
Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
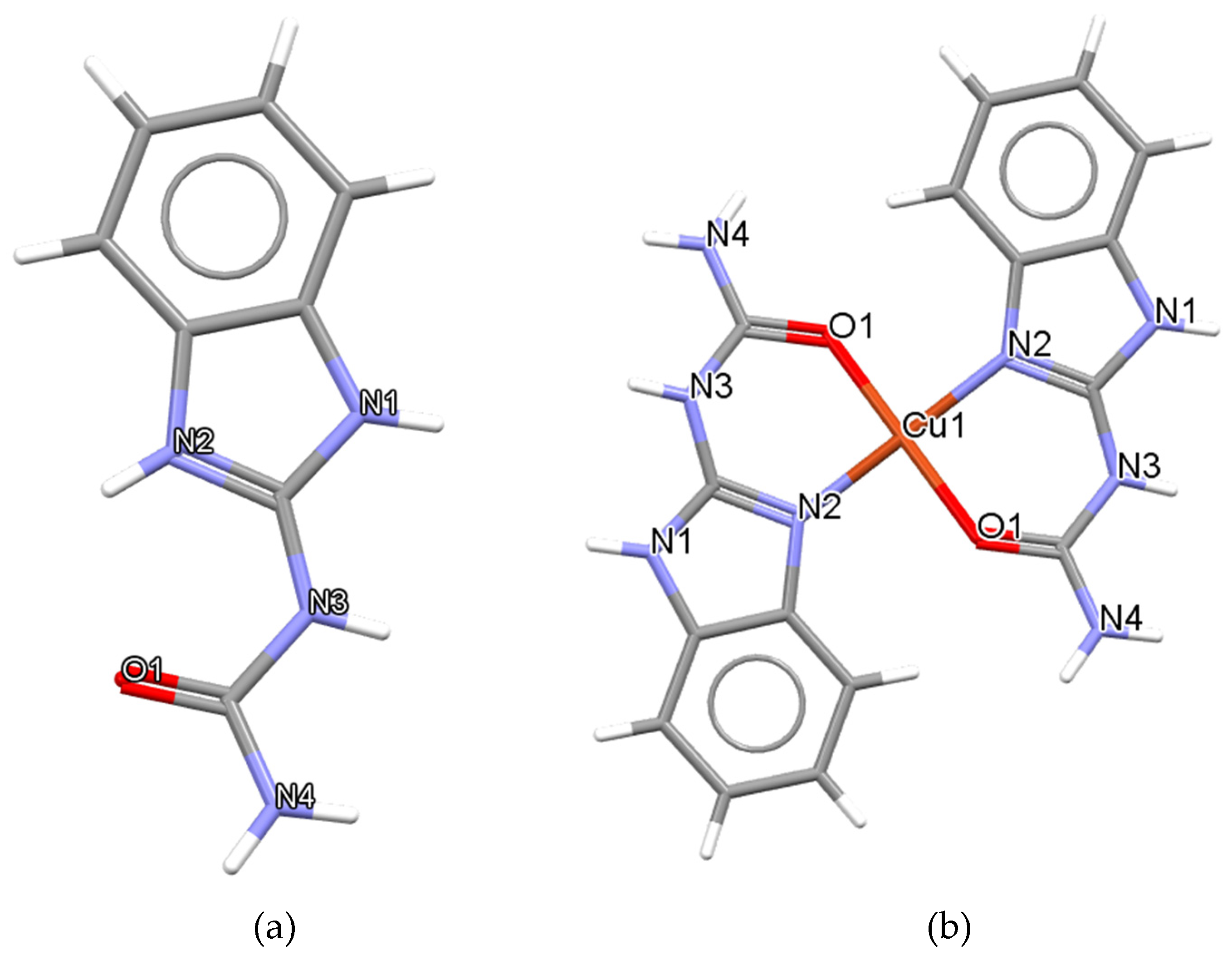
2. Computational Methods
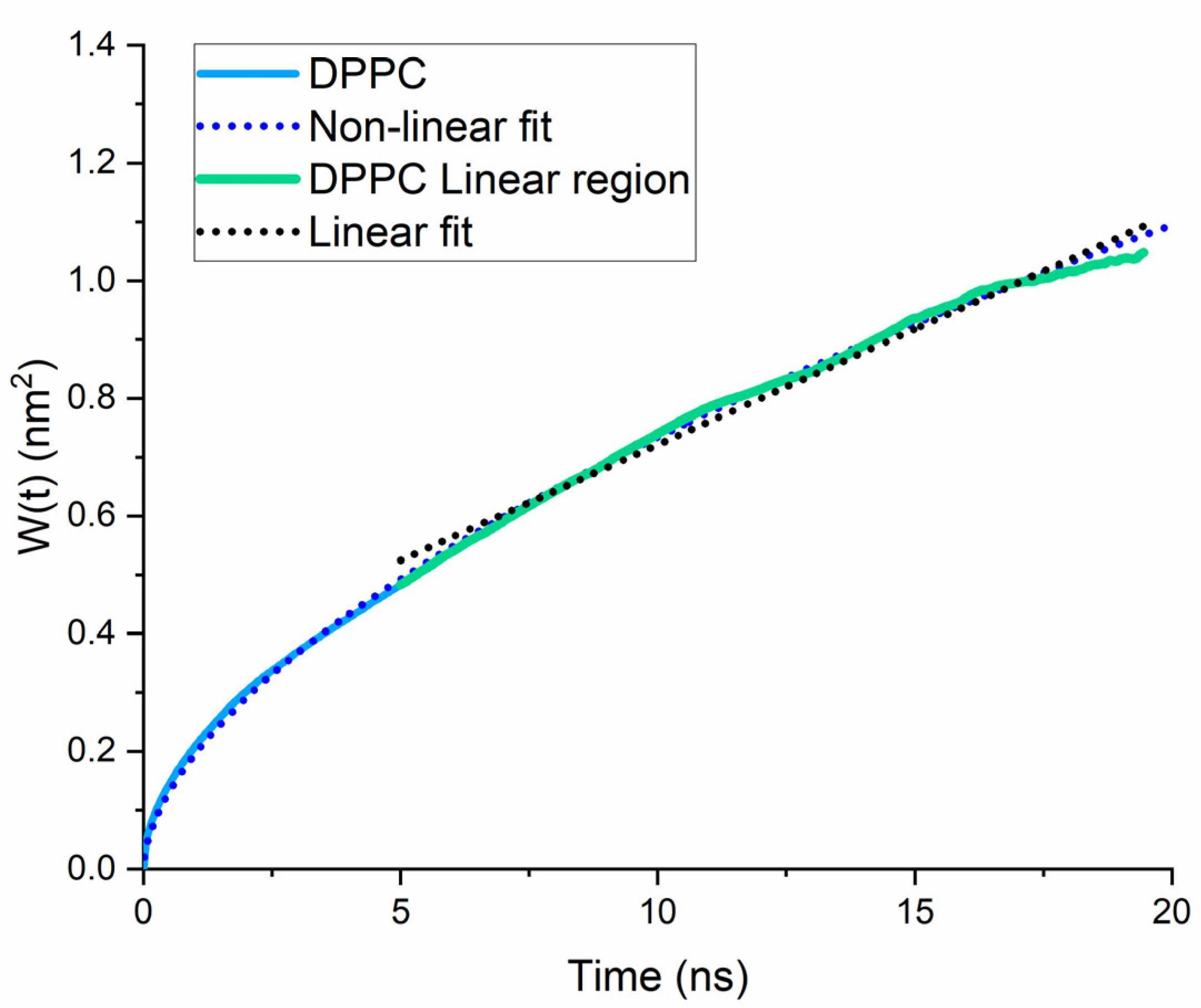
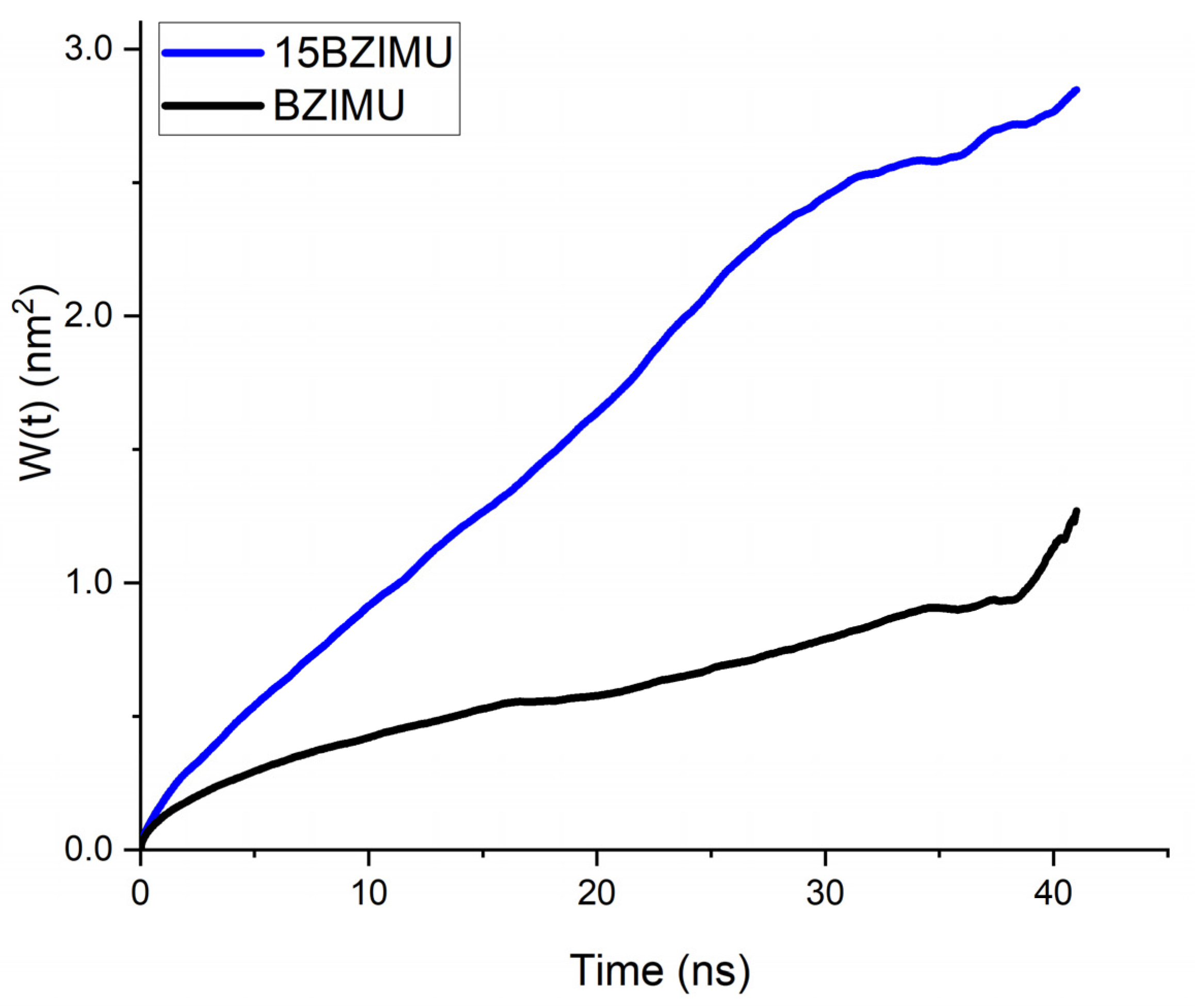
2.1. Lateral Diffusion Coefficient
2.2. Umbrella Sampling
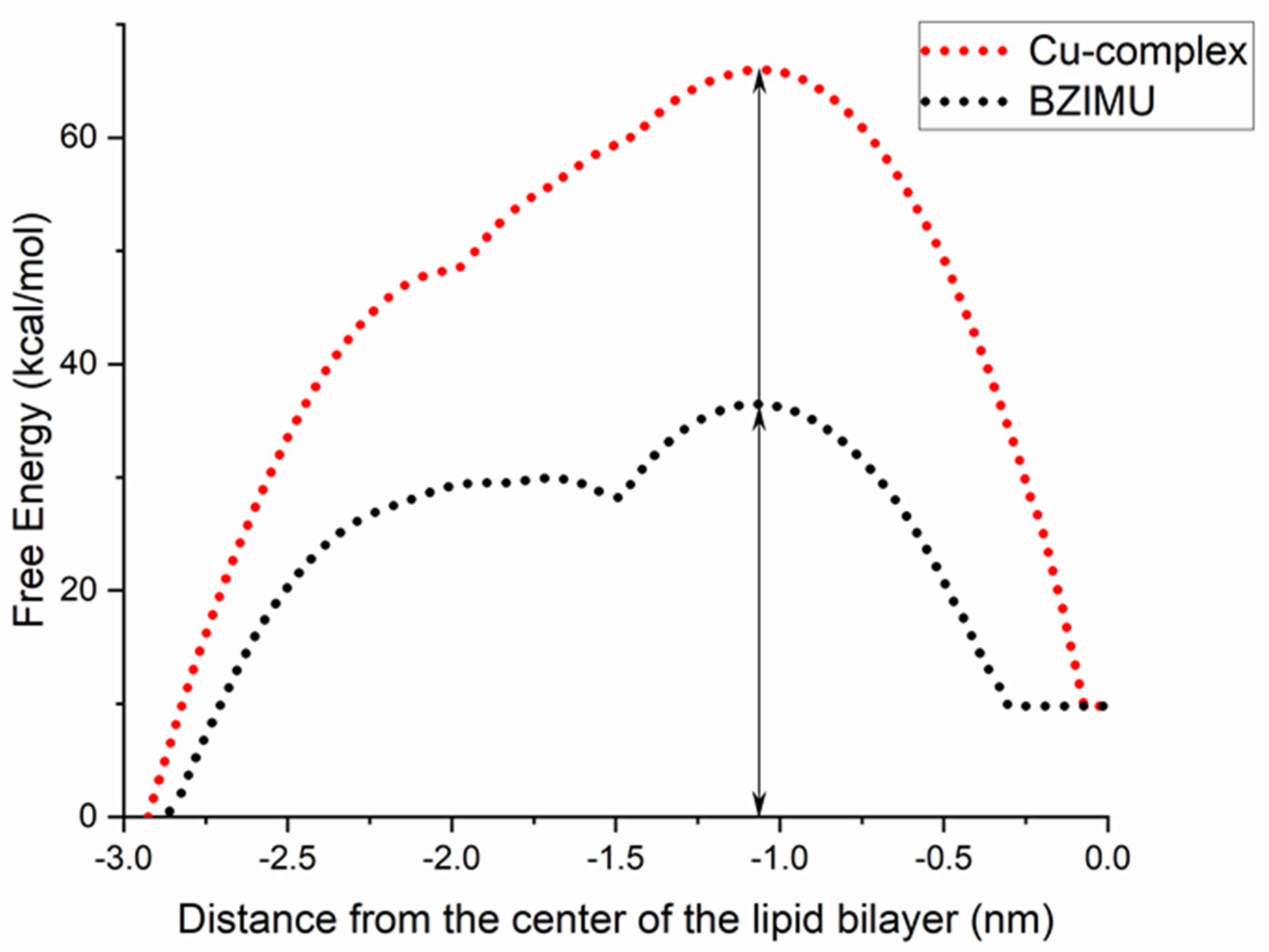
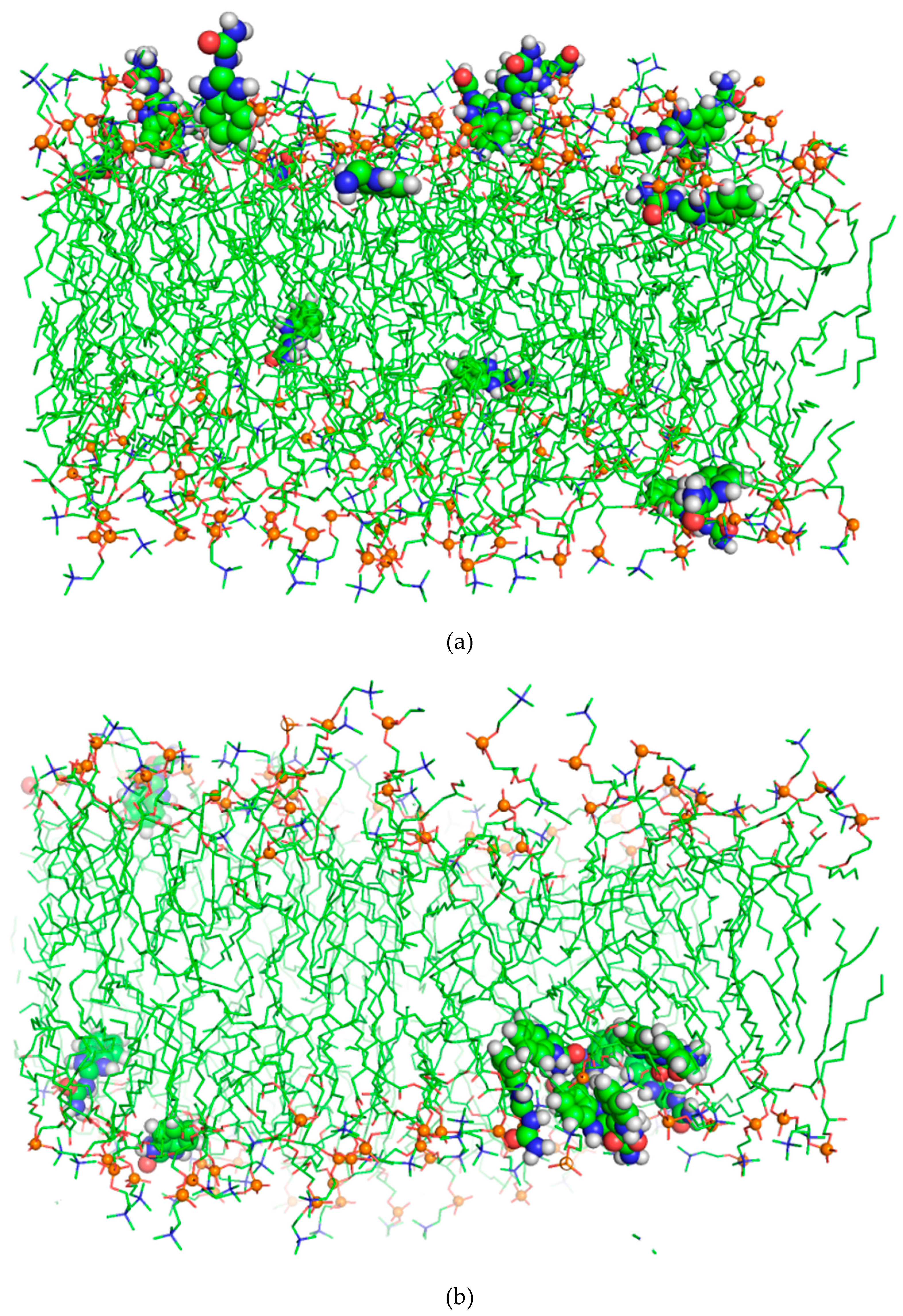
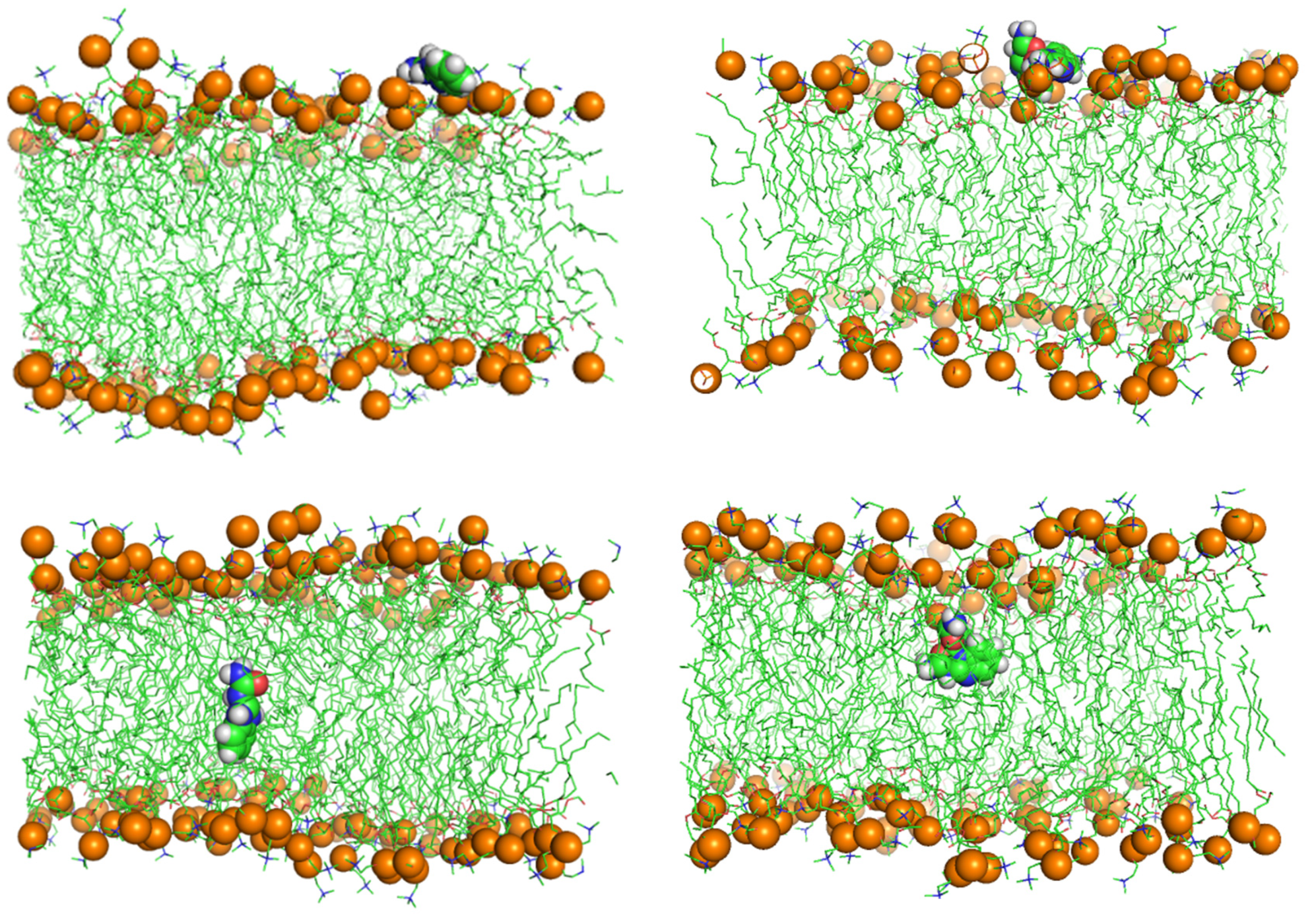
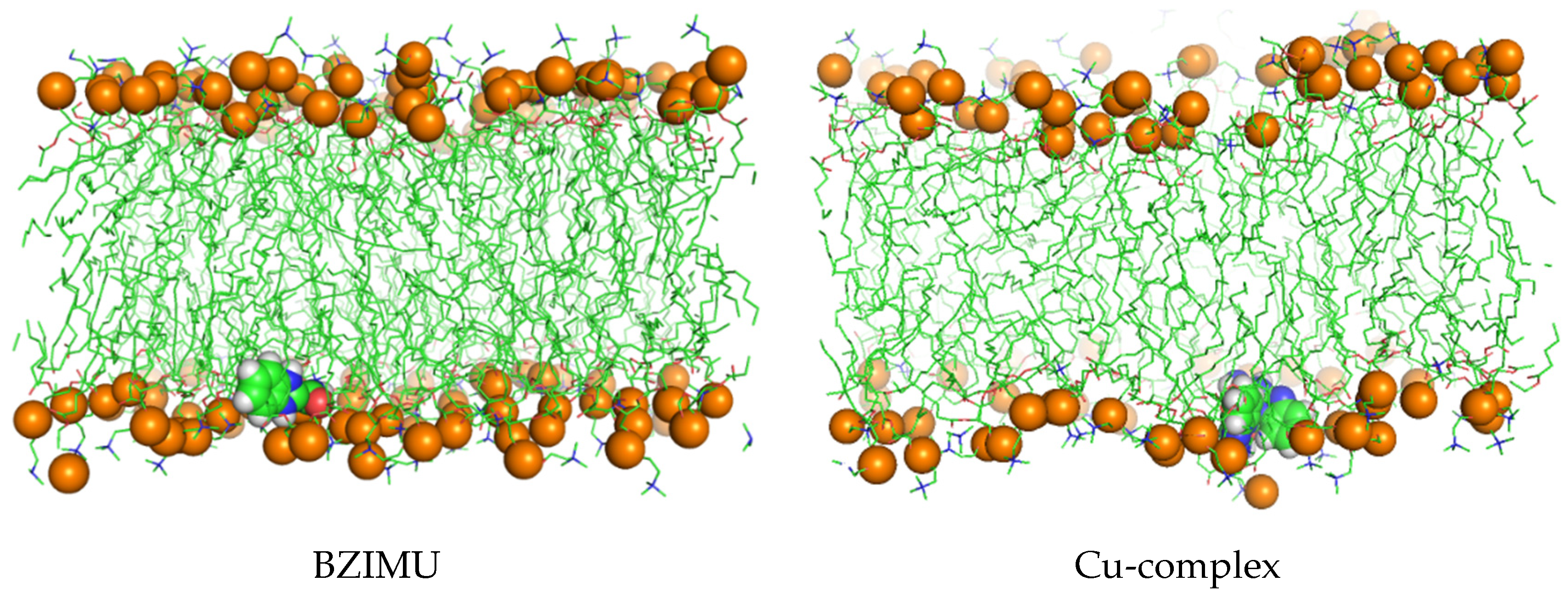
3. Results and Discussion
BZIMU Cluster Formation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Argüello, J.M.; Raimunda, D.; González-Guerrero, M. Metal Transport across Biomembranes: Emerging Models for a Distinct Chemistry. J. Biol. Chem. 2012, 287, 13510–13517. [Google Scholar] [CrossRef] [Green Version]
- Zehra, S.; Tabassum, S.; Arjmand, F. Biochemical Pathways of Copper Complexes: Progress over the Past 5 Years. Drug Discov. Today 2021, 26, 1086–1096. [Google Scholar] [CrossRef]
- Festa, R.A.; Thiele, D.J. Copper: An Essential Metal in Biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef] [Green Version]
