
Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis


 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of the Schistosoma Core Proteome
2.2. Subtractive Proteomics Approach
2.3. Prediction of Epitopes
2.3.1. Prediction of CTL Epitopes
2.3.2. Prediction of HTL Epitopes
2.3.3. Prediction of LBL Epitopes
2.4. World Population Coverage
2.5. MEV Construction
2.6. Structural Analysis of the Vaccine Construct
2.7. 3D Structure Prediction and Validation
2.8. Prediction of the B Cell Epitopes of the Vaccine
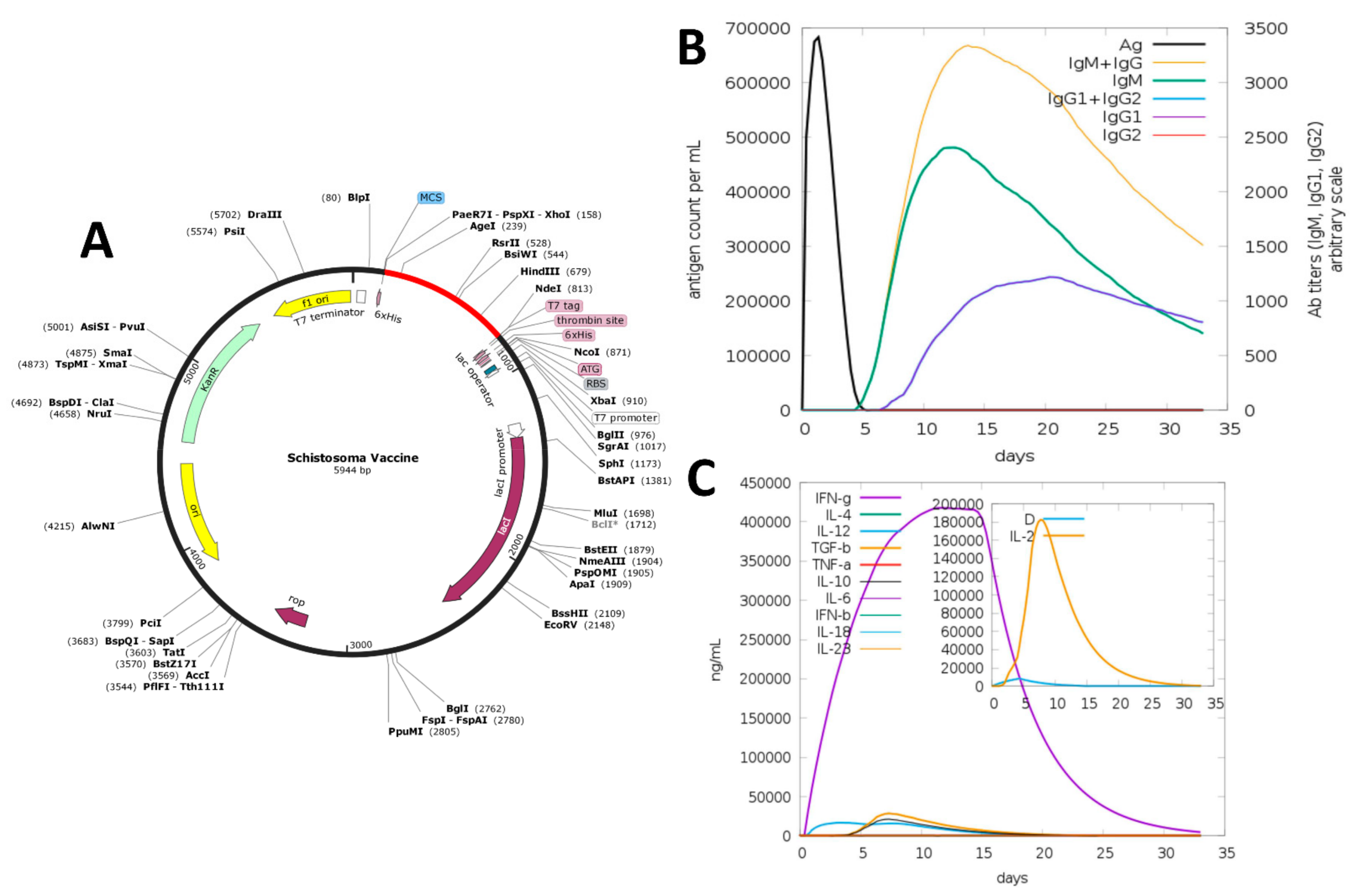
