a]pyrene and Ethanol of Obese Zebrafish Larvae. Biomolecules 2018, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Karunakaran, U.; Suma, E.; Chung, S.M.; Won, K.C. The Role of CD36 in Type 2 Diabetes Mellitus: β-Cell Dysfunction and Beyond. Diabetes Metab. J. 2020, 44, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Miura, K. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Dattaroy, D.; Seth, R.; Das, S.; Alhasson, F.; Chandrashekaran, V.; Michelotti, G.; Fan, D.; Nagarkatti, M.; Nagarkatti, P.; Diehl, A.M.; et al. Sparstolonin B attenuates early liver inflammation in experimental NASH by modulating TLR4 trafficking in lipid rafts via NADPH oxidase activation. Am. J. Physiol. Liver Physiol. 2016, 310, G510–G525. [Google Scholar] [CrossRef] [Green Version]
- Di Liddo, R.; Bertalot, T.; Schuster, A.; Schrenk, S.; Tasso, A.; Zanusso, I.; Conconi, M.T.; Schäfer, K.H. Anti-inflammatory activity of Wnt signaling in enteric nervous system: In vitro preliminary evidences in rat primary cultures. J. Neuroinflammation 2015, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, F.; Li, S.; Cai, J.; Shi, J.; Jiang, Y. TLR4 Influences Hepatitis B Virus Related Hepatocellular Carcinoma by Regulating the Wnt/β-Catenin Pathway. Cell. Physiol. Biochem. 2017, 42, 469–479. [Google Scholar] [CrossRef]
- Kim, M.-H.; Lee, M.-K. The Incretins and Pancreatic β-Cells: Use of Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Polypeptide to Cure Type 2 Diabetes Mellitus. Korean Diabetes J. 2010, 34, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Yokomori, H.; Ando, W. Spatial expression of glucagon-like peptide 1 receptor and caveolin-1 in hepatocytes with macrovesicular steatosis in non-alcoholic steatohepatitis. BMJ Open Gastroenterol. 2020, 7, e000370. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. WNT signalling is both an inducer and effector of glucagon-like peptide. Diabetologia 2008, 51, 1768–1770. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Luo, R.; Li, J.; Wang, D.; Zhang, Y.; Liu, L.; Zhang, N.; Xu, X.; Lu, B.; Zhao, K. β-catenin promotes NLRP3 inflammasome activation via increasing the association between NLRP3 and ASC. Mol. Immunol. 2020, 121, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, X.; Yuan, X.; Hong, J.; Bhat, O.; Li, G.; Li, P.-L.; Guo, J. NLRP3 Inflammasome Formation and Activation in Nonalcoholic Steatohepatitis: Therapeutic Target for Antimetabolic Syndrome Remedy FTZ. Oxidative Med. Cell. Longev. 2018, 2018, 2901871. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2015, 27, 84–95. [Google Scholar] [CrossRef]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic Free Cholesterol Accumulates in Obese, Diabetic Mice and Causes Nonalcoholic Steatohepatitis. Gastroenterology 2011, 141, 1393–1403.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.-M.; Ho, S.-L.; Jeng, Y.-M.; Lai, Y.-S.; Chen, Y.-H.; Lu, S.-C.; Chen, H.-L.; Chang, P.-Y.; Hu, R.-H.; Lee, P.-H. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J. Inflamm. 2019, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Kang, Q.; Chen, A. Curcumin eliminates oxidized LDL roles in activating hepatic stellate cells by suppressing gene expression of lectin-like oxidized LDL receptor. Lab. Investig. 2009, 89, 1275–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.C.; Vossio, S.; Vacca, F.; Snijder, B.; Larios, J.; Schaad, O.; Guex, N.; Kuznetsov, D.; Martin, O.; Chambon, M.; et al. Wnt directs the endosomal flux of LDL -derived cholesterol and lipid droplet homeostasis. EMBO Rep. 2015, 16, 741–752. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karabicici, M.; Azbazdar, Y.; Iscan, E.; Ozhan, G. Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases. Membranes 2021, 11, 844. https://doi.org/10.3390/membranes11110844
Karabicici M, Azbazdar Y, Iscan E, Ozhan G. Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases. Membranes. 2021; 11(11):844. https://doi.org/10.3390/membranes11110844
Chicago/Turabian StyleKarabicici, Mustafa, Yagmur Azbazdar, Evin Iscan, and Gunes Ozhan. 2021. "Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases" Membranes 11, no. 11: 844. https://doi.org/10.3390/membranes11110844
APA StyleKarabicici, M., Azbazdar, Y., Iscan, E., & Ozhan, G. (2021). Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases. Membranes, 11(11), 844. https://doi.org/10.3390/membranes11110844