
Nano-Microparticle Platforms in Developing Next-Generation Vaccines

 ,
,  ,
, 

Abstract
1. Introduction
2. Classical Vaccines
3. Nucleic Acid Vaccines
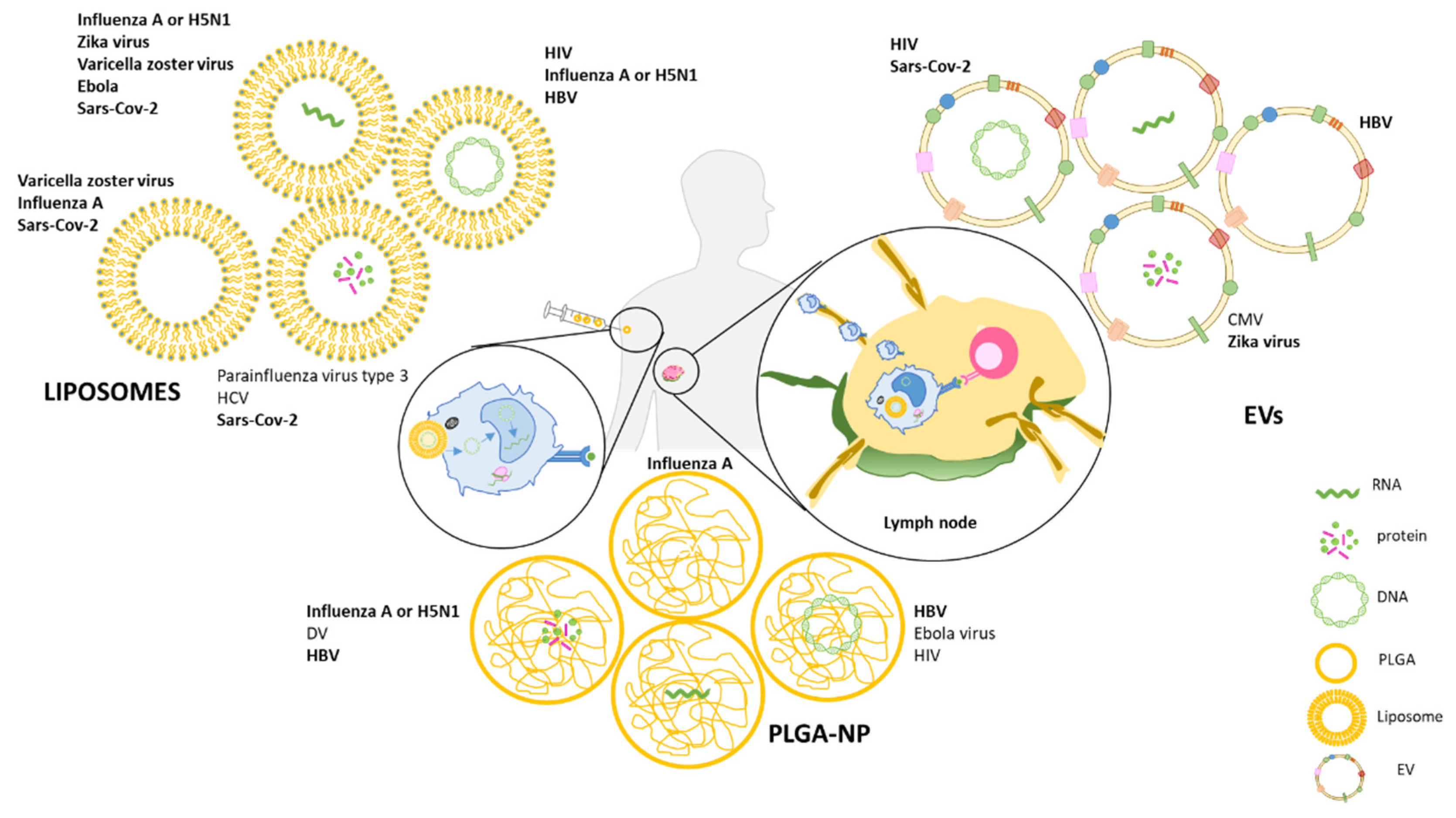
4. Next-Generation Vaccines Based on Nano/Microparticle Delivery Systems
4.1. PLGA
4.1.1. Protein—Based PLGA Viral Vaccines
4.1.2. PLGA in DNA Vaccines
4.1.3. PLGA in mRNA Vaccines
4.1.4. PLGA as Adjuvant in Vaccine Formulations
4.1.5. PLGA NP in Inverse Vaccination
4.2. Liposomes
4.2.1. Protein-Based Liposome Vaccines
4.2.2. Liposomes in DNA Vaccines
4.2.3. Liposomes in mRNA-Based Vaccines
4.2.4. Liposomes as Adjuvants in Vaccine Formulations
4.2.5. Liposomes in Inverse Vaccination
4.3. EVs as Delivery Systems in Vaccines
4.3.1. Protein-Based EV Vaccines
4.3.2. EVs in DNA-Based vaccines
4.3.3. EVs as Adjuvants in Vaccine Formulation
4.4. Limitations of Nano/Microparticle-Based Vaccines
5. COVID-19 Vaccines Based on Nano/Microparticle Platforms
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owen, J.A.; Punt, J.; Stranford, S.A.; Jones, P.P.; Kuby, J. Kuby Immunology, 7th ed.; W.H. Freeman: New York, NY, USA, 2013. [Google Scholar]
- Peng, J.; Thakur, A.; Zhang, S.; Dong, Y.; Wang, X.; Yuan, R.; Zhang, K.; Guo, X. Expressions of MiR-181a and MiR-20a in RPMI8226 Cell Line and Their Potential as Biomarkers for Multiple Myeloma. Tumor Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H.; Lee, A.; Santin, E. Microbiome and Pathogen Interaction with the Immune System. Poult. Sci. 2020, 99. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive Immunity. J. Allergy Clin. Immunol. 2010, 125. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like Receptor Control of the Adaptive Immune Responses. Nat. Immunol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.M.; Metkar, S.S.; Froelich, C.J. Cytotoxic Granule-Mediated Apoptosis: Unraveling the Complex Mechanism. Curr. Opin. Immunol. 2003, 15. [Google Scholar] [CrossRef]
- Pradeu, T.; Du Pasquier, L. Immunological Memory: What’s in a Name? Immunol. Rev. 2018, 283. [Google Scholar] [CrossRef]
- Loomis, R.J.; Johnson, P.R. Emerging Vaccine Technologies. Vaccines 2015, 3, 429–447. [Google Scholar] [CrossRef]
- Willis, N.J. Edward Jenner and the Eradication of Smallpox. Scott. Med. J. 1997. [Google Scholar] [CrossRef]
- Berche, P. Louis Pasteur, from Crystals of Life to Vaccination. Clin. Microbiol. Infect. 2012. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Principles of Vaccination. In Pink Book Webinar Series; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020; Chapter 1; pp. 1–7. [Google Scholar]
- Mok, D.Z.L.; Chan, K.R. The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses 2020, 12, 520. [Google Scholar] [CrossRef]
- World Health Organization. Module 2: Types of vaccines and adverse reactions. In Vaccine Safety Basics Learning Manual; World Health Organization: Geneva, Switzerland, 2013; pp. 38–60. [Google Scholar]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major Findings and Recent Advances in Virus–like Particle (VLP)-Based Vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Syomin, B.V.; Ilyin, Y.V. Virus-Like Particles as an Instrument of Vaccine Production. Mol. Biol. 2019, 53. [Google Scholar] [CrossRef]
- Foged, C.; Hansen, J.; Agger, E.M. License to Kill: Formulation Requirements for Optimal Priming of CD8 + CTL Responses with Particulate Vaccine Delivery Systems. Eur. J. Pharm. Sci. 2012, 45. [Google Scholar] [CrossRef]
- Park, W.H.; Schroder, M.C. Diphtheria Toxin-Antitoxin and Toxoid. Am. J. Public Health Nations Health 1932, 22, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, T.R. The Mechanisms of Action of Vaccines Containing Aluminum Adjuvants: An in Vitro vs in Vivo Paradigm. Springerplus 2015, 4. [Google Scholar] [CrossRef]