- Balsano, C.; Porcu, C.; Sideri, S. Is Copper a New Target to Counteract the Progression of Chronic Diseases? Metallomics 2018, 10, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Li, M.; Guo, Z.; Jin, C.; Zhang, F.; Ou, C.; Xie, Y.; Tan, S.; Wang, Z.; Zheng, S.; et al. TPP-Related Mitochondrial Targeting Copper (II) Complex Induces P53-Dependent Apoptosis in Hepatoma Cells through ROS-Mediated Activation of Drp1. Cell Commun. Signal. 2019, 17, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, G.; Ganguly, S. Structure Activity Relationship (SAR) Study of Benzimidazole Scaffold for Different Biological Activities: A Mini-Review. Eur. J. Med. Chem. 2015, 97, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.L.; Cassano, R.; Costanzo, P.; Herrera Cano, N.; Maiuolo, L.; Nardi, M.; Nicoletta, F.P.; Oliverio, M.; Procopio, A. Green Synthesis of Privileged Benzimidazole Scaffolds Using Active Deep Eutectic Solvent. Molecules 2019, 24, 2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyraz, M.; Sari, M.; Banti, C.N.; Hadjikakou, S.K. Synthesis, Characterization and Biological Activities of Copper(II) Complex of 2-Benzimidazolyl-Urea and the Nitrate Salt of 2-Benzimidazolyl-Urea. J. Mol. Struct. 2017, 1146, 809–813. [Google Scholar] [CrossRef]
