
Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein

 ,
,  , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Generation of IGIP-H1 Influenza Plasmids
2.3. Generation of IGIP-Influenza Viruses by Reverse Genetics
2.4. In Vitro Growth Kinetics
2.5. Mouse Studies
2.6. Hemagglutination Inhibition Assays
2.7. Virus Neutralization Assays
2.8. Virus Titration
2.9. Histopathology Examination
2.10. Influenza Antigen Microarray
2.11. Graphs/Statistical Analyses
3. Results
3.1. IGIP-H1att and IGIP-H1caLen Viruses Show Differences in Virus Yield
3.2. Studies in DBA/2 J Mice Showed Improved Safety of the IGIP-H1att in Comparison to the Isogenic H1att Virus
3.3. Efficacy of the IGIP-H1att in DBA/2 J Mice
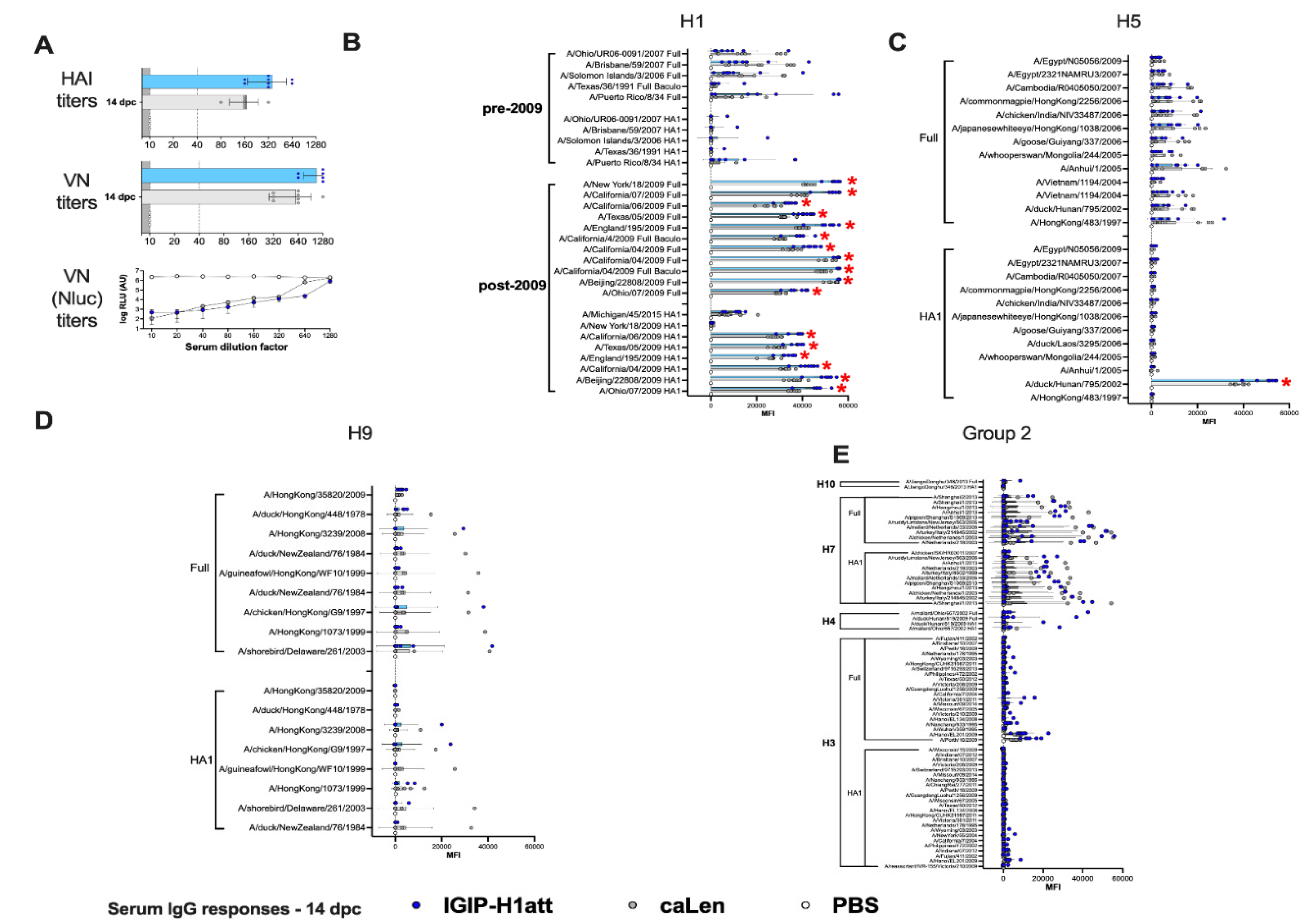
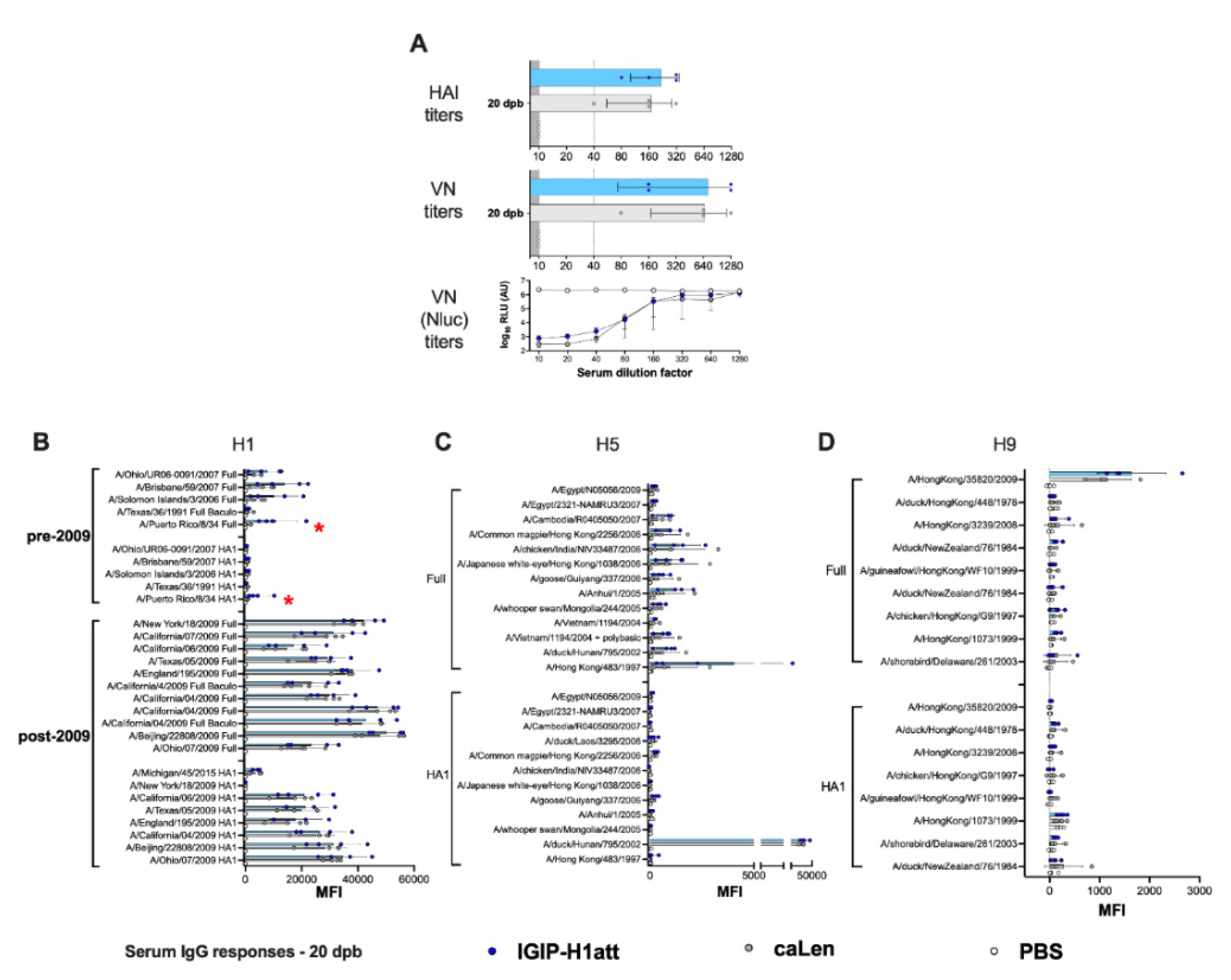
3.4. Qualitatively Different Humoral Responses Are Produced by the IGIP-H1att Virus Compared to the H1caLen Virus in DBA/2J Mice
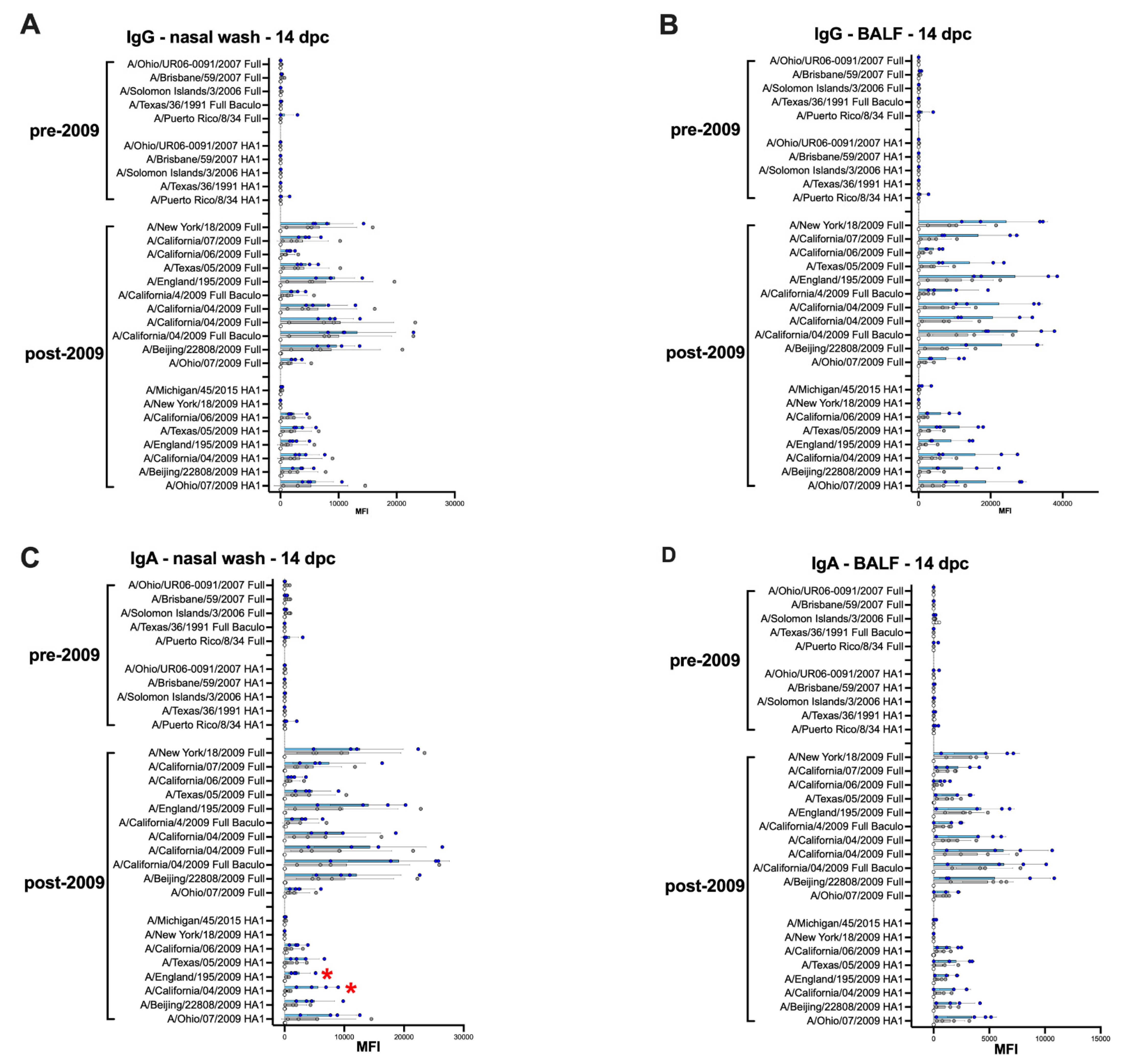
3.5. Average Higher Anti-H1 HA Mucosal IgG and IgA Responses in the IGIP-H1att Group Compared to the H1caLen Group at 14 dpc
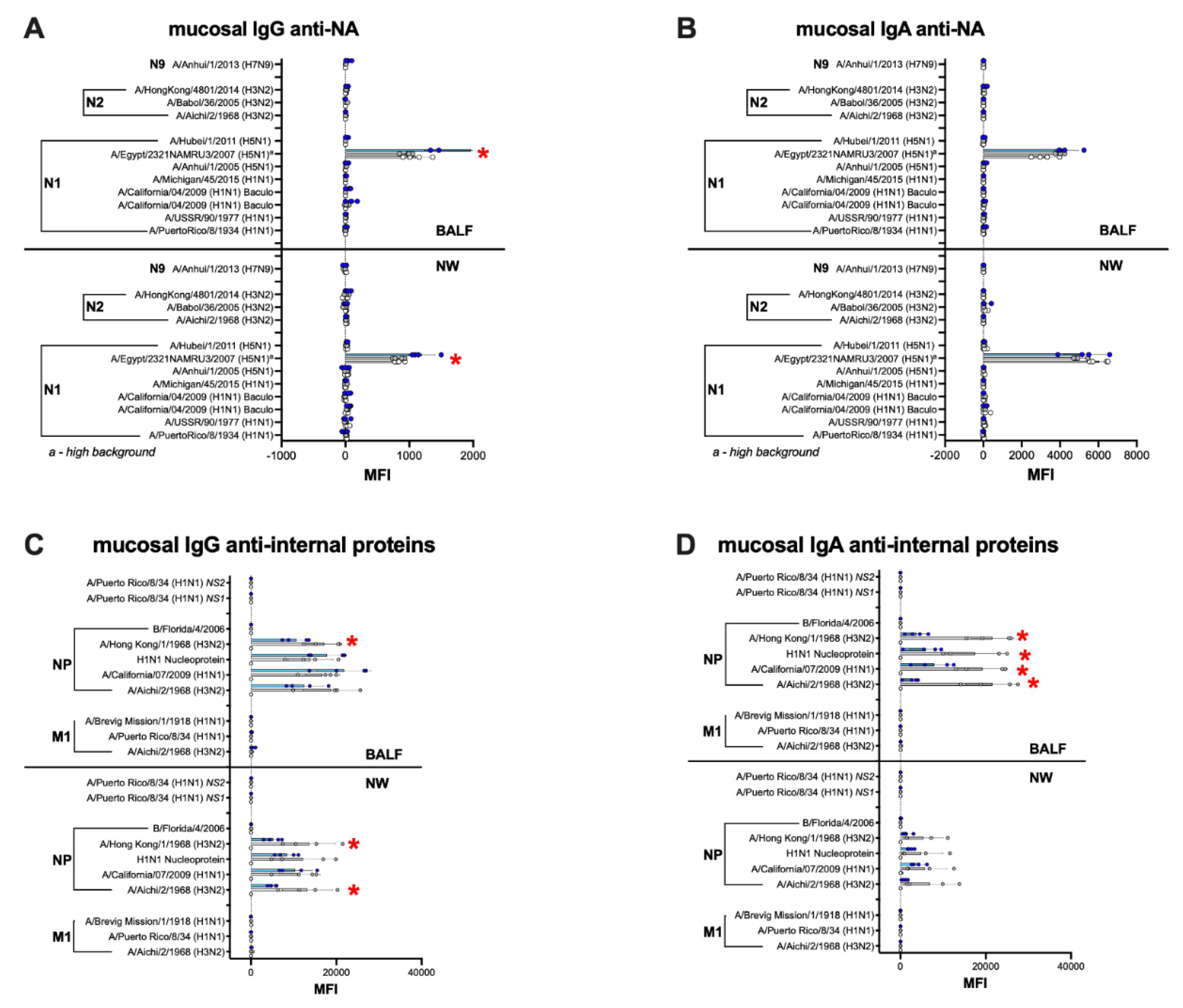
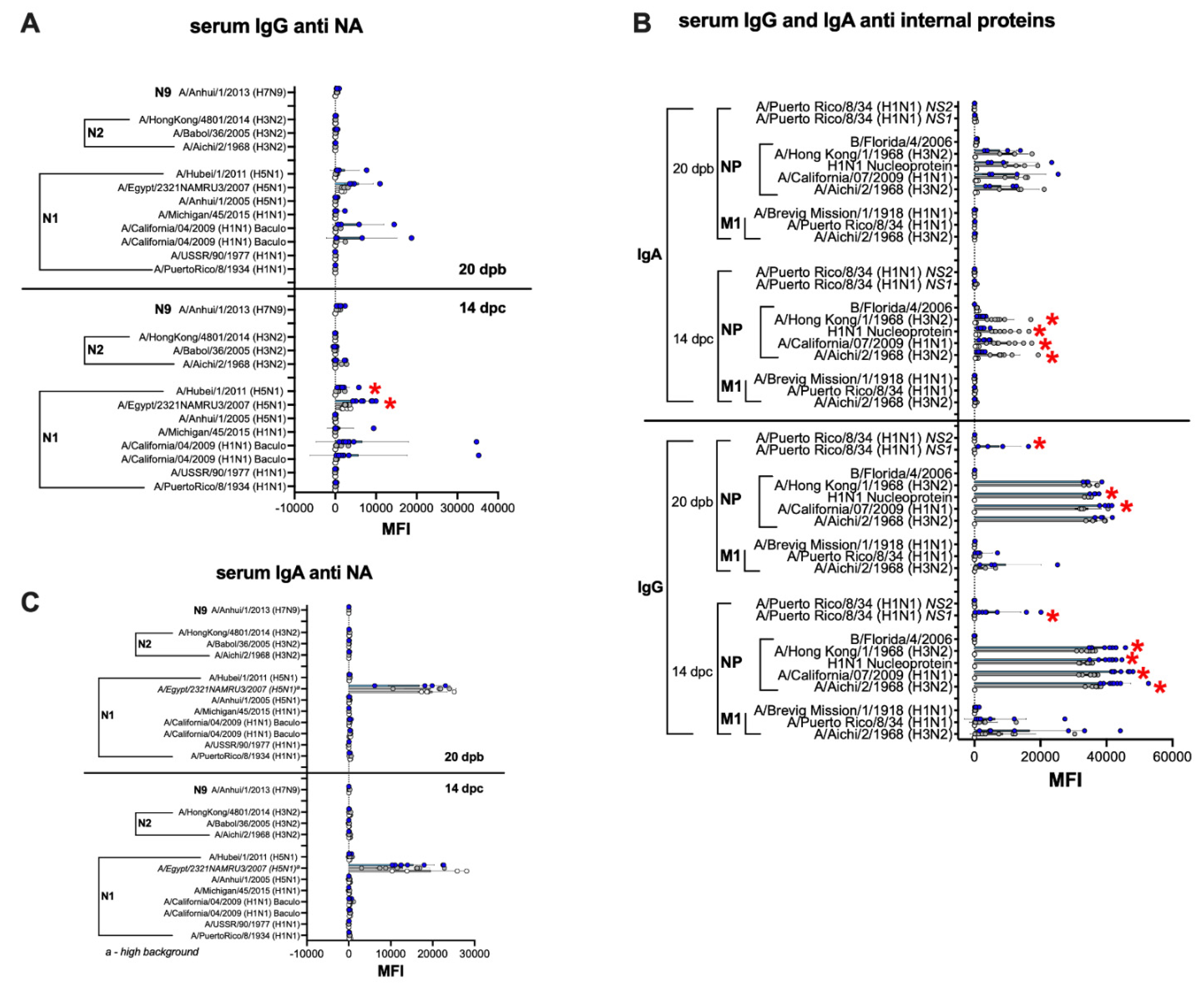
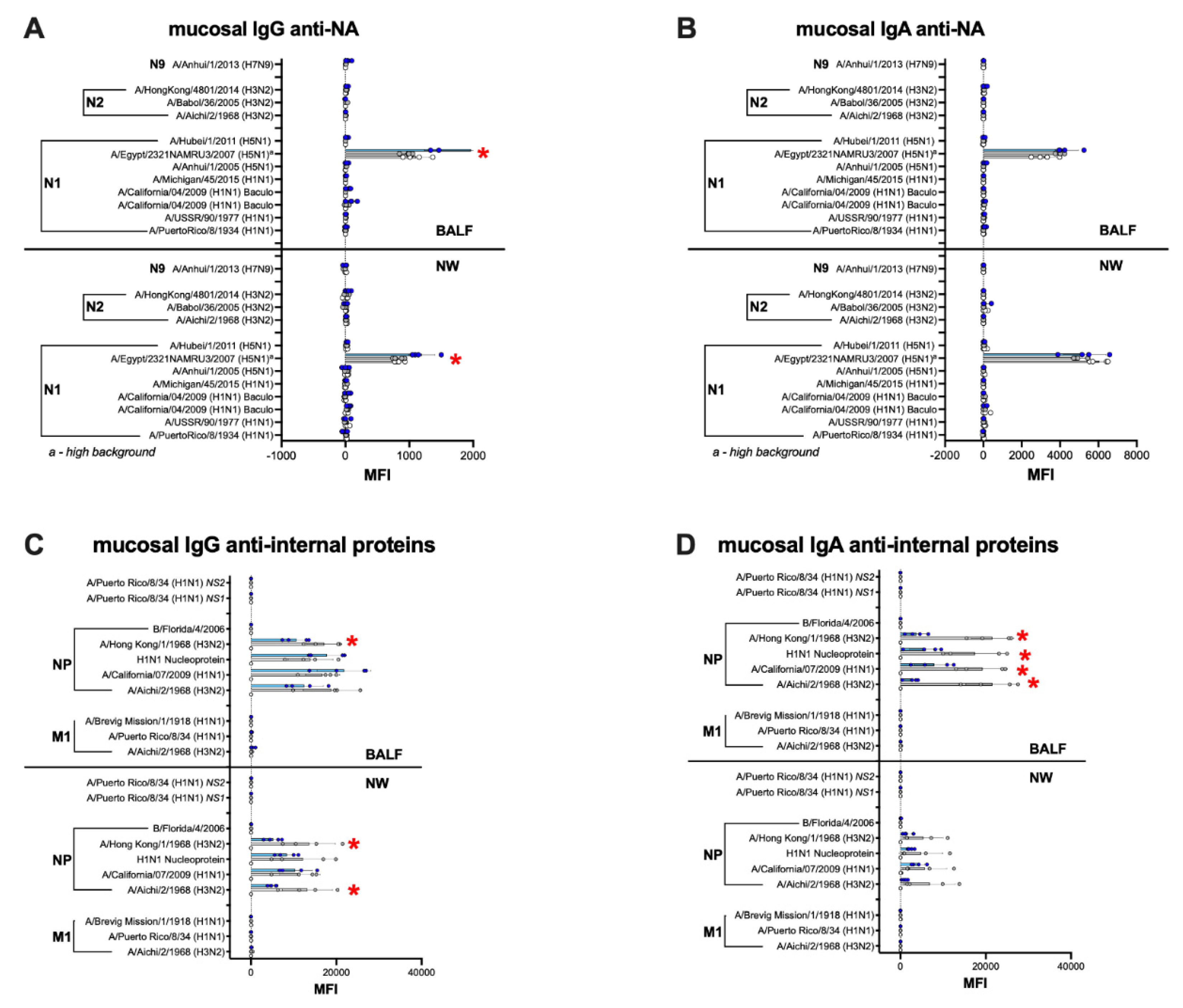
3.6. Humoral and Mucosal Responses against the NA and Internal Proteins Are Consistent with Anti-HA Response Patterns in the IGIP-H1att and H1caLen Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Girard, M.P.; Cherian, T.; Pervikov, Y.; Kieny, M.P. A review of vaccine research and development: Human acute respiratory infections. Vaccine 2005, 23, 5708–5724. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef]
- Paul Glezen, W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza B: A structured literature review. Am. J. Public Health 2013, 103, e43–e51. [Google Scholar] [CrossRef]