2.9. In Silico Cloning and Codon Adaptation
2.10. Immune Simulation
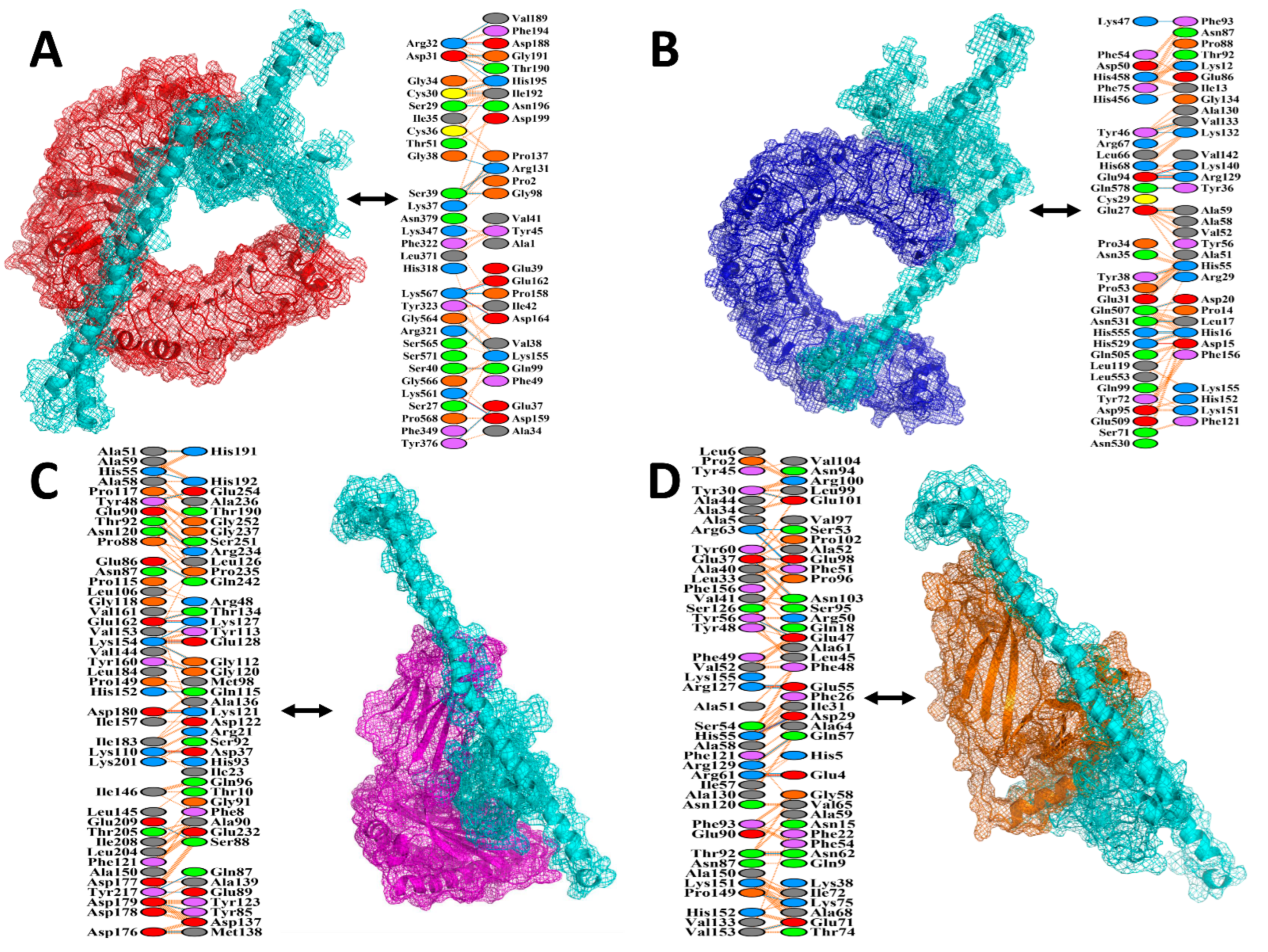
2.11. Protein–Protein Docking
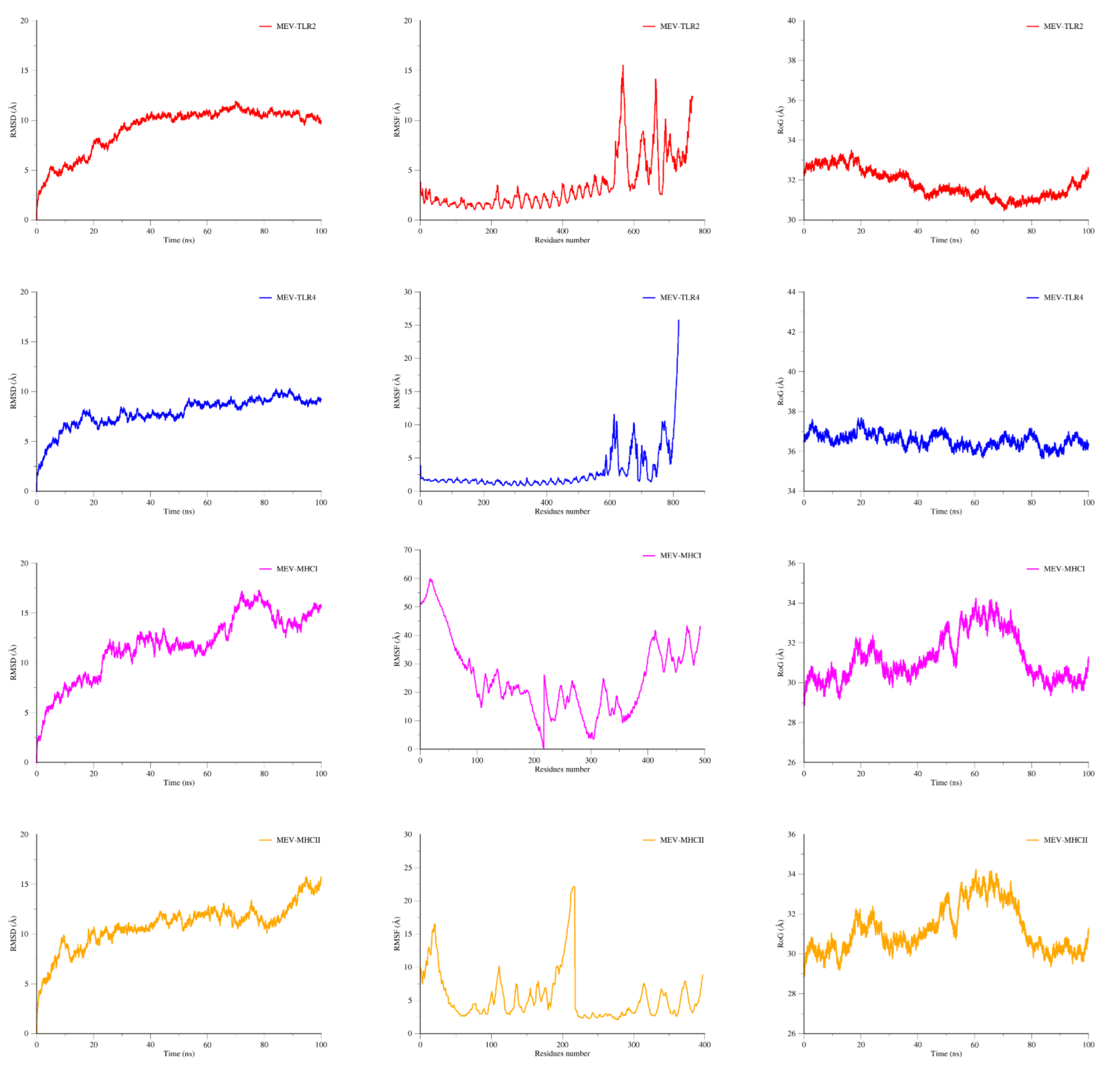
2.12. MD Simulations
2.13. MMGBSA Binding Energy Analysis
3. Results
3.1. Core Proteome Analysis
3.2. Identification of Vaccine Candidates
3.3. Epitopes Prediction
3.4. World Population Coverage
3.5. Construction of the MEV
3.6. Physiochemical and Structural Analysis of the Vaccine Construct
3.7. Prediction of B Cell Epitopes of the MEV
3.8. In Silico Cloning
3.9. Immune Simulation
3.10. Protein–Protein Docking
3.11. MD Simulation
3.12. MMGBSA Binding Energy Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffmann, K.F.; Brindley, P.J.; Berriman, M. Halting harmful helminths. Science 2014, 346, 168–169. [Google Scholar] [CrossRef]
- Frimpong-Boateng, K. Infectious Disease and Cancer in Africa-A Medical and Demographical Reality. A Keynote Lecture Presented at the “Global Health and Molecular Medicine: Our Common Future”, Hannover. Retrieved. 2017. Available online: http://www.ourcommonfuture.de/fileadmin/user_upload/dateien/Reden/Frimbong_Boateng.pdf (accessed on 23 April 2021).