- Banti, C.N.; Poyraz, M.; Sainis, I.; Sari, M.; Rossos, G.; Kourkoumelis, N.; Hadjikakou, S.K. The Periodic Table of Urea Derivative: Small Molecules of Zinc(II) and Nickel(II) of Diverse Antimicrobial and Antiproliferative Applications. Mol. Divers. 2020, 24, 31–43. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Ragab, F.A.F.; Ramla, M.M.; El Diwani, H.I. Novel Benzimidazole–Pyrimidine Conjugates as Potent Antitumor Agents. Eur. J. Med. Chem. 2010, 45, 2336–2344. [Google Scholar] [CrossRef]
- Wang, W.; Kong, D.; Cheng, H.; Tan, L.; Zhang, Z.; Zhuang, X.; Long, H.; Zhou, Y.; Xu, Y.; Yang, X.; et al. New Benzimidazole-2-Urea Derivates as Tubulin Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4250–4253. [Google Scholar] [CrossRef]
- Perković, I.; Antunović, M.; Marijanović, I.; Pavić, K.; Ester, K.; Kralj, M.; Vlainić, J.; Kosalec, I.; Schols, D.; Hadjipavlou-Litina, D.; et al. Novel Urea and Bis-Urea Primaquine Derivatives with Hydroxyphenyl or Halogenphenyl Substituents: Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2016, 124, 622–636. [Google Scholar] [CrossRef]