- Gasparini, R.; Amicizia, D.; Lai, P.L.; Panatto, D. Clinical and socioeconomic impact of seasonal and pandemic influenza in adults and the elderly. Hum. Vaccines Immunother. 2012, 8, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Lopez, C.E.; Legge, K.L. Influenza A Virus Vaccination: Immunity, Protection, and Recent Advances Toward A Universal Vaccine. Vaccines 2020, 8, 434. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Kawaoka, Y. Current and future influenza vaccines. Nat. Med. 2019, 25, 212–220. [Google Scholar] [CrossRef]
- Grohskopf, L.A.; Alyanak, E.; Broder, K.R.; Blanton, L.H.; Fry, A.M.; Jernigan, D.B.; Atmar, R.L. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2020–2021 Influenza Season. MMWR Recomm Rep. 2020, 69, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Corthesy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Kawaguchi, A.; Ainai, A.; Tamura, S.; Ito, R.; Multihartina, P.; Setiawaty, V.; Pangesti, K.N.; Odagiri, T.; Tashiro, M.; et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc. Natl. Acad. Sci. USA 2015, 112, 7809–7814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008, 8, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Austin, A.S.; Haas, K.M.; Naugler, S.M.; Bajer, A.A.; Garcia-Tapia, D.; Estes, D.M. Identification and characterization of a novel regulatory factor: IgA-inducing protein. J. Immunol. 2003, 171, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Estes, D.M. Regulation of IgA responses in cattle, humans and mice. Vet. Immunol. Immunopathol. 2010, 138, 312–317. [Google Scholar] [CrossRef]
- Ghendon, Y.Z.; Polezhaev, F.I.; Lisovskaya, K.V.; Medvedeva, T.E.; Alexandrova, G.I.; Klimov, A.I. Recombinant cold-adapted attenuated influenza A vaccines for use in children: Molecular genetic analysis of the cold-adapted donor and recombinants. Infect. Immun. 1984, 44, 730–733. [Google Scholar] [CrossRef] [Green Version]