- Gordon, C.A.; Kurscheid, J.; Williams, G.M.; Clements, A.C.; Li, Y.; Zhou, X.-N.; Utzinger, J.; McManus, D.P.; Gray, D.J. Asian schistosomiasis: Current status and prospects for control leading to elimination. Trop. Med. Infect. Dis. 2019, 4, 40. [Google Scholar] [CrossRef]
- Wu, W.; Wang, W.; Huang, Y.X. New insight into praziquantel against various developmental stages of schistosomes. Parasitol. Res. 2011, 109, 1501–1507. [Google Scholar] [CrossRef]
- Kurup, R.; Hunjan, G.S. Epidemiology and control of Schistosomiasis and other intestinal parasitic infections among school children in three rural villages of south Saint Lucia. J. Vector Borne Dis. 2010, 47, 228. [Google Scholar]
- Tebeje, B.M.; Harvie, M.; You, H.; Loukas, A.; McManus, D.P. Schistosomiasis vaccines: Where do we stand? Parasites Vectors 2016, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Biritwum, N.-K.; Woods, G.; Velleman, Y.; Fleming, F.; Stothard, J.R. Tailoring water, sanitation, and hygiene (WASH) targets for soil-transmitted helminthiasis and schistosomiasis control. Trends Parasitol. 2018, 34, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Hybridization may give some parasites a leg up. Science 2018, 361, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Unfilled vials. Science 2016, 351, 16–19. [Google Scholar] [CrossRef]
- Chew, S.Y.; Than, L.T.L. Vulvovaginal candidosis: Contemporary challenges and the future of prophylactic and therapeutic approaches. Mycoses 2016, 59, 262–273. [Google Scholar] [CrossRef]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479, 379–392. [Google Scholar] [CrossRef]
- Chang, S.; Dunn, J.; Heidari, M.; Lee, L.; Song, J.; Ernst, C.; Ding, Z.; Bacon, L.; Zhang, H. Genetics and vaccine efficacy: Host genetic variation affecting Marek’s disease vaccine efficacy in White Leghorn chickens. Poult. Sci. 2010, 89, 2083–2091. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a peptide-based vaccine against Shigella sonnei: A subtractive reverse vaccinology based approach. Biologicals 2017, 50, 87–99. [Google Scholar] [CrossRef]
- Saadi, M.; Karkhah, A.; Nouri, H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017, 51, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Shahid, F.; Ashfaq, U.A.; Javaid, A.; Khalid, H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect. Genet. Evol. 2020, 80, 104199. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Khalid, H.; Ashfaq, U.A. Exploring HCV genome to construct multi-epitope based subunit vaccine to battle HCV infection: Immunoinformatics based approach. J. Biomed. Inform. 2020, 108, 103498. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Rehman, A.; Ashfaq, U.A.; Awan, M.Q.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Ahmad, S.; Shahid, F.; Tahir ul Qamar, M.; Abbasi, S.W.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A.; Ullah Saeed, H.F. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef]
- Yadav, S.; Prakash, J.; Shukla, H.; Das, K.C.; Tripathi, T.; Dubey, V.K. Design of a multi-epitope subunit vaccine for immune-protection against Leishmania parasite. Pathog. Glob. Health 2020, 114, 471–481. [Google Scholar] [CrossRef]
- Kar, P.P.; Srivastava, A. Immuno-informatics analysis to identify novel vaccine candidates and design of a multi-epitope based vaccine candidate against Theileria parasites. Front. Immunol. 2018, 9, 2213. [Google Scholar] [CrossRef] [PubMed]
- Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahbar, M.R.; Ghasemi, Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol. Biosyst. 2017, 13, 699–713. [Google Scholar] [CrossRef]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Mamede, L.D.; de Paula, K.G.; de Oliveira, B.; Dos Santos, J.S.C.; Cunha, L.M.; Junior, M.C.; Jung, L.R.C.; Taranto, A.G.; de Oliveira Lopes, D.; Leclercq, S.Y. Reverse and structural vaccinology approach to design a highly immunogenic multi-epitope subunit vaccine against Streptococcus pneumoniae infection. Infect. Genet. Evol. 2020, 85, 104473. [Google Scholar] [CrossRef]
- Mahmood, M.; Javaid, A.; Shahid, F.; Ashfaq, U.A. Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect. Genet. Evol. 2021, 91, 104795. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Rahman, N.; Ajmal, A.; Ali, F.; Rastrelli, L. Core proteome mediated therapeutic target mining and multi-epitope vaccine design for Helicobacter pylori. Genomics 2020, 112, 3473–3483. [Google Scholar] [CrossRef]
- Solanki, V.; Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Madden, T. The BLAST sequence analysis tool. In The NCBI Handbook [Internet], 2nd ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2013. [Google Scholar]