- Glenny, A.T.; Buttle, G.A.H.; Stevens, M.F. Rate of Disappearance of Diphtheria Toxoid Injected into Rabbits and Guinea—Pigs: Toxoid Precipitated with Alum. J. Pathol. Bacteriol. 1931, 34, 267–275. [Google Scholar] [CrossRef]
- Ko, E.J.; Kang, S.M. Immunology and Efficacy of MF59-Adjuvanted Vaccines. Hum. Vaccines Immunother. 2018, 14. [Google Scholar] [CrossRef]
- Campbell, J.D. Development of the CpG Adjuvant 1018: A Case Study. Methods Mol. Biol. 2017, 1494. [Google Scholar] [CrossRef]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Donnelly, J.J.; Ulmer, J.B.; Liu, M.A. DNA Vaccines. Dev. Biol. Stand. 1997, 15, 617–648. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and MRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Robinson, H.L.; Pertmer, T.M. DNA Vaccines for Viral Infections: Basic Studies and Applications. Adv. Virus Res. 2000, 55. [Google Scholar] [CrossRef]
- Sheets, R.L.; Stein, J.; Manetz, T.S.; Duffy, C.; Nason, M.; Andrews, C.; Kong, W.P.; Nabel, G.J.; Gomez, P.L. Biodistribution of DNA Plasmid Vaccines against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile Virus Is Similar, without Integration, despite Differing Plasmid Backbones or Gene Inserts. Toxicol. Sci. 2006, 91, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ledwith, B.J.; Manam, S.; Troilo, P.J.; Barnum, A.B.; Pauley, C.J.; Griffiths, T.G.; Harper, L.B.; Beare, C.M.; Bagdon, W.J.; Nichols, W.W. Plasmid DNA Vaccines: Investigation of Integration into Host Cellular DNA Following Intramuscular Injection in Mice. Intervirology 2000, 43. [Google Scholar] [CrossRef]
- Houston, R.; Moxon, S.; Nogué, F.; Papadopoulou, N.; Ramon, M.; Waigmann, E. Assessment of the Potential Integration of the DNA Plasmid Vaccine CLYNAV into the Salmon Genome. EFSA J. 2017, 15, 1–15. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines-a New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17. [Google Scholar] [CrossRef]
- Forni, G.; Mantovani, A.; Forni, G.; Mantovani, A.; Moretta, L.; Rappuoli, R.; Rezza, G.; Bagnasco, A.; Barsacchi, G.; Bussolati, G.; et al. COVID-19 Vaccines: Where We Stand and Challenges Ahead. Cell Death Differ. 2021, 28. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-Amplifying RNA Vaccines for Infectious Diseases. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-Amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, C.; O’hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of Action of MRNA-Based Vaccines. Expert Rev. Vaccines 2017, 16. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. In Current Topics in Microbiology and Immunology; Springer: Berlin, Germany, 2020; pp. 1–40. [Google Scholar]
- Sun, B.; Wang, X.; Ji, Z.; Li, R.; Xia, T. NLRP3 Inflammasome Activation Induced by Engineered Nanomaterials. Small 2013, 9. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1Β-Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current Advances in Research and Clinical Applications of PLGA-Based Nanotechnology. Expert Rev. Mol. Diagn. 2009, 9. [Google Scholar] [CrossRef]
- Allahyari, M.; Mohit, E. Peptide/Protein Vaccine Delivery System Based on PLGA Particles. Hum. Vaccines Immunother. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, Long-Acting PLGA Formulations: Analyzing PLGA and Understanding Microparticle Formation. J. Control. Release 2019, 304. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Control. Release 2012, 161. [Google Scholar] [CrossRef]
- Cui, F.-d.; Tao, A.-j.; Cun, D.-m.; Zhang, L.-q.; Shi, K. Preparation of Insulin Loaded PLGA-Hp55 Nanoparticles for Oral Delivery. J. Pharm. Sci. 2007, 96. [Google Scholar] [CrossRef]
- Tamura, T.; Kita, T.; Nakagawa, T.; Endo, T.; Kim, T.S.; Ishihara, T.; Mizushima, Y.; Higaki, M.; Ito, J. Drug Delivery to the Cochlea Using PLGA Nanoparticles. Laryngoscope 2005, 115. [Google Scholar] [CrossRef]