- Hasegawa, M.; Nishigaki, N.; Washio, Y.; Kano, K.; Harris, P.A.; Sato, H.; Mori, I.; West, R.I.; Shibahara, M.; Toyoda, H.; et al. Discovery of Novel Benzimidazoles as Potent Inhibitors of TIE-2 and VEGFR-2 Tyrosine Kinase Receptors. J. Med. Chem. 2007, 50, 4453–4470. [Google Scholar] [CrossRef] [PubMed]
- Puckett, C.A.; Ernst, R.J.; Barton, J.K. Exploring the Cellular Accumulation of Metal Complexes. Dalton Trans. 2010, 39, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Arnesano, F.; Losacco, M.; Natile, G. An Updated View of Cisplatin Transport. Eur. J. Inorg. Chem. 2013, 2013, 2701–2711. [Google Scholar] [CrossRef]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving Cellular Uptake of Therapeutic Entities through Interaction with Components of Cell Membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyraz, M.; Berber, H.; Banti, C.N.; Kourkoumelis, N.; Manos, M.J.; Hadjikakou, S.K. Synthesis Characterization and Biological Activity of Mixed Ligand Silver(I) Complex of 2-Benzimidazolylurea and Triphenylphosphine. Polyhedron 2017, 128, 95–103. [Google Scholar] [CrossRef]
