Application of Hollow Fibre-Liquid Phase Microextraction Technique for Isolation and Pre-Concentration of Pharmaceuticals in Water

 , , ,
, , ,
Abstract
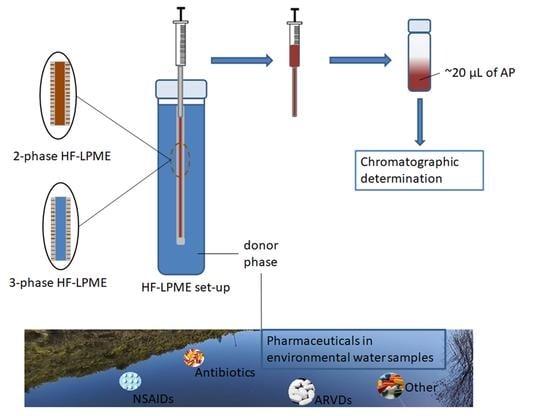
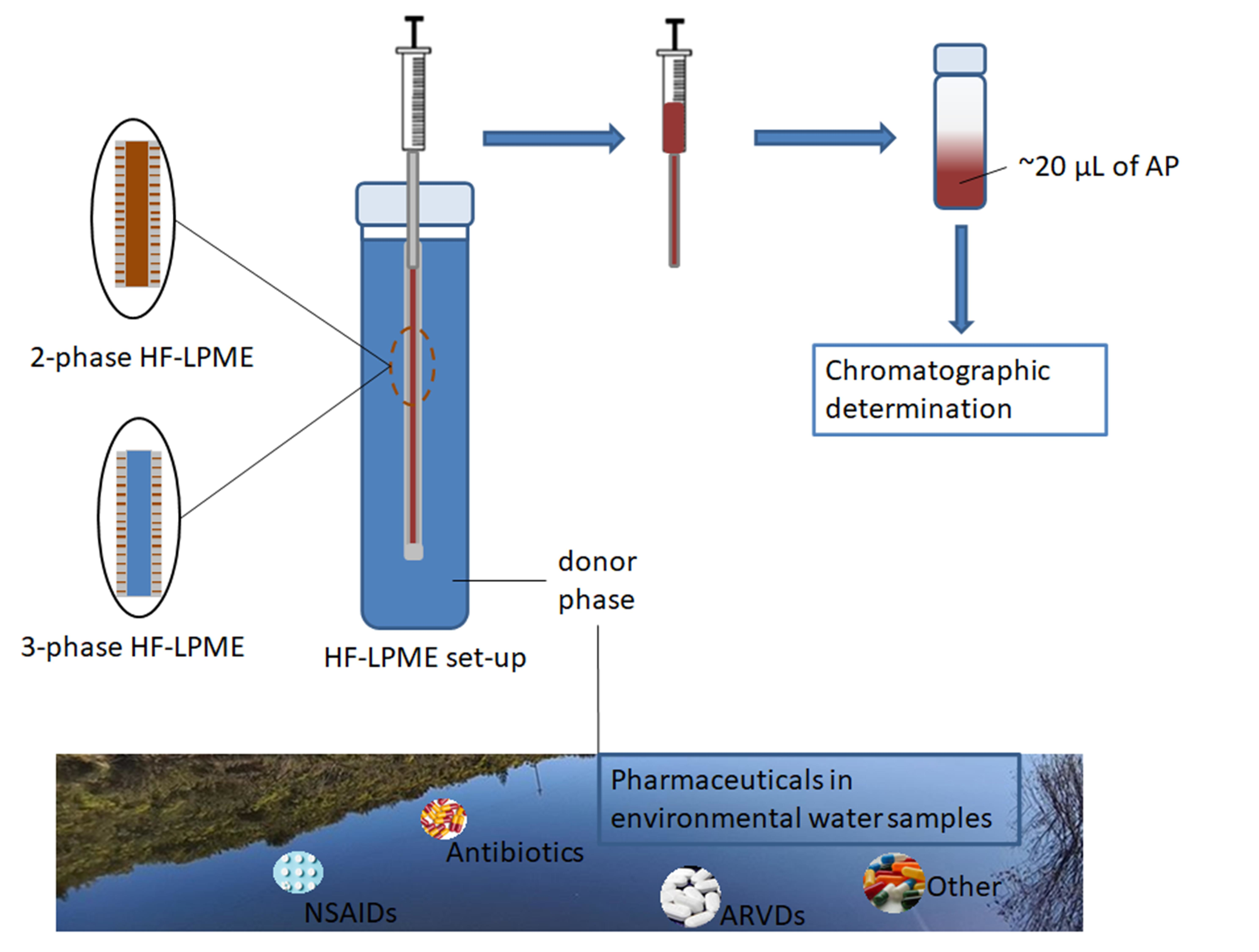
1. Introduction
2. Experimental Set-up, Modes, and Theoretical Principles
2.1. Modes, Principles, and Theory of HF-LPME
2.2. Pros and Cons
2.3. Carrier-Mediated HF-LPME
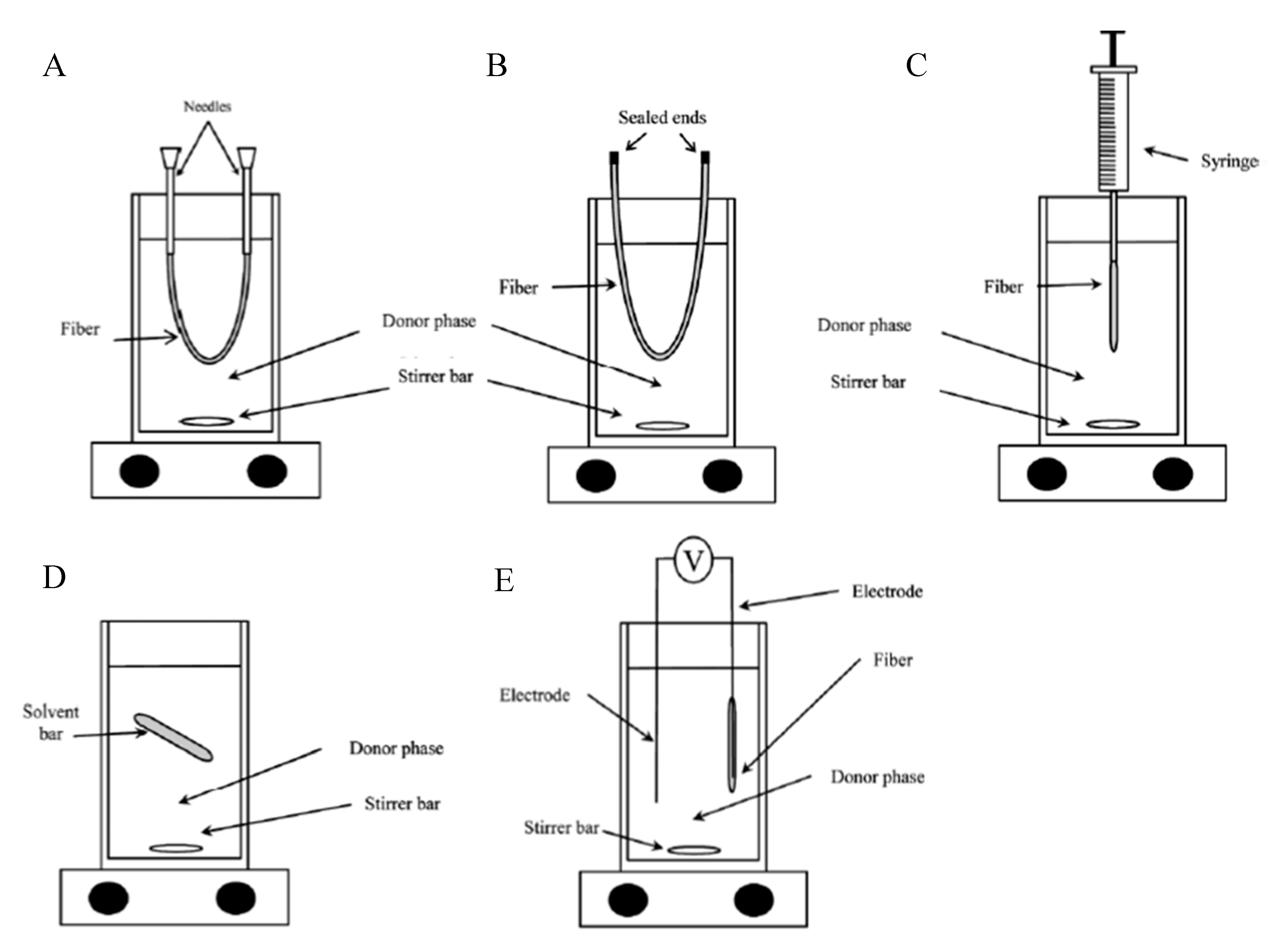
2.4. HF-LPME Experimental Set-up
3. Critical Parameters Affecting the Extraction Process
3.1. Supported Liquid Membrane
3.2. Sample and Acceptor Phase pH
3.3. Extraction Time
3.4. Stirring Rate
3.5. Temperature
3.6. Ionic Strength
3.7. Matrix Effects
4. Performance of HF-LPME in the Analysis of Pharmaceuticals in Water
5. Improvements of HF-LPME Based Methods for Pharmaceutical Analysis in Water
5.1. Advances in Supported Liquid Membrane
5.2. Application of Green Solvents in the Extraction Process
5.3. Inclusion of Solid Sorbents
5.4. Automated and Continuous Flow HF-LPME
6. Environmental Monitoring of Pharmaceuticals Using HF-LPME
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Sik, Y.; Kim, K.; Kwon, E.E.; Fai, Y. Occurrences and removal of pharmaceuticals and personal care products (PPCPs) in drinking water and water/sewage treatment plants: A review. Sci. Total Environ. 2017, 596–597, 303–320. [Google Scholar] [CrossRef]
- Nkoom, M.; Lu, G.; Liu, J. Occurrence and ecological risk assessment of pharmaceuticals and personal care products in Taihu Lake, China: A review. Environ. Sci. Process. Impacts 2018, 20, 1640–1648. [Google Scholar] [CrossRef]
- Madikizela, L.M.; Ncube, S.; Chimuka, L. Analysis, occurrence and removal of pharmaceuticals in African water resources: A current status. J. Environ. Manag. 2020, 253, 109741. [Google Scholar] [CrossRef]
- Patel, M.; Kumar, R.; Kishor, K.; Mlsna, T.; Pittman, C.U.; Mohan, D. Pharmaceuticals of emerging concern in aquatic systems: Chemistry, occurrence, effects, and removal methods. Chem. Rev. 2019, 119, 3510–3673. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, P.; Caroli, S.; Caracciolo, A.B. Pharmaceuticals as priority water contaminants. Toxicol. Environ. Chem. 2010, 92, 549–565. [Google Scholar] [CrossRef]
- Soliman, M.A.; Pedersen, J.A.; Suffet, I.H. Rapid gas chromatography-mass spectrometry screening method for human pharmaceuticals, hormones, antioxidants and plasticizers in water. J. Chromatogr. A 2004, 1029, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fernandez, V.; Rocca, L.M.; Tomai, P.; Salvatore, F.; Gentili, A. Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry. Anal. Chim. Acta 2017, 983, 9–41. [Google Scholar] [CrossRef] [PubMed]
- Mahugo-Santana, C.; Sosa-Ferrera, Z.; Torres-Padrón, M.E.; Santana-Rodríguez, J.J. Application of new approaches to liquid-phase microextraction for the determination of emerging pollutants. TrAC Trends Anal. Chem. 2011, 30, 731–748. [Google Scholar] [CrossRef]
- Larsson, E.; Al-Hamimi, S.; Jönsson, J.Å. Behaviour of nonsteroidal anti-inflammatory drugs and eight of their metabolites during wastewater treatment studied by hollow fibre liquid phase microextraction and liquid chromatography mass spectrometry. Sci. Total Environ. 2014, 485–486, 300–308. [Google Scholar] [CrossRef]
- Sagristà, E.; Larsson, E.; Ezoddin, M.; Hidalgo, M.; Salvadó, V.; Jonsson, J.Å. Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography—mass spectrometry. J. Chromatogr. A 2010, 1217, 6153–6158. [Google Scholar] [CrossRef]
- Quintana, J.B.; Rodil, R.; Reemtsma, T. Suitability of hollow fibre liquid-phase microextraction for the determination of acidic pharmaceuticals in wastewater by liquid chromatography – electrospray tandem mass spectrometry without matrix effects. J. Chromatogr. A 2004, 1061, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bozorgzadeh, E.; Shariati, S.; Esmaeilnejad, A. Central Composite Design for Optimizing Hollow Fiber Liquid Phase Microextraction of Carbamazepine from Aqueous and Biological Samples. J. Anal. Chem. 2020, 75, 154–160. [Google Scholar] [CrossRef]
- Li, Q.; Jing, S.; Zhang, J.; Zhang, L.; Ran, C.; Du, C.; Jiang, Y. Hollow fiber-protected liquid-phase microextraction followed by high performance liquid chromatography for simultaneously screening multiple trace level β-blockers in environmental water samples. Anal. Methods 2015, 7, 6251–6259. [Google Scholar] [CrossRef]