- Pena, L.; Vincent, A.L.; Ye, J.; Ciacci-Zanella, J.R.; Angel, M.; Lorusso, A.; Gauger, P.C.; Janke, B.H.; Loving, C.L.; Perez, D.R. Modifications in the polymerase genes of a swine-like triple-reassortant influenza virus to generate live attenuated vaccines against 2009 pandemic H1N1 viruses. J. Virol. 2011, 85, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Cardenas Garcia, S.; Liu, J.; Santos, J.; Carnaccini, S.; Geiger, G.; Ferreri, L.; Rajao, D.; Perez, D.R. Alternative Strategy for a Quadrivalent Live Attenuated Influenza Virus Vaccine. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Isakova-Sivak, I.; Chen, L.M.; Matsuoka, Y.; Voeten, J.T.; Kiseleva, I.; Heldens, J.G.; den Bosch, H.; Klimov, A.; Rudenko, L.; Cox, N.J.; et al. Genetic bases of the temperature-sensitive phenotype of a master donor virus used in live attenuated influenza vaccines: A/Leningrad/134/17/57 (H2N2). Virology 2011, 412, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; Muench, H. A simple method for estimating fifty percent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Ferreri, L.M.; Ortiz, L.; Geiger, G.; Barriga, G.P.; Poulson, R.; Gonzalez-Reiche, A.S.; Crum, J.A.; Stallknecht, D.; Moran, D.; Cordon-Rosales, C.; et al. Improved detection of influenza A virus from blue-winged teals by sequencing directly from swab material. Ecol. Evol. 2019, 9, 6534–6546. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.J.S.; Abente, E.J.; Obadan, A.O.; Thompson, A.J.; Ferreri, L.; Geiger, G.; Gonzalez-Reiche, A.S.; Lewis, N.S.; Burke, D.F.; Rajao, D.S.; et al. Plasticity of Amino Acid Residue 145 Near the Receptor Binding Site of H3 Swine Influenza A Viruses and Its Impact on Receptor Binding and Antibody Recognition. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Sorrell, E.M.; Cai, Y.; Shao, H.; Xu, K.; Pena, L.; Hickman, D.; Song, H.; Angel, M.; Medina, R.A.; et al. Variations in the hemagglutinin of the 2009 H1N1 pandemic virus: Potential for strains with altered virulence phenotype? PLoS Pathog. 2010, 6, e1001145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golde, W.T.; Gollobin, P.; Rodriguez, L.L. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. 2005, 34, 39–43. [Google Scholar] [CrossRef]