- Higaki, M.; Ishihara, T.; Izumo, N.; Takatsu, M.; Mizushima, Y. Treatment of Experimental Arthritis with Poly(D, L-Lactic/Glycolic Acid) Nanoparticles Encapsulating Betamethasone Sodium Phosphate. Ann. Rheum. Dis. 2005, 64. [Google Scholar] [CrossRef]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Maincent, P.; Kawashima, Y.; Lehr, C.M. Biodegradable Nanoparticles for Targeted Drug Delivery in Treatment of Inflammatory Bowel Disease. J. Pharmacol. Exp. Ther. 2001, 299, 775–781. [Google Scholar]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous Inverse Vaccination with PLGA Particles Loaded with a MOG Peptide and IL-10 Decreases the Severity of Experimental Autoimmune Encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles Target Distinct Dendritic Cell Populations According to Their Size. Eur. J. Immunol. 2008, 38. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Mutation and Epistasis in Influenza Virus Evolution. Viruses 2018, 10, 407. [Google Scholar] [CrossRef]
- Tukhvatulin, A.; Dzharullaeva, A.; Erokhova, A.; Zemskaya, A.; Balyasin, M.; Ozharovskaia, T.; Zubkova, O.; Shevlyagina, N.; Zhukhovitsky, V.; Fedyakina, I.; et al. Adjuvantation of an Influenza Hemagglutinin Antigen with Tlr4 and Nod2 Agonists Encapsulated in Poly(D,l-Lactide-Co-Glycolide) Nanoparticles Enhances Immunogenicity and Protection against Lethal Influenza Virus Infection in Mice. Vaccines 2020, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-s.; Li, I.-H.; Hong, P.-d.; Yeh, M.-k. Evaluation of Protective Efficacy Using a Nonstructural Protein NS1 in DNA Vaccine- Loaded Microspheres against Dengue 2 Virus. Int. J. Nanomed. 2013, 8. [Google Scholar] [CrossRef]
- Metz, S.W.; Thomas, A.; Brackbill, A.; Xianwen, Y.; Stone, M.; Horvath, K.; Miley, M.J.; Luft, C.; DeSimone, J.M.; Tian, S.; et al. Nanoparticle Delivery of a Tetravalent E Protein Subunit Vaccine Induces Balanced, Type-Specific Neutralizing Antibodies to Each Dengue Virus Serotype. PLoS Negl. Trop. Dis. 2018, 12. [Google Scholar] [CrossRef]
- Zhu, J.; Qin, F.; Ji, Z.; Fei, W.; Tan, Z.; Hu, Y.; Zheng, C. Mannose-Modified PLGA Nanoparticles for Sustained and Targeted Delivery in Hepatitis B Virus Immunoprophylaxis. AAPS PharmSciTech 2020, 21. [Google Scholar] [CrossRef]
- Thomas, C.; Gupta, V.; Ahsan, F. Influence of Surface Charge of PLGA Particles of Recombinant Hepatitis B Surface Antigen in Enhancing Systemic and Mucosal Immune Responses. Int. J. Pharm. 2009, 379. [Google Scholar] [CrossRef]
- Chong, C.S.W.; Cao, M.; Wong, W.W.; Fischer, K.P.; Addison, W.R.; Kwon, G.S.; Tyrrell, D.L.; Samuel, J. Enhancement of T Helper Type 1 Immune Responses against Hepatitis B Virus Core Antigen by PLGA Nanoparticle Vaccine Delivery. J. Control. Release 2005, 102. [Google Scholar] [CrossRef]
- Roopngam, P.; Liu, K.; Mei, L.; Zheng, Y.; Zhu, X.; Tsai, H.I.; Huang, L. Hepatitis C Virus E2 Protein Encapsulation into Poly D, L-Lactic-Co-Glycolide Microspheres Could Induce Mice Cytotoxic T-Cell Response. Int. J. Nanomed. 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, M.; Sankian, M.; Sadri, K.; Tafaghodi, M. Robust Mucosal and Systemic Responses against HTLV-1 by Delivery of Multi-Epitope Vaccine in PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2018, 133. [Google Scholar] [CrossRef]
- Roy, M.J.; Wu, M.S.; Barr, L.J.; Fuller, J.T.; Tussey, L.G.; Speller, S.; Culp, J.; Burkholder, J.K.; Swain, W.F.; Dixon, R.M.; et al. Induction of Antigen-Specific CD8+ T Cells, T Helper Cells, and Protective Levels of Antibody in Humans by Particle-Mediated Administration of a Hepatitis B Virus DNA Vaccine. Vaccine 2000, 19. [Google Scholar] [CrossRef]
- He, X.W.; Wang, F.; Jiang, L.; Li, J.; Liu, S.K.; Xiao, Z.Y.; Jin, X.Q.; Zhang, Y.N.; He, Y.; Li, K.; et al. Induction of Mucosal and Systemic Immune Response by Single-Dose Oral Immunization with Biodegradable Microparticles Containing DNA Encoding HBsAg. J. Gen. Virol. 2005, 86. [Google Scholar] [CrossRef]