- Zhang, H.; Du, Z.; Ji, Y.; Mei, M. Simultaneous trace determination of acidic non-steroidal anti-inflammatory drugs in purified water, tap water, juice, soda and energy drink by hollow fiber-based liquid-phase microextraction and ultra-high pressure liquid chromatography coupled to tandem. Talanta 2013, 109, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhang, Z.; Shao, X.; Yao, C.; Wu, X.; Yang, L.; Zhu, J.; Zhang, D. Hollow- fiber-supported liquid-phase microextraction using an ionic liquid as the extractant for the pre-concentration of bisphenol A, 17-β-estradiol, estrone and diethylstilbestrol from water samples with HPLC detection. Water Sci. Technol. 2014, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Mlunguza, N.Y.; Ncube, S.; Mahlambi, N.P.; Chimuka, L.; Madikizela, L.M. Determination of selected antiretroviral drugs in wastewater, surface water and aquatic plants using hollow fibre liquid phase microextraction and liquid chromatography - tandem mass spectrometry. J. Hazard. Mater. 2020, 382, 121067. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Liquid-phase microextraction and capillary electrophoresis of acidic drugs. Electrophoresis 2000, 21, 579–585. [Google Scholar] [CrossRef]
- Vasskog, T.; Anderssen, T.; Pedersen-bjergaard, S.; Kallenborn, R.; Jensen, E. Occurrence of selective serotonin reuptake inhibitors in sewage and receiving waters at Spitsbergen and in Norway. J. Chromatogr. A 2008, 1185, 194–205. [Google Scholar] [CrossRef]
- Tajik, M.; Yamini, Y.; Esrafili, A.; Ebrahimpour, B. Automated hollow fiber microextraction based on two immiscible organic solvents for extraction two hormonal drugs. J. Pharm. Biomed. Anal. 2014. [Google Scholar] [CrossRef]
- Pedersen-bjergaard, S.; Rasmussen, K.E. Liquid-liquid-liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal. Chem. 1999, 71, 2650–2656. [Google Scholar] [CrossRef]
- Bello-lópez, M.Á.; Ramos-payán, M.; Antonio, J.; Fernández-torres, R.; Callejón, M. Analytical Applications of Hollow Fiber Liquid Phase Microextraction (HF-LPME): A Review. Anal. Lett. 2012, 45, 804–830. [Google Scholar] [CrossRef]
- Han, D.; Row, K.H. Trends in liquid-phase microextraction, and its application to environmental and biological samples. Microchim. Acta 2012, 176, 1–22. [Google Scholar] [CrossRef]
- Khan, W.A.; Arain Balal, M.; Yamini, Y.; Shah, N.; Gul, T.; Pedersen-bjergaard, S.; Tajik, M. Hollow fiber-based liquid phase microextraction followed by analytical instrumental techniques for quantitative analysis of heavy metal ions and pharmaceuticals. J. Pharm. Anal. 2020, 10, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Sarafraz-yazdi, A.; Amiri, A. Liquid-phase microextraction. Trends Anal. Chem. 2010, 29, 1–14. [Google Scholar] [CrossRef]
- Pedersen-bjergaard, S.; Rasmussen, K.E. Liquid-phase microextraction with porous hollow fibers, a miniaturized and highly flexible format for liquid–liquid extraction. J. Chromatogr. A 2008, 1184, 132–142. [Google Scholar] [CrossRef]
- Gjelstad, A.; Yamini, Y.; Esrafili, A. Three-phase hollow fi ber liquid-phase microextraction and parallel artificial liquid membrane extraction. Trends Anal. Chem. 2019, 113, 25–31. [Google Scholar] [CrossRef]
- Ghambarian, M.; Yamini, Y.; Esrafili, A. Developments in hollow fiber based liquid-phase microextraction: Principles and applications. Microchim. Acta 2012, 177, 271–294. [Google Scholar] [CrossRef]
- Chimuka, L.; Msagati, T.A.M.; Cukrowska, E.; Tutu, H. Critical parameters in a supported liquid membrane extraction technique for ionizable organic compounds with a stagnant acceptor phase. J. Chromatogr. A 2010, 1217, 2318–2325. [Google Scholar] [CrossRef]
- Jalili, V.; Barkhordari, A.; Ghiasvand, A. New extraction media in microextraction techniques. A review of reviews. Microchem. J. 2020, 153, 104386. [Google Scholar] [CrossRef]
- Kokosa, J.M. Selecting an extraction solvent for a greener liquid phase microextraction (LPME) mode-based analytical method. Trends Anal. Chem. 2019, 118, 238–247. [Google Scholar] [CrossRef]
- Rutkowska, M.; Płotka-wasylka, J.; Sajid, M.; Andruch, V. Liquid – phase microextraction: A review of reviews. Microchem. J. 2019, 149, 103989. [Google Scholar] [CrossRef]
- Yamini, Y.; Rezazadeh, M.; Seidi, S. Liquid-phase microextraction - The different principles and configurations. Trends Anal. Chem. 2019, 112, 264–272. [Google Scholar] [CrossRef]