- Nakajima, R.; Supnet, M.; Jasinskas, A.; Jain, A.; Taghavian, O.; Obiero, J.; Milton, D.K.; Chen, W.H.; Grantham, M.; Webby, R.; et al. Protein Microarray Analysis of the Specificity and Cross-Reactivity of Influenza Virus Hemagglutinin-Specific Antibodies. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.; Zhou, H.; Kemble, G.; Jin, H. The cold adapted and temperature sensitive influenza A/Ann Arbor/6/60 virus, the master donor virus for live attenuated influenza vaccines, has multiple defects in replication at the restrictive temperature. Virology 2008, 380, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauger, P.C.; Loving, C.L.; Khurana, S.; Lorusso, A.; Perez, D.R.; Kehrli, M.E., Jr.; Roth, J.A.; Golding, H.; Vincent, A.L. Live attenuated influenza A virus vaccine protects against A(H1N1)pdm09 heterologous challenge without vaccine associated enhanced respiratory disease. Virology 2014, 471–473, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loving, C.L.; Lager, K.M.; Vincent, A.L.; Brockmeier, S.L.; Gauger, P.C.; Anderson, T.K.; Kitikoon, P.; Perez, D.R.; Kehrli, M.E., Jr. Efficacy in pigs of inactivated and live attenuated influenza virus vaccines against infection and transmission of an emerging H3N2 similar to the 2011–2012 H3N2v. J. Virol. 2013, 87, 9895–9903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Pica, N.; Iyer, A.; Ramos, I.; Bouvier, N.M.; Fernandez-Sesma, A.; Garcia-Sastre, A.; Lowen, A.C.; Palese, P.; Steel, J. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J. Virol. 2011, 85, 12825–12829. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, B.; Blazejewska, P.; Hessmann, M.; Bruder, D.; Geffers, R.; Mauel, S.; Gruber, A.D.; Schughart, K. Host genetic background strongly influences the response to influenza a virus infections. PLoS ONE 2009, 4, e4857. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.R.; Rolfes, M.A.; Flannery, B.; Prasad, P.; O’Halloran, A.; Garg, S.; Fry, A.M.; Singleton, J.A.; Patel, M.; Reed, C.; et al. Effects of Influenza Vaccination in the United States During the 2018–2019 Influenza Season. Clin. Infect. Dis. 2020, 71, e368–e376. [Google Scholar] [CrossRef]
- Dawood, F.S.; Chung, J.R.; Kim, S.S.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; et al. Interim Estimates of 2019–20 Seasonal Influenza Vaccine Effectiveness—United States, February 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Rolfes, M.A.; Flannery, B.; Chung, J.R.; O’Halloran, A.; Garg, S.; Belongia, E.A.; Gaglani, M.; Zimmerman, R.K.; Jackson, M.L.; Monto, A.S.; et al. Effects of Influenza Vaccination in the United States During the 2017–2018 Influenza Season. Clin. Infect. Dis. 2019, 69, 1845–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endsley, M.A.; Njongmeta, L.M.; Shell, E.; Ryan, M.W.; Indrikovs, A.J.; Ulualp, S.; Goldblum, R.M.; Mwangi, W.; Estes, D.M. Human IgA-inducing protein from dendritic cells induces IgA production by naive IgD+ B cells. J. Immunol. 2009, 182, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P.; Rennert, P.; Browning, J. BAFF AND APRIL: A tutorial on B cell survival. Annu. Rev. Immunol. 2003, 21, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Ambrose, C. The TNF family members BAFF and APRIL: The growing complexity. Cytokine Growth Factor Rev. 2003, 14, 311–324. [Google Scholar] [CrossRef]