- Yang, H.W.; Ye, L.; Guo, X.D.; Yang, C.; Compans, R.W.; Prausnitz, M.R. Ebola Vaccination Using a DNA Vaccine Coated on PLGA-PLL/ΓPGA Nanoparticles Administered Using a Microneedle Patch. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, B.; Yu, X.; Zha, X.; Zhang, X.; Wang, X.; Jin, Y.; Wu, Y.; Chen, Y.; Shan, Y.; et al. Controlled Release of PEI/DNA Complexes from PLGA Microspheres as a Potent Delivery System to Enhance Immune Response to HIV Vaccine DNA Prime/MVA Boost Regime. Eur. J. Pharm. Biopharm. 2008, 68. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, E.M.; Tiboni, M.; Soliman, M.E. Biocompatibility, Biodegradation and Biomedical Applications of Poly(Lactic Acid)/Poly(Lactic-Co-Glycolic Acid) Micro and Nanoparticles. J. Pharm. Investig. 2019, 49. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of Mrna-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef]
- Sharifnia, Z.; Bandehpour, M.; Hamishehkar, H.; Mosaffa, N.; Kazemi, B.; Zarghami, N. In-Vitro Transcribed Mrna Delivery Using Plga/Pei Nanoparticles into Human Monocyte-Derived Dendritic Cells. Iran. J. Pharm. Res. 2019, 18. [Google Scholar] [CrossRef]
- Seth, A.; Ritchie, F.K.; Wibowo, N.; Lua, L.H.L.; Middelberg, A.P.J. Non-Carrier Nanoparticles Adjuvant Modular Protein Vaccine in a Particle-Dependent Manner. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Yang, T.; Liu, Y.; Chen, X.; Liu, Q.; Jia, J.; Ma, G. Immunopotentiator-Loaded Polymeric Microparticles as Robust Adjuvant to Improve Vaccine Efficacy. Pharm. Res. 2015, 32. [Google Scholar] [CrossRef]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells in Vivo. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Waldmann, H.; Adams, E.; Fairchild, P.J.; Cobbold, S. Infectious Tolerance and the Long-Term Acceptance of Transplanted Tissue. Immunol. Rev. 2006, 212. [Google Scholar] [CrossRef]
- Capurso, N.A.; Look, M.; Jeanbart, L.; Nowyhed, H.; Abraham, C.; Craft, J.; Fahmy, T.M. Development of a Nanoparticulate Formulation of Retinoic Acid That Suppresses Th17 Cells and Upregulates Regulatory T Cells. Self Nonself Immune Recognit. Signal. 2010, 1. [Google Scholar] [CrossRef]
- Li, P.Y.; Bearoff, F.; Zhu, P.; Fan, Z.; Zhu, Y.; Fan, M.; Cort, L.; Kambayashi, T.; Blankenhorn, E.P.; Cheng, H. PEGylation Enables Subcutaneously Administered Nanoparticles to Induce Antigen-Specific Immune Tolerance. J. Control. Release 2021, 331. [Google Scholar] [CrossRef]
- Zhu, P.; Li, X.Y.; Wang, H.K.; Jia, J.F.; Zheng, Z.H.; Ding, J.; Fan, C.M. Oral Administration of Type-II Collagen Peptide 250-270 Suppresses Specific Cellular and Humoral Immune Response in Collagen-Induced Arthritis. Clin. Immunol. 2007, 122. [Google Scholar] [CrossRef]
- Keijzer, C.; Slütter, B.; van der Zee, R.; Jiskoot, W.; van Eden, W.; Broere, F. PLGA, PLGA-TMC and TMC-TPP Nanoparticles Differentially Modulate the Outcome of Nasal Vaccination by Inducing Tolerance or Enhancing Humoral Immunity. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Gregoriadis, G. Immunological Adjuvants: A Role for Liposomes. Immunol. Today 1990, 11. [Google Scholar] [CrossRef]
- Frézard, F. Liposomes: From Biophysics to the Design of Peptide Vaccines. Brazilian J. Med. Biol. Res. 1999, 32. [Google Scholar] [CrossRef]