- Carasek, E.; Morés, L.; Merib, J. Basic principles, recent trends and future directions of microextraction techniques for the analysis of aqueous environmental samples. Trends Environ. Anal. Chem. 2018, 19, e00060. [Google Scholar] [CrossRef]
- Zorita, S.; Mårtensson, L.; Mathiasson, L. Occurrence and removal of pharmaceuticals in a municipal sewage treatment system in the south of Sweden. Sci. Total Environ. 2009, 407, 2760–2770. [Google Scholar] [CrossRef]
- Chimuka, L.; Cukrowska, E.; Michel, M.; Buszewski, B. Advances in sample preparation using membrane-based liquid-phase microextraction techniques. TrAC Trends Anal. Chem. 2011, 30, 1781–1792. [Google Scholar] [CrossRef]
- Esrafili, A.; Baharfar, M.; Tajik, M.; Yamini, Y. Two-phase hollow fiber liquid-phase microextraction. Trends Anal. Chem. 2018, 108, 314–322. [Google Scholar] [CrossRef]
- Poliwoda, A.; Krzyzak, M.; Wieczorek, P.P. Supported liquid membrane extraction with single hollow fiber for the analysis of fluoroquinolones from environmental surface water samples. J. Chromatogr. A 2010, 1217, 3590–3597. [Google Scholar] [CrossRef]
- Wen, X.; Tu, C.; Lee, H.K. Two-step liquid-liquid-liquid microextraction of nonsteroidal antiinflammatory drugs in wastewater. Anal. Chem. 2004, 76, 228–232. [Google Scholar] [CrossRef]
- Mlunguza, N.Y.; Ncube, S.; Mahlambi, P.N.; Luke, C.; Madikizela, L.M. Optimization and application of hollow fiber liquid-phase microextraction and microwave-assisted extraction for the analysis of non-steroidal anti-inflammatory drugs in aqueous and plant samples. Environ. Monit. Assess. 2020, 192, 557. [Google Scholar] [CrossRef]
- Cui, S.; Ouyang, G.; Duan, G.; Hou, J.; Luan, T.; Zhang, X. The mass transfer dynamics of hollow fiber liquid-phase microextraction and its application for rapid analysis of biological samples. J. Chromatogr. A 2012, 1266, 10–16. [Google Scholar] [CrossRef]
- Ho, T.S.; Vasskog, T.; Anderssen, T.; Jensen, E.; Einar, K.; Pedersen-bjergaard, S. 25,000-fold pre-concentration in a single step with liquid-phase microextraction. Anal. Chim. Acta 2007, 592, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Alexovi, M.; Horstkotte, B.; Solich, P.; Sabo, J. Automation of static and dynamic non-dispersive liquid phase microextraction. Part 2: Approaches based on impregnated membranes and porous supports. Anal. Chim. Acta 2016, 907, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Seidi, S.; Rezazadeh, M.; Yamini, Y. Pharmaceutical applications of liquid-phase microextraction. Trends Anal. Chem. 2018, 108, 296–305. [Google Scholar] [CrossRef]
- Shariati, S.; Yamini, Y.; Esrafili, A. Carrier mediated hollow fiber liquid phase microextraction combined with HPLC—UV for preconcentration and determination of some tetracycline antibiotics. J. Chromatogr. B 2009, 877, 393–400. [Google Scholar] [CrossRef]
- De los Ríos, A.P.; Hernández-Fernández, F.J.; Tomás-Alonso, F.; Palacios, J.M.; Víllora, G. Stability studies of supported liquid membranes based on ionic liquids: Effect of surrounding phase nature. Desalination 2009, 245, 776–782. [Google Scholar] [CrossRef]
- Basheer, C.; Kamran, M.; Ashraf, M.; Kee, H. Enhancing liquid-phase microextraction efficiency through chemical reactions. Trends Anal. Chem. 2019, 118, 426–433. [Google Scholar] [CrossRef]
- Yamini, Y.; Reimann, C.T.; Vatanara, A.; Jonsson, J.A. Extraction and preconcentration of salbutamol and terbutaline from aqueous samples using hollow fiber supported liquid membrane containing anionic carrier. J. Chromatogr. A 2006, 1124, 57–67. [Google Scholar] [CrossRef]
- Carasek, E.; Merib, J. Membrane-based microextraction techniques in analytical chemistry: A review. Anal. Chim. Acta 2015, 880, 8–25. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Electrokinetic migration across artificial liquid membranes: New concept for rapid sample preparation of biological fluids. J. Chromatogr. A 2006, 1109, 183–190. [Google Scholar] [CrossRef]
- Asadi, M.; Dadfarnia, S.; Mohammad, A.; Shabani, H. Hollow fiber liquid phase microextraction method combined with high-performance liquid chromatography for simultaneous separation and determination of ultra-trace amounts of naproxen and nabumetone in cow milk, water, and biological samples. Food Anal. Methods 2016, 9, 2762–2772. [Google Scholar] [CrossRef]