- Shenoy, P.J.; Vin, H.M. Cello: A disk scheduling framework for next generation operating systems. Acm. Sigmetrics Perform. Eval. Rev. 1998, 26, 44–55. [Google Scholar] [CrossRef]
- Deng, W.; Nickle, D.C.; Learn, G.H.; Maust, B.; Mullins, J.I. ViroBLAST: A stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics 2007, 23, 2334–2336. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI–multi FASTA ProtParam interface. Bioinformation 2016, 12, 74. [Google Scholar] [CrossRef]
- Ahmad, B.; Ashfaq, U.A.; Rahman, M.-u.; Masoud, M.S.; Yousaf, M.Z. Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: An immuno-informatics approach. Microb. Pathog. 2019, 132, 243–253. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.; Consortium, O.S.D.D. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Zaharieva, N.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. VaxiJen dataset of bacterial immunogens: An update. Curr. Comput. Aided Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. BloodJ. Am. Soc. Hematol. 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Rishi, V.L.; Rui, Z.; Asha, D.V.; Bing, X. CD4⁺ T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 2013, 8, 1–15. [Google Scholar] [CrossRef]
- Karkhah, A.; Saadi, M.; Nouri, H.R. In silico analyses of heat shock protein 60 and calreticulin to designing a novel vaccine shifting immune response toward T helper 2 in atherosclerosis. Comput. Biol. Chem. 2017, 67, 244–254. [Google Scholar] [CrossRef]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.K.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, 1–10. [Google Scholar]
- Cooper, M.D. The early history of B cells. Nat. Rev. Immunol. 2015, 15, 191–197. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.K.; Rahman, M.M. Overlapping CD8+ and CD4+ T-cell epitopes identification for the progression of epitope-based peptide vaccine from nucleocapsid and glycoprotein of emerging Rift Valley fever virus using immunoinformatics approach. Infect. Genet. Evol. 2017, 56, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Nain, Z.; Abdulla, F.; Rahman, M.M.; Karim, M.M.; Khan, M.S.A.; Sayed, S.B.; Mahmud, S.; Rahman, S.R.; Sheam, M.M.; Haque, Z. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2019, 1–18. [Google Scholar] [CrossRef]
- Ong, E.; He, Y.; Yang, Z. Epitope promiscuity and population coverage of Mycobacterium tuberculosis protein antigens in current subunit vaccines under development. Infect. Genet. Evol. 2020, 80, 104186. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. Proteom. Protoc. Handb. 2005, 571–607. [Google Scholar]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, 1–9. [Google Scholar] [CrossRef]
- Deléage, G. ALIGNSEC: Viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics 2017, 33, 3991–3992. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Lengths, M.; Angles, M. Limitations of structure evaluation tools errat. Quick Guidel. Comput. Drug Des. 2018, 16, 75. [Google Scholar]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, G.; Hempel, D.C.; Jahn, D. SnapGene Viewer. Available online: https://www.snapgene.com/snapgene-viewer/ (accessed on 12 April 2021).
- Pritam, M.; Singh, G.; Swaroop, S.; Singh, A.K.; Pandey, B.; Singh, S.P. A cutting-edge immunoinformatics approach for design of multi-epitope oral vaccine against dreadful human malaria. Int. J. Biol. Macromol. 2020, 158, 159–179. [Google Scholar] [CrossRef]
- Banerjee, S.; Majumder, K.; Gutierrez, G.J.; Gupta, D.; Mittal, B. Immuno-Informatics Approach for Multi-Epitope Vaccine Designing against SARS-CoV-2. In BioRxiv; 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7386484/ (accessed on 22 May 2021).
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883. [Google Scholar] [CrossRef]
- Ohto, U.; Yamakawa, N.; Akashi-Takamura, S.; Miyake, K.; Shimizu, T. Structural analyses of human Toll-like receptor 4 polymorphisms D299G and T399I. J. Biol. Chem. 2012, 287, 40611–40617. [Google Scholar] [CrossRef]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef]
- Kirksey, T.J.; Pogue-Caley, R.R.; Frelinger, J.A.; Collins, E.J. The structural basis for the increased immunogenicity of two HIV-reverse transcriptase peptide variant/class I major histocompatibility complexes. J. Biol. Chem. 1999, 274, 37259–37264. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Protein Crystallogr. 2017, 627–641. [Google Scholar]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. Ccp4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.v.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, Version 5.1.19. Available online: https://plasma-gate.weizmann.ac.il/Grace/ (accessed on 22 May 2021).