- Solorzano, A.; Ye, J.; Perez, D.R. Alternative live-attenuated influenza vaccines based on modifications in the polymerase genes protect against epidemic and pandemic flu. J. Virol. 2010, 84, 4587–4596. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, J.W.; Whitehead, G.; Berman, K.G.; Tekippe, E.M.; Gilmour, M.I.; Larkin, J.E.; Quackenbush, J.; Schwartz, D.A. Genetic basis of murine antibacterial defense to streptococcal lung infection. Immunogenetics 2007, 59, 713–724. [Google Scholar] [CrossRef]
- Santos, J.J.S.; Finch, C.; Sutton, T.; Obadan, A.; Aguirre, I.; Wan, Z.; Lopez, D.; Geiger, G.; Gonzalez-Reiche, A.S.; Ferreri, L.; et al. Development of an Alternative Modified Live Influenza B Virus Vaccine. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Abente, E.J.; Rajao, D.S.; Santos, J.; Kaplan, B.S.; Nicholson, T.L.; Brockmeier, S.L.; Gauger, P.C.; Perez, D.R.; Vincent, A.L. Comparison of Adjuvanted-Whole Inactivated Virus and Live-Attenuated Virus Vaccines against Challenge with Contemporary, Antigenically Distinct H3N2 Influenza A Viruses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Nachbagauer, R.; Zhu, L.; Huang, Y.; Xie, X.; Jin, S.; Zhang, A.; Wan, Y.; Hirsh, A.; Tian, D.; et al. Induction of Broadly Cross-Reactive Stalk-Specific Antibody Responses to Influenza Group 1 and Group 2 Hemagglutinins by Natural H7N9 Virus Infection in Humans. J. Infect. Dis. 2017, 215, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Fouchier, R.A.M.; Eichelberger, M.C.; Webby, R.J.; Shaw-Saliba, K.; Wan, H.; Wilson, P.C.; Compans, R.W.; Skountzou, I.; Monto, A.S. NAction! How Can Neuraminidase-Based Immunity Contribute to Better Influenza Virus Vaccines? mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiers, W.; De Filette, M.; El Bakkouri, K.; Schepens, B.; Roose, K.; Schotsaert, M.; Birkett, A.; Saelens, X. M2e-based universal influenza A vaccine. Vaccine 2009, 27, 6280–6283. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Asthagiri Arunkumar, G.; Liu, W.C.; Stadlbauer, D.; Albrecht, R.A.; Pavot, V.; Aramouni, M.; Lambe, T.; Gilbert, S.C.; Krammer, F. Vaccination With Viral Vectors Expressing Chimeric Hemagglutinin, NP and M1 Antigens Protects Ferrets Against Influenza Virus Challenge. Front. Immunol. 2019, 10, 2005. [Google Scholar] [CrossRef] [PubMed]