- Cui, S.; Tan, S.; Ouyang, G.; Pawliszyn, J. Automated polyvinylidene difluoride hollow fiber liquid-phase microextraction of flunitrazepam in plasma and urine samples for gas chromatography/tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Lambropoulou, D.A.; Albanis, T.A. Application of hollow fiber liquid phase microextraction for the determination of insecticides in water. J. Chromatogr. A 2005, 1072, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Salvatierra-stamp, V.C.; Muñiz-valencia, R.; Jose, M.; Ceballos-magaña, S.G. Hollow fiber liquid phase microextraction combined with liquid chromatography-tandem mass spectrometry for the analysis of emerging contaminants in water samples. Microchem. J. 2018, 140, 87–95. [Google Scholar] [CrossRef]
- Es’haghi, Z. Determination of widely used non-steroidal anti-inflammatory drugs in water samples by in situ derivatization, continuous hollow fiber liquid-phase microextraction and gas chromatography-flame ionization detector. Anal. Chim. Acta 2009, 641, 83–88. [Google Scholar] [CrossRef]
- Payan, M.R.; Ángel, M.; López, B.; Fernández-torres, R.; Callejón, M.; Luis, J.; Ariza, G. Application of hollow fiber-based liquid-phase microextraction (HF-LPME) for the determination of acidic pharmaceuticals in wastewaters. Talanta 2010, 82, 854–858. [Google Scholar] [CrossRef]
- Payán, M.R.; Ángel, M.; López, B.; Fernández-torres, R.; Mochón, M.C.; Antonio, J. Hollow fiber-based liquid phase microextraction (HF-LPME) as a new approach for the HPLC determination of fluoroquinolones in biological and environmental matrices. J. Pharm. Biomed. Anal. 2011, 55, 332–341. [Google Scholar] [CrossRef]
- Tong, F.; Zhang, Y.; Chen, F.; Li, Y.; Ma, G.; Chen, Y.; Liu, K.; Dong, J.; Ye, J.; Chu, Q. Hollow-fiber liquid-phase microextraction combined with capillary electrophoresis for trace analysis of sulfonamide compounds. J. Chromatogr. B 2013, 942–943, 134–140. [Google Scholar] [CrossRef]
- Payán, M.R.; Ángel, M.; López, B.; Fernández-torres, R.; Navarro, M.V.; Mochón, M.C. Hollow fiber-based liquid phase microextraction (HF-LPME) for a highly sensitive HPLC determination of sulfonamides and their main metabolites. J. Chromatogr. B 2011, 879, 197–204. [Google Scholar] [CrossRef]
- Liu, M.; Qiu, B.; Jin, X.; Zhang, L.; Chen, G.; Chen, X. Determination of estrogens in wastewater using microextraction followed by HPLC. J. Sep. Sci. 2008, 31, 622–628. [Google Scholar] [CrossRef]
- Msagati, T.A.M.; Mamba, B.B. Development of supported liquid membrane techniques for the monitoring of trace levels of organic pollutants in wastewaters and water purification systems. Phys. Chem. Earth 2011, 36, 1167–1177. [Google Scholar] [CrossRef]
- Da Silva, G.S.; Lima, D.L.D.; Esteves, V.I. Salicylic acid determination in estuarine and riverine waters using hollow fiber liquid-phase microextraction and capillary zone electrophoresis. Environ. Sci. Pollut. Res. 2017, 24, 15748–15755. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.; Petersson, E.; Rylander, M.; Jonsson, J.A. Continuous flow hollow fiber liquid-phase microextraction and monitoring of NSAID pharmaceuticals in a sewage treatment plant effluent. Anal. Methods 2009, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ezoddin, M.; Jönsson, J.Å.; Kyani, A. Equilibrium sampling through membrane based on a hollow fiber for determination of naproxen and diclofenac in sludge slurry using Taguchi orthogonal array experimental design. Desalin. Water Treat. 2014, 52, 2472–2480. [Google Scholar] [CrossRef]
- Villar Navarro, M.; Ramos Payán, M.; Fernández-Torres, R.; Bello-López, M.A.; Callejón Mochón, M.; Guiráum Pérez, A. Capillary electrophoresis determination of nonsteroidal anti-inflammatory drugs in wastewater using hollow fiber liquid-phase microextraction. Electrophoresis 2011, 32, 2107–2113. [Google Scholar] [CrossRef]
- Payán, M.R.; Ángel, M.; López, B.; Fernández-torres, R.; Luis, J.; Bernal, P.; Mochón, M.C. HPLC determination of ibuprofen, diclofenac and salicylic acid using hollow fiber-based liquid phase microextraction (HF-LPME). Anal. Chim. Acta 2009, 653, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Larsson, E.; Yamini, Y.; Jonsson, J.Å. Hollow fiber liquid phase microextraction as a preconcentration and clean-up step after pressurized hot water extraction for the determination of non-steroidal anti-inflammatory drugs in sewage sludge. J. Chromatogr. A 2011, 1218, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Manso, J.; Larsson, E.; Jonsson, J.Å. Determination of 4-isobutylacetophenone and other transformation products of anti-inflammatory drugs in water and sludge from five wastewater treatment plants in Sweden by hollow fiber liquid phase microextraction and gas chromatography—mass spectrometr. Talanta 2014, 125, 87–93. [Google Scholar] [CrossRef]