- Tanaka, Y.; Taneichi, M.; Kasai, M.; Kakiuchi, T.; Uchida, T. Liposome-Coupled Antigens Are Internalized by Antigen-Presenting Cells via Pinocytosis and Cross- Presented to CD8+ T Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Senchi, K.; Matsunaga, S.; Hasegawa, H.; Kimura, H.; Ryo, A. Development of Oligomannose-Coated Liposome-Based Nasal Vaccine against Human Parainfluenza Virus Type 3. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Filskov, J.; Mikkelsen, M.; Hansen, P.R.; Christensen, J.P.; Thomsen, A.R.; Andersen, P.; Bukh, J.; Agger, E.M. Broadening CD4+ and CD8+ T Cell Responses against Hepatitis C Virus by Vaccination with NS3 Overlapping Peptide Panels in Cross-Priming Liposomes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Kobayashi, N.; Taneichi, M.; Uchida, T.; Akatsuka, T. Coupling to the Surface of Liposomes Alters the Immunogenicity of Hepatitis C Virus-Derived Peptides and Confers Sterile Immunity. Biochem. Biophys. Res. Commun. 2013, 430. [Google Scholar] [CrossRef]
- Ohno, S.; Kohyama, S.; Taneichi, M.; Moriya, O.; Hayashi, H.; Oda, H.; Mori, M.; Kobayashi, A.; Akatsuka, T.; Uchida, T.; et al. Synthetic Peptides Coupled to the Surface of Liposomes Effectively Induce SARS Coronavirus-Specific Cytotoxic T Lymphocytes and Viral Clearance in HLA-A*0201 Transgenic Mice. Vaccine 2009, 27. [Google Scholar] [CrossRef]
- Qiao, C.; Liu, J.; Yang, J.; Li, Y.; Weng, J.; Shao, Y.; Zhang, X. Enhanced Non-Inflammasome Mediated Immune Responses by Mannosylated Zwitterionic-Based Cationic Liposomes for HIV DNA Vaccines. Biomaterials 2016, 85. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.E.; Zamorano, P.; Wilkowsky, S.; Torrá, F.; Ferreri, L.; Dominguez, M.; Florin-Christensen, M. Delivery of Recombinant Vaccines against Bovine Herpesvirus Type 1 GD and Babesia Bovis MSA-2c to Mice Using Liposomes Derived from Egg Yolk Lipids. Vet. J. 2013, 196. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Wang, B.; Zeng, S.; Qi, F.; Lu, C.; Kimura, Y.; Liu, B. Oral Vaccination with a Liposome-Encapsulated Influenza DNA Vaccine Protects Mice against Respiratory Challenge Infection. J. Med. Virol. 2014, 86. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Guo, L.; Zhang, S.; Xu, B.; Gao, Y.; Hu, Y.; Hou, J.; Bai, B.; Shen, H.; Mao, P. DNA-Based Vaccination against Hepatitis B Virus Using Dissolving Microneedle Arrays Adjuvanted by Cationic Liposomes and CpG ODN. Drug Deliv. 2016, 23. [Google Scholar] [CrossRef]
- Dong, L.; Liu, F.; Fairman, J.; Hong, D.K.; Lewis, D.B.; Monath, T.; Warner, J.F.; Belser, J.A.; Patel, J.; Hancock, K.; et al. Cationic Liposome-DNA Complexes (CLDC) Adjuvant Enhances the Immunogenicity and Cross-Protective Efficacy of a Pre-Pandemic Influenza A H5N1 Vaccine in Mice. Vaccine 2012, 30. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-Based Vectors for Therapeutic MRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019, 25. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jeklenec, A.; Langer, R.; Blankschtein, D. MRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Deliv. 2016, 7. [Google Scholar] [CrossRef]
- Pascolo, S. Vaccination with Messenger RNA (MRNA). Handb. Exp. Pharmacol. 2008, 183. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Translation of Rabbit Globin MRNA Introduced by Liposomes into Mouse Lymphocytes. Nature 1978, 274. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of Virus-specific Cytotoxic T Lymphocytes in Vivo by Liposome-entrapped MRNA. Eur. J. Immunol. 1993, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, Y.; Jin, Y.; Zhang, G.; Wong, J.; Sun, L.Q.; Wang, M. Prophylactic, Therapeutic and Immune Enhancement Effect of Liposome-Encapsulated PolyICLC on Highly Pathogenic H5N1 Influenza Infection. J. Gene Med. 2011, 13. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, Q.; Durnell, L.A.; et al. Intramuscular Delivery of Replicon RNA Encoding ZIKV-117 Human Monoclonal Antibody Protects against Zika Virus Infection. Mol. Ther. Methods Clin. Dev. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-Modified MRNA Immunization Elicits Influenza Virus Hemagglutinin Stalk-Specific Antibodies. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Monslow, M.A.; Elbashir, S.; Sullivan, N.L.; Thiriot, D.S.; Ahl, P.; Smith, J.; Miller, E.; Cook, J.; Cosmi, S.; Thoryk, E.; et al. Immunogenicity Generated by MRNA Vaccine Encoding VZV GE Antigen Is Comparable to Adjuvanted Subunit Vaccine and Better than Live Attenuated Vaccine in Nonhuman Primates. Vaccine 2020, 38. [Google Scholar] [CrossRef]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified MRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs from Ebola Virus Disease. J. Infect. Dis. 2018, 217. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An MRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Wui, S.R.; Kim, K.S.; Ryu, J.I.; Ko, A.; Do, H.T.T.; Lee, Y.J.; Kim, H.J.; Lim, S.J.; Park, S.A.; Cho, Y.J.; et al. Efficient Induction of Cell-Mediated Immunity to Varicella-Zoster Virus Glycoprotein E Co-Lyophilized with a Cationic Liposome-Based Adjuvant in Mice. Vaccine 2019, 37. [Google Scholar] [CrossRef] [PubMed]
- Rosenkrands, I.; Vingsbo-Lundberg, C.; Bundgaard, T.J.; Lindenstrøm, T.; Enouf, V.; van der Werf, S.; Andersen, P.; Agger, E.M. Enhanced Humoral and Cell-Mediated Immune Responses after Immunization with Trivalent Influenza Vaccine Adjuvanted with Cationic Liposomes. Vaccine 2011, 29. [Google Scholar] [CrossRef] [PubMed]
- Wørzner, K.; Sheward, D.J.; Schmidt, S.T.; Hanke, L.; Zimmermann, J.; McInerney, G.; Hedestam, G.B.K.; Murrell, B.; Christensen, D.; Pedersen, G.K. Adjuvanted SARS-CoV-2 Spike Protein Elicits Neutralizing Antibodies and CD4 T Cell Responses after a Single Immunization in Mice. EBioMedicine 2021, 63. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Arankalle, V.A. Differential Immune Responses in Mice Immunized with Recombinant Neutralizing Epitope Protein of Hepatitis e Virus Formulated with Liposome and Alum Adjuvants. Viral Immunol. 2016, 29. [Google Scholar] [CrossRef] [PubMed]
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Nowakowska, D.; Plasencia, A.; Stanton, V.P.; Sherr, D.H.; et al. Tolerogenic Nanoparticles Suppress Central Nervous System Inflammation. Proc. Natl. Acad. Sci. USA 2020, 117. [Google Scholar] [CrossRef]
- Vives-Pi, M.; Rodríguez-Fernández, S.; Pujol-Autonell, I. How Apoptotic β-Cells Direct Immune Response to Tolerance or to Autoimmune Diabetes: A Review. Apoptosis 2015, 20. [Google Scholar] [CrossRef]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine Is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ. 2016, 23. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Autonell, I.; Mansilla, M.J.; Rodriguez-Fernandez, S.; Cano-Sarabia, M.; Navarro-Barriuso, J.; Ampudia, R.M.; Rius, A.; Garcia-Jimeno, S.; Perna-Barrull, D.; Caceres, E.M.; et al. Liposome-Based Immunotherapy against Autoimmune Diseases: Therapeutic Effect on Multiple Sclerosis. Nanomedicine 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Autonell, I.; Serracant-Prat, A.; Cano-Sarabia, M.; Ampudia, R.M.; Rodriguez-Fernandez, S.; Sanchez, A.; Izquierdo, C.; Stratmann, T.; Puig-Domingo, M.; Maspoch, D.; et al. Use of Autoantigen-Loaded Phosphatidylserine-Liposomes to Arrest Autoimmunity in Type 1 Diabetes. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Benne, N.; van Duijn, J.; Lozano Vigario, F.; Leboux, R.J.T.; van Veelen, P.; Kuiper, J.; Jiskoot, W.; Slütter, B. Anionic 1,2-Distearoyl-Sn-Glycero-3-Phosphoglycerol (DSPG) Liposomes Induce Antigen-Specific Regulatory T Cells and Prevent Atherosclerosis in Mice. J. Control. Release 2018, 291. [Google Scholar] [CrossRef]
- Kawakita, A.; Shirasaki, H.; Yasutomi, M.; Tokuriki, S.; Mayumi, M.; Naiki, H.; Ohshima, Y. Immunotherapy with Oligomannose-Coated Liposomes Ameliorates Allergic Symptoms in a Murine Food Allergy Model. Allergy Eur. J. Allergy Clin. Immunol. 2012, 67. [Google Scholar] [CrossRef]
- Jesus, S.; Soares, E.; Cruz, M.T.; Borges, O. Exosomes as Adjuvants for the Recombinant Hepatitis B Antigen: First Report. Eur. J. Pharm. Biopharm. 2018, 133. [Google Scholar] [CrossRef] [PubMed]