- Turner, P.; McLennan, A.; Bates, A.; White, M. BIOS Instant Notes in Molecular Biology; Taylor & Francis Group: New York, NY, USA, 2007. [Google Scholar]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Azam, S.S.; Shamim, A. An insight into the exploration of druggable genome of Streptococcus gordonii for the identification of novel therapeutic candidates. Genomics 2014, 104, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a multi-epitope vaccine against Mycobacteroides abscessus by pangenome-reverse vaccinology. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Samson, O.O.; Olaleka, F.S.; Ofelia, O.G. Selection of T cell epitopes from S. mansoni Sm23 protein as a vaccine construct, using Immunoinformatics approach. J. Comput. Biol. Bioinform. Res. 2018, 8, 1–11. [Google Scholar]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef]
- ul Ain, Q.; Ahmad, S.; Azam, S.S. Subtractive proteomics and immunoinformatics revealed novel B-cell derived T-cell epitopes against Yersinia enterocolitica: An etiological agent of Yersiniosis. Microb. Pathog. 2018, 125, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; Biddison, W.E.; Klausner, R.D. Class II MHC molecules can use the endogenous pathway of antigen presentation. Nature 1990, 343, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N. Immunology: The ins and outs of antigen processing and presentations. Nature 1986, 322, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.A.; Burrows, S.R.; Misko, I.S.; Moss, D.J.; Coupar, B.E.; Khanna, R. Targeting a polyepitope protein incorporating multiple class II-restricted viral epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: A novel approach to vaccine design. J. Virol. 1998, 72, 2246–2252. [Google Scholar] [CrossRef]
- Ivory, C.; Chadee, K. DNA vaccines: Designing strategies against parasitic infections. Genet. Vaccines Ther. 2004, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef] [PubMed]
- Kringelum, J.V.; Nielsen, M.; Padkjær, S.B.; Lund, O. Structural analysis of B-cell epitopes in antibody: Protein complexes. Mol. Immunol. 2013, 53, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Coler, R.N.; Baldwin, S.L.; Shaverdian, N.; Bertholet, S.; Reed, S.J.; Raman, V.S.; Lu, X.; DeVos, J.; Hancock, K.; Katz, J.M. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS ONE 2010, 5, e13677. [Google Scholar] [CrossRef] [PubMed]
- Onile, O.S.; Fadahunsi, A.I.; Adekunle, A.A.; Oyeyemi, B.F.; Anumudu, C.I. An immunoinformatics approach for the design of a multi-epitope subunit vaccine for urogenital schistosomiasis. PeerJ 2020, 8, e8795. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.A.; Ami, J.Q.; Faisal, K.; Chowdhury, R.; Ghosh, P.; Hossain, F.; Abd El Wahed, A.; Mondal, D. An immunoinformatic approach driven by experimental proteomics: In silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 2020, 13, 1–21. [Google Scholar] [CrossRef]
- Tarang, S.; Kesherwani, V.; LaTendresse, B.; Lindgren, L.; Rocha-Sanchez, S.M.; Weston, M.D. In silico design of a multivalent vaccine against Candida albicans. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef]