- Yudthavorasit, S.; Chiaochan, C. Simultaneous determination of multi-class antibiotic residues in water using carrier-mediated hollow-fiber liquid-phase microextraction coupled with ultra-high performance liquid chromatography tandem mass spectrometry. Microchim. Acta 2011, 172, 39–49. [Google Scholar] [CrossRef]
- Yang, L.; Shi, Y.; Li, J.; Luan, T. In situ derivatization and hollow-fiber liquid-phase microextraction to determine sulfonamides in water using UHPLC with fluorescence detection. J. Seperation Sci. 2017, 1–38. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.; Namies´nik, J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Tao, Y.; Liu, J.; Hu, X.; Li, H.; Wang, T.; Jiang, G. Hollow fiber supported ionic liquid membrane microextraction for determination of sulfonamides in environmental water samples by high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 6259–6266. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lee, H.K. Orthogonal array designs for the optimization of liquid-liquid-liquid microextraction of nonsteroidal anti-inflammatory drugs combined with high-performance liquid chromatography-ultraviolet detection. J. Chromatogr. A 2005, 1092, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Möder, M.; Schrader, S.; Popp, P. Semi-automated hollow-fibre membrane extraction, a novel enrichment technique for the determination of biologically active compounds in water samples. J. Chromatogr. A 2003, 985, 99–106. [Google Scholar] [CrossRef]
- Zhang, C.; Ye, L.; Xu, L. Orthogonal array design for the optimization of hollow fiber protected liquid-phase microextraction of salicylates from environmental waters. Anal. Chim. Acta 2011, 689, 219–225. [Google Scholar] [CrossRef]
- Seidi, S.; Alavi, L.; Jabbari, A.; Shanehsaz, M. Three-phase carrier-mediated hollow fiber microextraction based on deep eutectic solvent followed by HPLC–UV for determination of raloxifene and ethinylestradiol in pharmaceutical wastewater treatment plants. J. Iran. Chem. Soc. 2019, 16, 1007–1018. [Google Scholar] [CrossRef]
- Rajabi, M.; Ghassab, N.; Hemmati, M.; Asghari, A. Highly effective and safe intermediate based on deep eutectic medium for carrier less-three phase hollow fiber microextraction of antiarrhythmic agents in complex matrices. J. Chromatogr. B 2019, 1104, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Dimpe, K.M.; Nomngongo, P.N. Current sample preparation methodologies for analysis of emerging pollutants in different environmental matrices. Trends Anal. Chem. 2016, 82, 199–207. [Google Scholar] [CrossRef]
- Ocana-Gonzalez, A.J.; Fernandez-Torres, R.; Bello-Lopez, M.A.; Ramos-Payan, M. New developments in microextraction techniques in bioanalysis. A review. Anal. Chim. Acta 2016, 905, 8–23. [Google Scholar] [CrossRef]
- Spietelun, A.; Marcinkowski, Ł.; De, M.; Namie, J. Green aspects, developments and perspectives of liquid phase microextraction techniques. Talanta 2014, 119, 34–45. [Google Scholar] [CrossRef]
- Tabani, H.; Nojavan, S.; Alexoviˇ, M. Recent developments in green membrane-based extraction techniques for pharmaceutical and biomedical analysis. J. Pharm. Biomed. Anal. 2018, 160, 244–267. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Liquid-phase microextraction techniques within the framework of green chemistry. TrAC Trends Anal. Chem. 2010, 29, 617–628. [Google Scholar] [CrossRef]
- Hao, Z.; Vilt, M.E.; Wang, Z.; Zhang, W.; Winston Ho, W.S. Supported liquid membranes with feed dispersion for recovery of Cephalexin. J. Memb. Sci. 2014, 468, 423–431. [Google Scholar] [CrossRef]
- Vilt, M.E.; Ho, W.S.W. In situ removal of Cephalexin by supported liquid membrane with strip dispersion. J. Memb. Sci. 2011, 367, 71–77. [Google Scholar] [CrossRef]
- Bhosale, V.K.; Chana, H.K.; Kamble, S.P.; Kulkarni, P.S. Separation of nitroaromatics from wastewater by using supported ionic liquid membranes. J. Water Process Eng. 2019, 32, 100925. [Google Scholar] [CrossRef]
- Wang, H.; Wu, W.; Wei, D.; Guo, Z.; Wang, S. Hollow fiber supported ionic liquid membrane microextraction for preconcentration of kanamycin sulfate with electrochemiluminescence detection. J. Electroanal. Chem. 2014, 735, 136–141. [Google Scholar] [CrossRef]
- Kamaz, M.; Vogler, R.J.; Jebur, M.; Sengupta, A.; Wickramasinghe, R. πElectron induced separation of organic compounds using supported ionic liquid membranes. Sep. Purif. Technol. 2020, 236, 116237. [Google Scholar] [CrossRef]