- Bensikaddour, H.; Snoussi, K.; Lins, L.; Van Bambeke, F.; Tulkens, P.M.; Brasseur, R.; Goormaghtigh, E.; Mingeot-Leclercq, M.-P. Interactions of Ciprofloxacin with DPPC and DPPG: Fluorescence Anisotropy, ATR-FTIR and 31P NMR Spectroscopies and Conformational Analysis. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 2535–2543. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella Sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Orsi, M.; Essex, J.W. Permeability of Drugs and Hormones through a Lipid Bilayer: Insights from Dual-Resolution Molecular Dynamics. Soft Matter 2010, 6, 3797–3808. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr. Sect. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. G_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Stachura, S.S.; Malajczuk, C.J.; Kuprusevicius, E.; Mancera, R.L. Influence of Bilayer Size and Number in Multi-Bilayer DOPC Simulations at Full and Low Hydration. Langmuir 2019, 35, 2399–2411. [Google Scholar] [CrossRef]
- Kneller, G.R.; Baczynski, K.; Pasenkiewicz-Gierula, M. Communication: Consistent Picture of Lateral Subdiffusion in Lipid Bilayers: Molecular Dynamics Simulation and Exact Results. J. Chem. Phys. 2011, 135, 141105. [Google Scholar] [CrossRef] [Green Version]
- Nagle, J.F.; Zhang, R.; Tristram-Nagle, S.; Sun, W.; Petrache, H.I.; Suter, R.M. X-Ray Structure Determination of Fully Hydrated L Alpha Phase Dipalmitoylphosphatidylcholine Bilayers. Biophys. J. 1996, 70, 1419–1431. [Google Scholar] [CrossRef] [Green Version]
- Pinisetty, D.; Moldovan, D.; Devireddy, R. The Effect of Methanol on Lipid Bilayers: An Atomistic Investigation. Ann. Biomed. Eng. 2006, 34, 1442–1451. [Google Scholar] [CrossRef]
- Hofsäß, C.; Lindahl, E.; Edholm, O. Molecular Dynamics Simulations of Phospholipid Bilayers with Cholesterol. Biophys. J. 2003, 84, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.K.; Abrams, C.F. Dynamics of Lipids, Cholesterol, and Transmembrane α-Helices from Microsecond Molecular Dynamics Simulations. J. Phys. Chem. B 2014, 118, 13590–13600. [Google Scholar] [CrossRef] [Green Version]