- Abbas, G.; Zafar, I.; Ahmad, S.; Azam, S.S. Immunoinformatics design of a novel multi-epitope peptide vaccine to combat multi-drug resistant infections caused by Vibrio vulnificus. Eur. J. Pharm. Sci. 2020, 142, 105160. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Durdagi, S.; ul Qamar, M.T.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Corradin, G.; Villard, V.; Kajava, A.V. Protein structure based strategies for antigen discovery and vaccine development against malaria and other pathogens. Endocr. Metab. Immune Disord. Drug Targets (Former. Curr. Drug Targets ImmuneEndocr. Metab. Disord.) 2007, 7, 259–265. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Messaoudi, A.; Belguith, H.; Hamida, J.B. Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theor. Biol. Med. Model. 2013, 10, 1–10. [Google Scholar] [CrossRef]
- Monath, T.P. Flaviviruses; Army Medical Research Inst Of Infectious Diseases Fort Detrick Md: Frederick, MD, USA, 1990. [Google Scholar]
- Gori, A.; Longhi, R.; Peri, C.; Colombo, G. Peptides for immunological purposes: Design, strategies and applications. Amino Acids 2013, 45, 257–268. [Google Scholar] [CrossRef]
- Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.; Bhattacharya, M.; Sharma, G.; Agoramoorthy, G.; Lee, S. Diabetes and COVID-19: A major challenge in pandemic period? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11409–11420. [Google Scholar] [PubMed]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef]
- Sanches, R.C.; Tiwari, S.; Ferreira, L.C.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.; Maia, E.H.; Taranto, A.G.; Kato, R.; Azevedo, V.A. Immunoinformatics design of multi-epitope peptide-based vaccine against Schistosoma mansoni using transmembrane proteins as a target. Front. Immunol. 2021, 12, 490. [Google Scholar] [CrossRef]
- Rahmani, A.; Baee, M.; Rostamtabar, M.; Karkhah, A.; Alizadeh, S.; Tourani, M.; Nouri, H.R. Development of a conserved chimeric vaccine based on helper T-cell and CTL epitopes for induction of strong immune response against Schistosoma mansoni using immunoinformatics approaches. Int. J. Biol. Macromol. 2019, 141, 125–136. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No | Protein Name | Subcellular Localization | Antigenicity | TMHMM Helices |
|---|---|---|---|---|
| TNN18811.1 | Calcium binding | Nuclear | 0.6922 | 0 |
| TNN05631.1 | Mycosubtilin synthase subunit C | Nuclear | 0.5741 | 0 |
| CTL Epitopes | Protein | Position | Corresponding Alleles | Antigenicity | Immunogenicity |
|---|---|---|---|---|---|
| IPDHLEGDI | Calcium binding | 32–40 | HLA-C*08:02 | 1.3173 | 0.16476 |
| HLA-B*51:01 | |||||
| HLA-B*53:01 | |||||
| FSPRRYRKL | Calcium binding | 152–160 | HLA-B*14:02 | 1.0381 | 0.01308 |
| HLA-C*07:02 | |||||
| HLA-E*01:01 | |||||
| HLA-B*08:01 | |||||
| HLA-C*06:02 | |||||
| EVEAVIEAY | Calcium binding | 285–293 | HLA-A*26:01 | 0.7113 | 0.34471 |
| HLA-A*25:01 | |||||
| HLA-A*01:01 | |||||
| HLA-B*35:01 | |||||
| FMAVFSHYI | Mycosubtilin synthase subunit C | 373–381 | HLA-A*02:01 | 1.1695 | 0.03765 |
| HLA-A*02:06 | |||||
| HLA-A*68:02 | |||||
| HLA-A*29:02 | |||||
| HLA-B*39:01 | |||||
| HLA-A*32:01 | |||||
| HLA-A*24:02 | |||||
| HLA-B*46:01 | |||||
| RSRERARKV | Mycosubtilin synthase subunit C | 463–471 | HLA-C*15:02 | 1.9596 | 0.12246 |
| HLA-C*06:02 | |||||
| HLA-A*30:01 | |||||
| HLA-C*07:01 | |||||
| RQLGFNVNL | Mycosubtilin synthase subunit C | 514–522 | HLA-B*48:01 | 1.5715 | 0.16947 |
| HLA-A*32:01 | |||||
| HLA-B*40:01 | |||||
| HLA-A*02:06 | |||||
| HLA-B*27:05 | |||||
| HLA-B*39:01 | |||||
| HLA-B*40:02 | |||||
| YENPYEHTF | Mycosubtilin synthase subunit C | 669–677 | HLA-B*18:01 | 1.1243 | 0.12737 |
| HLA-B*44:02 | |||||
| HLA-B*40:01 | |||||
| HLA-C*07:02 | |||||