- Jiang, B.; Dou, H.; Zhang, L.; Wang, B.; Sun, Y.; Yang, H.; Huang, Z.; Bi, H. Novel supported liquid membranes based on deep eutectic solvents for olefin-paraffin separation via facilitated transport. J. Memb. Sci. 2017, 536, 123–132. [Google Scholar] [CrossRef]
- Zeng, L.; Liu, Q.; Van der Bruggen, B.; Tang, K.; Yi, X.; Wang, G. An integrated separation process for recovery and enantioseparation of amlodipine from wastewater: Supported liquid membrane-aqueous/organic phase crystallization. Sep. Purif. Technol. 2020, 248, 117–121. [Google Scholar] [CrossRef]
- Liu, W.; Wei, Z.; Zhang, Q.; Wu, F.; Lin, Z.; Lu, Q.; Lin, F.; Chen, G. Novel multifunctional acceptor phase additive of water-miscible ionic liquid in hollow-fiber protected liquid phase microextraction. Talanta 2012, 88, 43–49. [Google Scholar] [CrossRef]
- Wang, J.; Huang, S.; Wang, P.; Yang, Y. Method development for the analysis of phthalate esters in tea beverages by ionic liquid hollow fibre liquid-phase microextraction and liquid chromatographic detection. Food Control 2016, 67, 278–284. [Google Scholar] [CrossRef]
- Khataei, M.M.; Yamini, Y.; Nazaripour, A.; Karimi, M. Novel generation of deep eutectic solvent as an acceptor phase in three- phase hollow fiber liquid phase microextraction for extraction and preconcentration of steroidal hormones from biological fl uids. Talanta 2018, 178, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Madikizela, L.M.; Tavengwa, N.T.; Tutu, H.; Chimuka, L. Green aspects in molecular imprinting technology: From design to environmental applications. Trends Environ. Anal. Chem. 2018, 17, 14–22. [Google Scholar] [CrossRef]
- Shishov, A.; Bulatov, A.; Locatelli, M.; Carradori, S.; Andruch, V. Application of deep eutectic solvents in analytical chemistry. A review. Microchem. J. 2017, 135, 33–38. [Google Scholar] [CrossRef]
- Zhang, Q.; De Oliveira Vigier, K.; Sebastien, R.; Jerome, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, R.; Chen, X.; Hu, S.; Bai, X. Three-phase hollow-fiber liquid-phase microextraction based on deep eutectic solvent as acceptor phase for extraction and preconcentration of main active compounds in a traditional Chinese medicinal formula. J. Sep. Sci. 2019, 42, 2187–2298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Chen, X.; Hu, S.; Bai, X. Deep eutectic solvent-based hollow fiber liquid-phase microextraction for quantification of Q-markers of cinnamic acid derivatives in traditional Chinese medicines and research of their plasma protein binding rates. Microchem. J. 2020, 155, 104696. [Google Scholar] [CrossRef]
- Sarafraz-yazdi, A.; Abedi, M.R.; Es, Z.; Branch, Q.; Azad, I. Pre-concentration and determination of β-blockers using carbon nanotube-assisted pseudo stirbar hollow fiber solid-/liquid-phase microextraction and high-performance liquid chromatography with fluorescence. J. Liq. Chromatogr. Relat. Technol. 2013, 36, 750–769. [Google Scholar] [CrossRef]
- Rezaeifar, Z.; Es’haghi, Z.; Rounaghi, G.H.; Chamsaz, M. Hyperbranchedpolyglycerol/graphene oxide nanocomposite reinforced hollow fiber solid/liquid phase microextraction for measurement of ibuprofen and naproxen in hair and waste water samples. J. Chromatogr. B 2016, 1029–1030, 81–87. [Google Scholar] [CrossRef]
- Song, X.; Shi, Y.; Chen, J. A novel extraction technique based on carbon nanotubes reinforced hollow fiber solid / liquid microextraction for the measurement of piroxicam and diclofenac combined with high performance liquid chromatography. Talanta 2012, 100, 153–161. [Google Scholar] [CrossRef]
- Pebdani, A.A.; Mohammad, A.; Shabani, H.; Dadfarnia, S.; Khodadoust, S. Solid phase microextraction of diclofenac using molecularly imprinted polymer sorbent in hollow fiber combined with fiber optic-linear array spectrophotometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 147, 26–30. [Google Scholar] [CrossRef]
- Barahona, F.; Albero, B.; Tadeo, J.L.; Martín-esteban, A. Molecularly imprinted polymer-hollow fiber microextraction of hydrophilic fluoroquinolone antibiotics in environmental waters and urine samples. J. Chromatogr. A 2019, 1587, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Shen, G.; Lee, H.K. A utomated hollow fiber-protected dynamic liquid-phase microextraction of pesticides for gas chromatography – mass spectrometric analysis. J. Chromatogr. A 2003, 985, 107–116. [Google Scholar] [CrossRef]
