
The Many Faces of Innate Immunity in SARS-CoV-2 Infection

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. SARS-CoV-2 and COVID-19 Pathogenesis
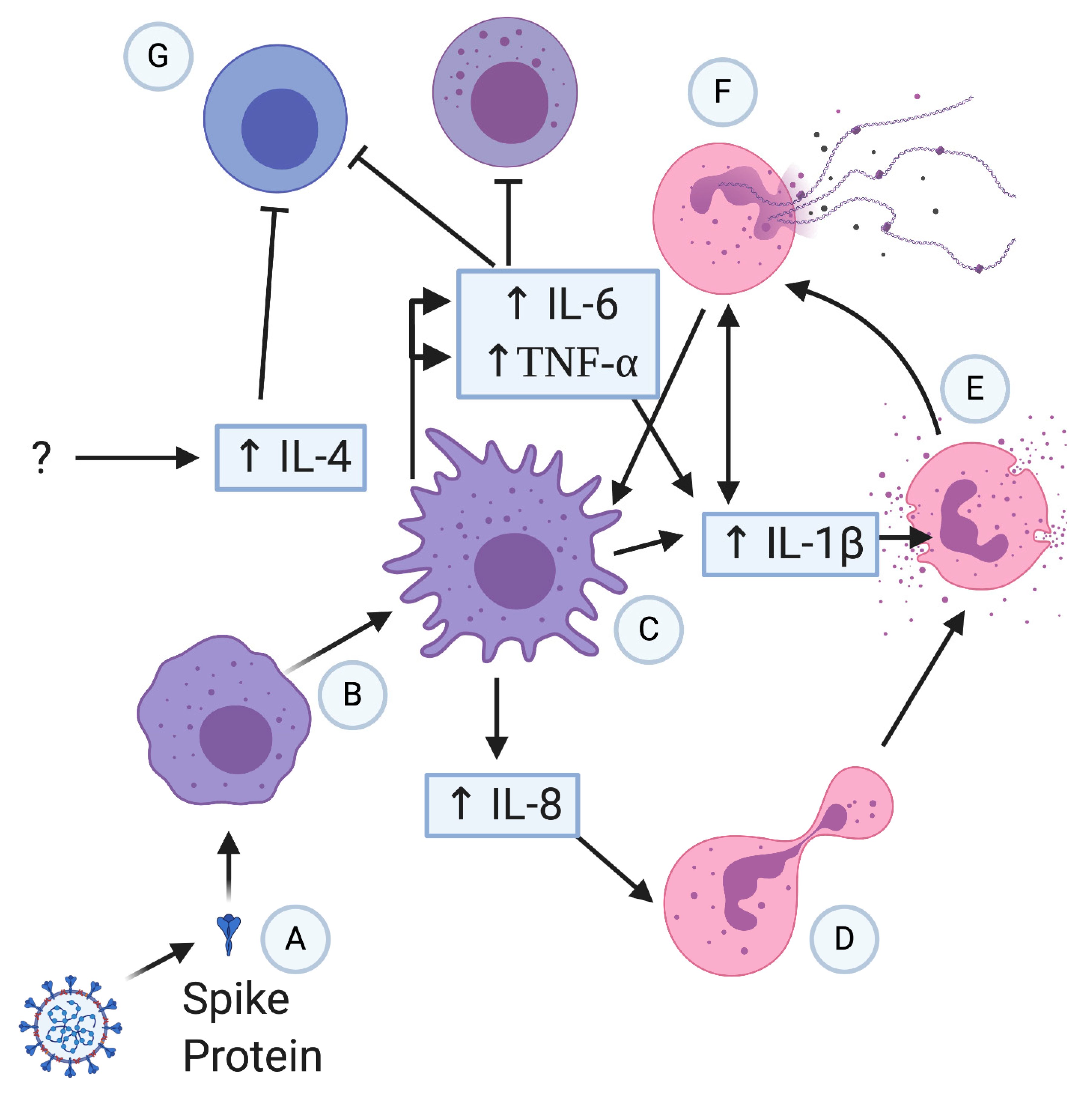
3. Innate Immune Cells & Their Role in COVID-19
3.1. Overview of the Interplay between the Innate Immune Responses and SARS-CoV-2
3.2. Overactivity of the Innate Immune System
3.3. Cytokines, Chemokines, and Hyperinflammation
3.4. Lymphopenia
3.5. Immunosuppression
4. Co-Morbid Conditions and Genetic Risks Stoke Immunopathology of SARS-CoV-2
4.1. Co-Morbidities: A Target for SARS-CoV-2
4.2. Genetic-Associated Risks: Influences of Age, Race, and Gender Disparities
5. Ongoing Therapeutic Approaches
6. Future Global Disease Surveillance
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 2 June 2021).