- Sabanovic, B.; Piva, F.; Cecati, M.; Giulietti, M. Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology 2021, 10, 94. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current Knowledge on Exosome Biogenesis and Release. Cell. Mol. Life Sci. 2018, 193–208. [Google Scholar] [CrossRef]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-Infected Human Endothelial Cells Can Stimulate Allogeneic CD4 + Memory T Cells by Releasing Antigenic Exosomes. J. Immunol. 2009, 182. [Google Scholar] [CrossRef]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.K.; Gould, S.J.; Sandefur, S.; Varthakavi, V. Exosomes and HIV Gag Bud from Endosome-like Domains of the T Cell Plasma Membrane. J. Cell Biol. 2006, 172. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Freed, E.O. HIV Type 1 Gag as a Target for Antiviral Therapy. AIDS Res. Hum. Retrovir. 2012, 28. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as Drug Carriers for Cancer Therapy and Challenges Regarding Exosome Uptake. Biomed. Pharmacother. 2020, 128. [Google Scholar] [CrossRef]
- Rojas, C.; Campos-Mora, M.; Cárcamo, I.; Villalón, N.; Elhusseiny, A.; Contreras-Kallens, P.; Refisch, A.; Gálvez-Jirón, F.; Emparán, I.; Montoya-Riveros, A.; et al. T Regulatory Cells-Derived Extracellular Vesicles and Their Contribution to the Generation of Immune Tolerance. J. Leukoc. Biol. 2020, 108. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.S.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 Expression on Extracellular Vesicles Derived from CD4+CD25+Foxp3+ T Cells Contributes to Their Regulatory Function. Eur. J. Immunol. 2013, 43. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014, 41. [Google Scholar] [CrossRef]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct Exosome Stimulation of Peripheral Human T Cells Detected by ELISPOT. Eur. J. Immunol. 2006, 36. [Google Scholar] [CrossRef]
- Martins, P.; Machado, D.; Theizen, T.H.; Guarnieri, J.P.O.; Bernardes, B.G.; Gomide, G.P.; Corat, M.A.F.; Abbehausen, C.; Módena, J.L.P.; Melo, C.F.O.R.; et al. Outer Membrane Vesicles from Neisseria Meningitidis (Proteossome) Used for Nanostructured Zika Virus Vaccine Production. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-Specific CTL Activity Elicited by in Vivo Engineered Exosomes Produced through DNA Inoculation. Int. J. Nanomed. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Anticoli, S.; Manfredi, F.; Chiozzini, C.; Arenaccio, C.; Olivetta, E.; Ferrantelli, F.; Capocefalo, A.; Falcone, E.; Ruggieri, A.; Federico, M. An Exosome-Based Vaccine Platform Imparts Cytotoxic T Lymphocyte Immunity Against Viral Antigens. Biotechnol. J. 2018, 13. [Google Scholar] [CrossRef]
- Polak, K.; Greze, N.; Lachat, M.; Merle, D.; Chiumento, S.; Bertrand-Gaday, C.; Trentin, B.; Mamoun, R.Z. Extracellular Vesicle-Based Vaccine Platform Displaying Native Viral Envelope Proteins Elicits a Robust Anti-SARS-CoV-2 Response in Mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tregoning, J.S.; Brown, E.S.; Cheeseman, H.M.; Flight, K.E.; Higham, S.L.; Lemm, N.M.; Pierce, B.F.; Stirling, D.C.; Wang, Z.; Pollock, K.M. Vaccines for COVID-19. Clin. Exp. Immunol. 2020, 202. [Google Scholar] [CrossRef]
- Bohn, M.K.; Hall, A.; Sepiashvili, L.; Jung, B.; Steele, S.; Adeli, K. Pathophysiology of COVID-19: Mechanisms Underlying Disease Severity and Progression. Physiology 2020, 35. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Battaglini, D.; Pelosi, P.; Rocco, P.R.M. Multiple Organ Dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev. Respir. Med. 2020, 14. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383. [Google Scholar] [CrossRef]
- World Health Organization. Draft Landscape of COVID-19 Candidate Vaccines. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 23 April 2021).