- Esrafili, A.; Yamini, Y.; Ghambarian, M. Analysis of trace amounts of chlorobenzenes in water samples: An approach towards the automation of dynamic hollow fiber liquid-phase microextraction. Microchim. Acta 2012, 176, 367–374. [Google Scholar] [CrossRef]
- Goh, S.X.L.; Lee, H.K. Automated bundled hollow fiber array-liquid-phase microextraction with liquid chromatography tandem mass spectrometric analysis of perfluorinated compounds in aqueous media. Anal. Chim. Acta 2018, 1019, 74–83. [Google Scholar] [CrossRef]
- Daniels, K.D.; Park, M.; Huang, Z.; Jia, A.; Flores, G.S.; Lee, H.K.; Snyder, S.A.; Daniels, K.D.; Park, M.; Huang, Z.; et al. A review of extraction methods for the analysis of pharmaceuticals in environmental waters. Crit. Rev. Environ. Sci. Technol. 2020, 1–29. [Google Scholar] [CrossRef]
- Sharifi, V.; Abbasi, A.; Nosrati, A. Application of hollow fiber liquid phase microextraction and dispersive liquid e liquid microextraction techniques in analytical toxicology. J. Food Drug Anal. 2015, 24, 264–276. [Google Scholar] [CrossRef]
- Venson, R.; Korb, A.; Cooper, G. A review of the application of hollow-fiber liquid-phase microextraction in bioanalytical methods – A systematic approach with focus on forensic toxicology. J. Chromatogr. B 2019, 1108, 32–53. [Google Scholar] [CrossRef]
- Lee, J.; Lee, H.K.; Rasmussen, K.E.; Pedersen-Bjergaard, S. Environmental and bioanalytical applications of hollow fiber membrane liquid-phase microextraction: A review. Anal. Chim. Acta 2008, 624, 253–268. [Google Scholar] [CrossRef]
- Madikizela, L.M.; Tavengwa, N.T.; Chimuka, L. Status of pharmaceuticals in African water bodies: Occurrence, removal and analytical methods. J. Environ. Manag. 2017, 193, 211–220. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmaceuticals | Matrix, Volume and Its pH | Supported Liquid Membrane + Carrier Molecule Composition | Acceptor Phase, Its Volume and pH | Stirring Rate (rpm), Extraction Time (min) | Analytical Technique | Detection Limits (ng L−1) | Reference |
|---|---|---|---|---|---|---|---|
| Naproxen and nabumetone | 9 mL WWTP influent and tap water, pH 3 | - | 14 μL of 1-undecanol | 600, 20 | LC-FLD | 1.3–2.9 | [50] |
| Ibuprofen, naproxen, and ketoprofen | 20 mL tap water, wastewater and surface water, pH 3.5 | 1-octanol | 4 μL of octanol | * 1 mL min−1, 20 | GC-FID | 1–2 | [54] |
| Amitriptyline, clomipramine, doxepin, mianserin and nortriptyline | 100 mL wastewater, pH 11.8 | di-n-hexyl ether | 20 µL of 10 mM formic acid | 800, 60 | LC-MS | 0.006–0.031 | [41] |
| Salbutamol and terbutaline | 11 mL environmental water (pH 11) | dihexyl ether + 20% (w/v) Aliquat 336 | 24 µL 1M NaBr | 50, 60 | LC-DAD | 500–2500 | [47] |
| 17-β-ethynylestradiol, 17-β-estradiol, estrone | 100 mL tap and sewage water | di-n-hexyl ether + 10% (w/v) TOPO | 10 µL of n-undecane | 1 100, 2 | GC-MS | 1.6–10 | [34] |
| Ketoprofen, naproxen, diclofenac and ibupprofen | 1 L WWTP effluent, pH 1.5–2 | di-n-hexyl ether | aqueous solution at pH 9.5 | ** 30 mL min−1, 45 | LC-DAD-FLD | 10–50 | [62] |
| 4 NSAIDs and 8 of their metabolites | 50 mL wastewater, pH 2 | di-n-hexyl ether + 5% (w/v) TOPO | 10 µL of 0.1 M ammonium carbonate, pH 9 | 660, 5 h | LC-MS | 7.1–89.3 µg L−1 | [9] |
| Carbamazepine | 12 mL wastewater, well and river waters, pH 8.9 | octanol | 25 µL octanol | 400, 48.5 | LC-DAD | 2800 | [12] |
| 6 β-blockers | 55 mL wastewater, pH 11.5 | heptanol | 25 µL heptanol | 800, 60 | LC-UV | 80–500 | [13] |
| 3 antiretroviral drugs | 10 mL surface and wastewater, pH 4 | dihexyl ether + 5%, (w/w) DEHPA | 22.6 μL of 0.4 mM HCl | 1000, 60 | LC-MS | 9–160 | [16] |
| 4 NSAIDs | 6 mL wastewater and surface water, pH 3 | dihexyl ether + 5%, (w/w) di-(2-ethylhexyl) phosphoric acid | 22.6 µL of aqueous solution (pH 10) | 900, 60 | LC-MS | 0.05–0.35 | [39] |
| 7 NSAIDs | 50 mL wastewater, pH 2 | Dihexyl ether | 30 µL of aqueous solution (pH 12) | 300, 20 | CE | 205–860 | [64] |
| Ibuprofen, naproxen, and ketoprofen | 2.5 mL pure water containing 250 µL 0.1 M HCl | Dihexyl ether | 25 µL of 10 mM NaOH | 400, 45 | CE | 5000 | [17] |
| Ibuprofen and clofibric acid | 4 mL of 0.1 M HCl wastewater solution | 1-octanol | 100 µL 0.01 M NaOH | 700, 40 | LC-UV | 15–100 | [38] |
| 5 sulfonamides | 4 mL river and wastewater (pH 4.5) | ionic liquid + 14% (w/v) TOPO | 25 µL aqueous solution (pH 13) | 300, 8 h | LC-DAD | 100–400 | [71] |