- van de Sandt, C.E.; Kreijtz, J.H.; de Mutsert, G.; Geelhoed-Mieras, M.M.; Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Osterhaus, A.D.; Fouchier, R.A.; Rimmelzwaan, G.F. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. J. Virol. 2014, 88, 1684–1693. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.Y.; Ha do, L.A.; Simmons, C.; de Jong, M.D.; Chau, N.V.; Schumacher, R.; Peng, Y.C.; McMichael, A.J.; Farrar, J.J.; Smith, G.L.; et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J. Clin. Investig. 2008, 118, 3478–3490. [Google Scholar] [CrossRef]
- Isakova-Sivak, I.; Matyushenko, V.; Kotomina, T.; Kiseleva, I.; Krutikova, E.; Donina, S.; Rekstin, A.; Larionova, N.; Mezhenskaya, D.; Sivak, K.; et al. Sequential Immunization with Universal Live Attenuated Influenza Vaccine Candidates Protects Ferrets against a High-Dose Heterologous Virus Challenge. Vaccines 2019, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Pleguezuelos, O.; James, E.; Fernandez, A.; Lopes, V.; Rosas, L.A.; Cervantes-Medina, A.; Cleath, J.; Edwards, K.; Neitzey, D.; Gu, W.; et al. Efficacy of FLU-v, a broad-spectrum influenza vaccine, in a randomized phase IIb human influenza challenge study. NPJ Vaccines 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA Gene | Backbone | Titer TCID50/mL | IGIP-H1 Segment 4 Stability | |||
|---|---|---|---|---|---|---|
| Segment | Modification | E1 (NGS) a | E5 (Sanger) b | |||
| IGIP-H1 | OH/04att | 5 × 106 | HA | HA SP | No change | t58cnon-syn(L9P) c |
| IGIP | No change | t86cSyn | ||||
| G4S | No change | No change | ||||
| Furin CS | No change | No change | ||||
| Tav2A | No change | No change | ||||
| GlucS | No change | No change | ||||
| PB2 | S265 (g821) | No change | No change | |||