- Nademi, Y.; Amjad Iranagh, S.; Yousefpour, A.; Mousavi, S.Z.; Modarress, H. Molecular Dynamics Simulations and Free Energy Profile of Paracetamol in DPPC and DMPC Lipid Bilayers. J. Chem. Sci. 2014, 126, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.; Zhou, G.; Wang, Y.; Xu, C.; Wang, X. Molecular Dynamics Simulations of the Permeation of Bisphenol A and Pore Formation in a Lipid Membrane. Sci. Rep. 2016, 6, 33399. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Lv, M.; Xiu, P.; Huynh, T.; Zhang, M.; Castelli, M.; Liu, Z.; Huang, Q.; Fan, C.; Fang, H.; et al. Destructive Extraction of Phospholipids from Escherichia Coli Membranes by Graphene Nanosheets. Nat. Nanotechnol. 2013, 8, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Ruano, L.; Cárdenas, G.; Nogueira, J.J. The Permeation Mechanism of Cisplatin Through a Dioleoylphosphocholine Bilayer. Comput. Theor. Chem. 2021, 22, 1251–1261. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, W.; Jia, F.; Ye, J.; Zhao, Y.; Luo, Q.; Zhu, Z.; Wang, F. Cisplatin-Induced Alteration on Membrane Composition of A549 Cells Revealed by ToF-SIMS. Surf. Interface Anal. 2020, 52, 256–263. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Duszyk, M.; Tuszynski, J.A. Chemotherapy Drugs Form Ion Pores in Membranes Due to Physical Interactions with Lipids. Chem. Biol. Drug Des. 2012, 80, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Ashrafuzzaman, M.; Khan, Z.; Alqarni, A.; Alanazi, M.; Alam, M.S. Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes. Membranes 2021, 11, 501. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossos, G.; Hadjikakou, S.K.; Kourkoumelis, N. Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative. Membranes 2021, 11, 743. https://doi.org/10.3390/membranes11100743
Rossos G, Hadjikakou SK, Kourkoumelis N. Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative. Membranes. 2021; 11(10):743. https://doi.org/10.3390/membranes11100743
Chicago/Turabian StyleRossos, Georgios, Sotiris K. Hadjikakou, and Nikolaos Kourkoumelis. 2021. "Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative" Membranes 11, no. 10: 743. https://doi.org/10.3390/membranes11100743
APA StyleRossos, G., Hadjikakou, S. K., & Kourkoumelis, N. (2021). Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative. Membranes, 11(10), 743. https://doi.org/10.3390/membranes11100743