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384. [Google Scholar] [CrossRef]
- Nojszewska, M.; Kalinowska, A.; Adamczyk-Sowa, M.; Kułakowska, A.; Bartosik-Psujek, H. COVID-19 MRNA Vaccines (Pfizer-BioNTech and Moderna) in Patients with Multiple Sclerosis: A Statement by a Working Group Convened by the Section of Multiple Sclerosis and Neuroimmunology of the Polish Neurological Society. Neurol. Neurochir. Pol. 2021, 55. [Google Scholar] [CrossRef]
- Knoll, M.D.; Wonodi, C. Oxford–AstraZeneca COVID-19 Vaccine Efficacy. Lancet 2021, 397. [Google Scholar] [CrossRef]


{kind=link}
| Antigen | Nano/Microparticle Platform | Disease | Animal/Human |
|---|---|---|---|
| Hemagglutinin (HA) | PLGA-NPs adjuvanted with MLPA and muramyl dipeptide (MDP) | Influenza A [51] | BALB/c and C57BL/6 mice |
| Nonstructural protein 1 (NS1) | PLGA/polyethylene glycol (PEG)-NPs | Dengue [52] | BALB/c mice |
| Hepatitis B surface antigen (HBsAg) | mannose-grafted PLGA-NPs | Hepatitis B virus [53] | Balb/c mice |
| Hepatitis B core antigen (HBcAg) | PLGA-NPs with monophospholipid A (MPLA) | chronic hepatitis B infection [53] | C57BL/6J mice |
| Insoluble form of E2 envelope glycoprotein subtype 1b of hepatitis C virus (HCV1b-E2) | PLGA microspheres | Hepatitis C virus (HCV) [57] | Balb/c mice |
| Plasmid DNA encoding HBsAg | PLGA-NPs | hepatitis B virus (HBV) [60] | Balb/c mice |
| M2e peptide (CapM2e) | PLGA-NPs | Influenza A [66] | Balb/c mice |
| Hemagglutinin-neuraminidase (HN) | oligomannose-coated liposome and Poly(I:C) as adjuvant | human parainfluenza virus type 3 (HPIV3) [79] | BALB/c mice |
| Mixture of peptides (pepmix) spanning the entire sequence of nonstructural protein 3 (NS3) | cationic liposomes (CAF09) | chronic hepatitis C virus (HCV) [80] | CB6F1 (C57BL/6 × BALB/c) and C3H mice |
| Four HLA-A*0201-restricted cytotoxic T lymphocytes (CTL) epitopes | Liposomes | Severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) [82] | HLA-A*0201 transgenic mice |
| Influenza virus nucleoprotein (NP) | Cholesterol/phosphatidylcholine/phosphatidylserine liposomes | Influenza virus [92] | Mice |
| Hemagglutinin (HA) | Lipid nanoparticles (LNPs) | Influenza virus [95] | Mice, rabbits, and ferrets |
| VZV gE antigen | LNPs | Varicella-zoster virus (VZV) [96] | Indian rhesus macaques |
| Ebola envelope glycoprotein | LNPs | Ebola virus [97] | Guinea pigs |
| SARS-CoV-2 spike protein | LNPs | severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [98] | Healthy human adults |
| Recombinant VZV glycoprotein E (gE) | Cationic liposomes with the TLR4 agonist de-O-acylated | Varicella zoster virus (VZV) [100] | BALB/c and C57BL/6 mice |
| Trivalent influenza vaccine (TIV) | Cationic liposome adjuvant system CAF01 | New influenza A (H1N1) [101] | BALB/c mice |
| Spike receptor binding domain (RBD) | Three different adjuvant systems: an aluminum hydroxide (AH), an oil-in-water squalene emulsion (SE) adjuvant resembling MF59™, a cationic liposome-based adjuvant (CAF®01) | Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [102] | C57Bl/6 mice |
| recombinant neutralizing epitope protein (rNEp), a part of the structural capsid protein open-reading-frame-2 (ORF-2) | Liposomes | Hepatitis E virus (HEV) [103] | Mice and rhesus macaques |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappellano, G.; Abreu, H.; Casale, C.; Dianzani, U.; Chiocchetti, A. Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines 2021, 9, 606. https://doi.org/10.3390/vaccines9060606
Cappellano G, Abreu H, Casale C, Dianzani U, Chiocchetti A. Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines. 2021; 9(6):606. https://doi.org/10.3390/vaccines9060606
Chicago/Turabian StyleCappellano, Giuseppe, Hugo Abreu, Chiara Casale, Umberto Dianzani, and Annalisa Chiocchetti. 2021. "Nano-Microparticle Platforms in Developing Next-Generation Vaccines" Vaccines 9, no. 6: 606. https://doi.org/10.3390/vaccines9060606
APA StyleCappellano, G., Abreu, H., Casale, C., Dianzani, U., & Chiocchetti, A. (2021). Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines, 9(6), 606. https://doi.org/10.3390/vaccines9060606