| PB1 | E391(g1195, g1197) | No change | No change | |||
| G581(g1766) | No change | No change | ||||
| T661(a2005, g2007) | No change | No change | ||||
| HA tag | No change | No change | ||||
| Other segments | N/A d | No change | Not performed | |||
| H1 | OH/04att | 2.32 × 107 | N/A | N/A | N/A | N/A |
| IGIP-H1 | caLen | 1 × 104 | N/A | N/A | N/A | N/A |
| H1 | caLen | 2 × 107 | N/A | N/A | N/A | N/A |
| Group | Virus Challenge? | HE | IHC |
|---|---|---|---|
| Mock-vaccinated | Yes | 4-4-4-4 | 3-3-3-3 |
| IGIP-H1att | Yes | 1-0-1-1 | 0-0-0-0 |
| H1-caLen | Yes | 1-2-0-1 | 0-0-0-0 |
| PBS | No | 0-0-0-0 | 0-0-0-0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cáceres, C.J.; Cardenas-Garcia, S.; Jain, A.; Gay, L.C.; Carnaccini, S.; Seibert, B.; Ferreri, L.M.; Geiger, G.; Jasinskas, A.; Nakajima, R.; et al. Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein. Vaccines 2021, 9, 703. https://doi.org/10.3390/vaccines9070703
Cáceres CJ, Cardenas-Garcia S, Jain A, Gay LC, Carnaccini S, Seibert B, Ferreri LM, Geiger G, Jasinskas A, Nakajima R, et al. Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein. Vaccines. 2021; 9(7):703. https://doi.org/10.3390/vaccines9070703
Chicago/Turabian StyleCáceres, C. Joaquín, Stivalis Cardenas-Garcia, Aarti Jain, L. Claire Gay, Silvia Carnaccini, Brittany Seibert, Lucas M. Ferreri, Ginger Geiger, Algimantas Jasinskas, Rie Nakajima, and et al. 2021. "Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein" Vaccines 9, no. 7: 703. https://doi.org/10.3390/vaccines9070703
APA StyleCáceres, C. J., Cardenas-Garcia, S., Jain, A., Gay, L. C., Carnaccini, S., Seibert, B., Ferreri, L. M., Geiger, G., Jasinskas, A., Nakajima, R., Rajao, D. S., Isakova-Sivak, I., Rudenko, L., Vincent, A. L., Davies, D. H., & Perez, D. R. (2021). Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein. Vaccines, 9(7), 703. https://doi.org/10.3390/vaccines9070703