| HLA-B*40:02 | |||||
| HLA-B*40:02 | |||||
| HLA-B*38:01 | |||||
| HLA-A*23:01 |
| HTL Epitopes | Protein | Position | Alleles | Antigenicity | IL4/IL10 | IFN |
|---|---|---|---|---|---|---|
| QDNRLLRLSKNKKSK | Calcium binding | 333–347 | HLA-DRB1*04:26 | 1.1906 | Inducer | Positive |
| HLA-DRB1*11:01 | ||||||
| HLA-DRB1*04:21 | ||||||
| HLA-DRB5*01:01 | ||||||
| HLA-DRB1*04:02 | ||||||
| RNFKLIRSRERARKV | Mycosubtilin synthase subunit C | 457–471 | HLA-DRB5*01:01 | 1.0836 | Inducer | Positive |
| HLA-DRB5*01:05 | ||||||
| HLA-DRB1*08:04 | ||||||
| HLA-DRB1*11:01 | ||||||
| HLA-DRB1*08:13 | ||||||
| HLA-DRB1*08:06 | ||||||
| NKLVGVLISLPAKHV | Mycosubtilin synthase subunit C | 680–694 | HLA-DRB1*01:01 | 0.8180 | Inducer | Positive |
| HLA-DRB1*04:04 | ||||||
| HLA-DRB1*09:01 | ||||||
| HLA-DRB5*01:01 | ||||||
| HLA-DRB1*15:01 | ||||||
| HLA-DRB1*12:01 |
| Peptide | Protein | Position | Score | Antigenicity | Immunogenicity |
|---|---|---|---|---|---|
| FIPDYVEDDLDGNG | Calcium binding | 358–371 | 0.89 | 1.6532 | 0.23092 |
| DCDDDDDDDDGILD | Calcium binding | 537–550 | 0.61 | 1.7767 | 0.29686 |
| DVTGIVFHNELDVK | Mycosubtilin synthase subunit C | 96–109 | 0.84 | 1.1856 | 0.4856 |
| LTEVIESYLNAHKY | Mycosubtilin synthase subunit C | 644–657 | 0.66 | 0.8240 | 0.05747 |
| Docking Statistics | MEV-TLR2 | MEV-TLR4 | MEV-MHC I | MEV-MHC II |
|---|---|---|---|---|
| Cluster size | 6 | 7 | 18 | 22 |
| HADDOCK score | 170.5 ± 17.0 | 84.7 ± 38.6 | 73.8 ± 18.3 | 86.3 ± 33.0 |
| RMSD from the overall Lowest Energy Structure | 13.2 ± 0.1 | 1.0 ± 0.8 | 3.8 ± 0.2 | 0.9 ± 0.6 |
| Restraints violation energy | 3336.6 ± 113.8 | 3416.1 ± 411.3 | 3071.8 ± 126.1 | 3073.3 ± 188.1 |
| Electrostatic energy | −388.8 ± 78.9 | −484.1 ± 54.5 | −527.2 ± 80.2 | −325.9 ± 41.6 |
| Van der Waals energy | −67.4 ± 7.5 | −97.6 ± 3.9 | −96.0 ± 13.9 | −104.6 ± 11.5 |
| Buried surface area | 2914.2 ± 152.2 | 3658.5 ± 91.0 | 4338.3 ± 271.5 | 4135.9 ± 163.5 |
| De-solvation energy | −18.0 ± 5.2 | −62.5 ± 9.6 | −32.0 ± 2.5 | −51.2 ± 5.2 |
| Z-score | −1.7 | −1.8 | −1.0 | −1.7 |
| Energies | MEV-TLR2 (kcal/mol) | MEV-TLR4 (kcal/mol) | MEV-MHC I (kcal/mol) | MEV-MHC II (kcal/mol) |
|---|---|---|---|---|
| vdW | −69.19 | −59.77 | −91.74 | −85.01 |
| Ele | −29.07 | −45.00 | −59.12 | −44.11 |
| Polar solvation | 46.68 | 51.2 | 33.22 | 44.7 |
| Non polar solvation | −34.66 | −37.9 | −31.61 | −39.71 |
| ∆Gas | −98.26 | −104.77 | −150.86 | −129.12 |
| ∆Solvation | 12.02 | 13.3 | 1.61 | 4.99 |
| ∆total | −86.24 | −91.47 | −149.25 | −124.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. https://doi.org/10.3390/vaccines9060658
Rehman A, Ahmad S, Shahid F, Albutti A, Alwashmi ASS, Aljasir MA, Alhumeed N, Qasim M, Ashfaq UA, Tahir ul Qamar M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines. 2021; 9(6):658. https://doi.org/10.3390/vaccines9060658
Chicago/Turabian StyleRehman, Abdur, Sajjad Ahmad, Farah Shahid, Aqel Albutti, Ameen S. S. Alwashmi, Mohammad Abdullah Aljasir, Naif Alhumeed, Muhammad Qasim, Usman Ali Ashfaq, and Muhammad Tahir ul Qamar. 2021. "Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis" Vaccines 9, no. 6: 658. https://doi.org/10.3390/vaccines9060658
APA StyleRehman, A., Ahmad, S., Shahid, F., Albutti, A., Alwashmi, A. S. S., Aljasir, M. A., Alhumeed, N., Qasim, M., Ashfaq, U. A., & Tahir ul Qamar, M. (2021). Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines, 9(6), 658. https://doi.org/10.3390/vaccines9060658