| Ketoprofen, naproxen, and clofibric acid | 10 mL of 0.01M HCl wastewater solution | 1-octanol | 5 µL of 0.5M NaOH | 73 rad s−1, 60 | LC-UV | 30–300 | [72] |
| 4 sulfonamides and their main metabolites | 50 mL wastewater, river and tap water, pH 4 | 1-octanol | 50 µL aqueous solution, pH 12 | 300, 6 h | LC-DAD-FLD | 0.3–33 | [58] |
| Sulphonamides | Water samples (pH 6) | 5% TOPO in hexylamine | 0.4 M H2SO4 | Continuous flow at 0.3 mL min−1 for 60 min | LC-DAD | <20 µg L−1 | [60] |
| Steroids | Water samples (pH 6) | n-undecane/di-n-hexyl ether (1:1 v/v) + 5% (w/v) TOPO | 0.4 M H2SO4 | Continuous flow at 0.1 mL min−1 for 60 min | LC-DAD | <2.4 | [60] |
| Salicylic acid, diclofenac, and ibuprofen | 50 mL wastewater, pH 2 | Dihexyl ether | 50 µL aqueous solution, pH 12.5 | 300, 15 | LC-MS | 20–300 | [55] |
| 8 fluoroquinolones | 50 mL wastewater, river water and tap water, pH 7 | 1-octanol | 50 µL aqueous solution, pH 12 | 300, 5.5 h | LC-DAD-FLD | 0.3–16 | [56] |
| 9 NSAIDs | 22 mL wastewater, pH 2 | 1-octanol | 20 µL of 10 mM ammonium carbonate | 500, 45 | LC-MS | 0.5–42 | [11] |
| Tetracycline, oxytetracycline, and doxycycline | 11 mL tap water, pH 9 | 1-octanol + 10% (w/v) aliquat-336 | 24 µL of 0.1 M H3PO4 and 1.0 M NaCl, pH 1.6 | 900, 35 | LC-UV/Vis | 500–1000 | [44] |
| Megestrol acetate and levonorgestrel | 20 mL water, pH not adjusted | n-dodecane | 25 µL methanol | 1000, 40 | LC-UV/Vis | 250 | [19] |
| 5 selective serotonin reuptake inhibitors and 4 of their metabolites | 1.1 L seawater and wastewater, pH 11.8 | Dihexyl ether | 20 µL aqueous solution, pH 2 | 800, 2 h | LC-MS | 0.017–0.618 | [18] |
| 4 fluoroquinolone antibiotics | 10 mL surface water (pH 6) | di-n-hexyl ether + 20% (w/w) DEHPA | 56.5 µL of 0.1 M HCl | 200, 2 h | LC-DAD | 10–20 | [37] |
| carbamazepine ibuprofen, phenazone, 17-α-ethinylestradiol | 5 mL water sample (pH 2) | 1-octanol | 17 µL 1-octanol | 1 000, 60 | GC-MS | 20–40 | [73] |
| diethylstilbestrol, dienestrol, and hexestrol (oestrogens) | 10 mL wastewater (pH 1.5) | 1-octanol | 10 µL of 0.5 M NaOH | 1 200, 40 | LC-UV/Vis | 250–500 | [59] |
| 8 sulfonamides | 8 mL wastewater, pH 3.5 | 1-octanol | 30 µL NaOH, pH 12.5 | 600, 75 | LC-FLD | 3.1–11.2 | [69] |
| Salicylic acid, para-aminosalicylic acid and acetylsalicylic acid | 10 mL sea and river water, pH 3 | 1-octanol | 15 µL purified water pH 6.2 | 1000, 45 | LC-UV/Vis | 600–1200 | [74] |
| 4 NSAIDs | 5 mL purified water, tap water, pH 1.5 | 1-octanol | 15 µL 1-octanol | 300, 20 | LC-MS | 500–1250 | [14] |
| 17-β-estradiol, estrone and diethylstilbestrol | 50 mL river water, pH 2 | Ionic liquid | 2.5 µL ionic liquid | 200, 8 h | LC-UV/Vis | 50–100 | [15] |
| 11 antibiotics | 20 mL river water, pH 8 | dihexyl ether + 20% (w/v) aliquat-336 | 20 µL acetic acid, pH 4 | 200, 60 | LC-MS | 10–250 | [68] |
| Raloxifene and ethinylestradiol | 17 mL pharmaceutical wastewater, pH 11 | 1-octanol + 0.04 g mL−1 CTAB | 20 µL deep eutectic solvent | 700, 42 | LC-UV | 5000–10,000 | [75] |
| 4 anti-arrhythmic agents | 10 mL pharmaceutical wastewater, pH 12.3 | ChCl:Ph-ETOH | 40 µL aqueous solution, pH 2.5 | 1100, 40 | LC-UV | 300–800 | [76] |
| 27 emerging contaminants included pharmaceuticals | 1000 mL river water, pH 7 | 1-octanol | 60 µL of 1-octanol | 100, 30 | LC-MS | 1.09–98.15 | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madikizela, L.M.; Pakade, V.E.; Ncube, S.; Tutu, H.; Chimuka, L. Application of Hollow Fibre-Liquid Phase Microextraction Technique for Isolation and Pre-Concentration of Pharmaceuticals in Water. Membranes 2020, 10, 311. https://doi.org/10.3390/membranes10110311
Madikizela LM, Pakade VE, Ncube S, Tutu H, Chimuka L. Application of Hollow Fibre-Liquid Phase Microextraction Technique for Isolation and Pre-Concentration of Pharmaceuticals in Water. Membranes. 2020; 10(11):311. https://doi.org/10.3390/membranes10110311
Chicago/Turabian StyleMadikizela, Lawrence Mzukisi, Vusumzi Emmanuel Pakade, Somandla Ncube, Hlanganani Tutu, and Luke Chimuka. 2020. "Application of Hollow Fibre-Liquid Phase Microextraction Technique for Isolation and Pre-Concentration of Pharmaceuticals in Water" Membranes 10, no. 11: 311. https://doi.org/10.3390/membranes10110311
APA StyleMadikizela, L. M., Pakade, V. E., Ncube, S., Tutu, H., & Chimuka, L. (2020). Application of Hollow Fibre-Liquid Phase Microextraction Technique for Isolation and Pre-Concentration of Pharmaceuticals in Water. Membranes, 10(11), 311. https://doi.org/10.3390/membranes10110311