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72,314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E.; Cyranoski, D.; Mallapaty, S.; Stoye, E.; Tollefson, J. The coronavirus pandemic in five powerful charts. Nature 2020, 579, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020, 8, 750–752. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.-C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Cheung, M.C.; Perera, R.; Ng, K.C.; Bui, C.H.T.; Ho, J.C.W.; Ng, M.M.T.; Kuok, D.I.T.; Shih, K.C.; Tsao, S.W.; et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: An analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 2020, 8, P687–P695. [Google Scholar] [CrossRef]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Prakash, A.; Avti, P.; Bhattacharyya, A.; Kaur, H.; Kumar, S.; Bansal, S.; Sharma, A.R.; et al. Update on the target structures of SARS-CoV-2: A systematic review. Indian J. Pharmacol. 2020, 52, 142–149. [Google Scholar] [CrossRef]
- Ulrich, H.; Pillat, M.M. CD147 as a Target for COVID-19 Treatment: Suggested Effects of Azithromycin and Stem Cell Engagement. Stem. Cell Rev. Rep. 2020, 16, 434–440. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv 2020. [Google Scholar] [CrossRef]
- Kutti Sridharan, G.; Kotagiri, R.; Chandiramani, V.H.; Mohan, B.P.; Vegunta, R.; Vegunta, R.; Rokkam, V.R.P. COVID-19 and Avoiding Ibuprofen. How Good Is the Evidence? Am. J. Ther. 2020, 27, e400–e402. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. Nasal ACE2 Levels and COVID-19 in Children. JAMA 2020, 323, 2386–2387. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA 2020, 323, 2427–2429. [Google Scholar] [CrossRef] [PubMed]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01815-18. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Broszeit, F.; Tzarum, N.; Zhu, X.; Nemanichvili, N.; Eggink, D.; Leenders, T.; Li, Z.; Liu, L.; Wolfert, M.A.; Papanikolaou, A.; et al. N-Glycolylneuraminic Acid as a Receptor for Influenza A Viruses. Cell Rep. 2019, 27, 3284–3294.e3286. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. RIG-I-like receptor regulation in virus infection and immunity. Curr. Opin. Virol. 2015, 12, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef]
- Maelfait, J.; Beyaert, R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 2012, 76, 33–45. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Targeted Immunosuppression Distinguishes COVID-19 from Influenza in Moderate and Severe Disease. MedRxiv 2020. [Google Scholar] [CrossRef]
- Schroeder, S.P.F.; Niemeyer, D.; Veith, T.; Richter, A.; Muth, D.; Goffinet, C.; Müller, M.A.; Drosten, C. Interferon antagonism by SARS-CoV-2: A functional study using reverse genetics. Lancet Microbe 2021, 2, e210–e218. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- McKechnie, J.L.; Blish, C.A. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe 2020, 27, 863–869. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.; Mathew, D.; Baxter, A.E.; Vella, L.; et al. Immunologic perturbations in severe COVID-19/SARS-CoV-2 infection. BioRxiv 2020. [Google Scholar] [CrossRef]
- Chang, F.Y.; Chen, H.C.; Chen, P.J.; Ho, M.S.; Hsieh, S.L.; Lin, J.C.; Liu, F.T.; Sytwu, H.K. Immunologic aspects of characteristics, diagnosis, and treatment of coronavirus disease 2019 (COVID-19). J. Biomed. Sci. 2020, 27, 72. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Song, J.-W.; Zhang, C.; Fan, X.; Meng, F.-P.; Xu, Z.; Xia, P.; Cao, W.-J.; Yang, T.; Dai, X.-P.; Wang, S.-Y.; et al. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat. Commun. 2020, 11, 3410. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renne, C.; Renne, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef]
- Golonka, R.M.; Saha, P.; Yeoh, B.S.; Chattopadhyay, S.; Gewirtz, A.T.; Joe, B.; Vijay-Kumar, M. Harnessing innate immunity to eliminate SARS-CoV-2 and ameliorate COVID-19 disease. Physiol. Genom. 2020, 52, 217–221. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e1003. [Google Scholar] [CrossRef]
- Zhou, J.; Chu, H.; Li, C.; Wong, B.H.; Cheng, Z.S.; Poon, V.K.; Sun, T.; Lau, C.C.; Wong, K.K.; Chan, J.Y.; et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: Implications for pathogenesis. J. Infect. Dis. 2014, 209, 1331–1342. [Google Scholar] [CrossRef]
- He, R.; Lu, Z.; Zhang, L.; Fan, T.; Xiong, R.; Shen, X.; Feng, H.; Meng, H.; Lin, W.; Jiang, W.; et al. The clinical course and its correlated immune status in COVID-19 pneumonia. J. Clin. Virol. 2020, 127, 104361. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, Z.; Diao, B.; Wang, R.; Wang, G.; Wang, C.; Tan, Y.; Liu, L.; Wang, C.; Liu, Y.; et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. MedRxiv 2020. [Google Scholar] [CrossRef]
- He, Z.; Zhao, C.; Dong, Q.; Zhuang, H.; Song, S.; Peng, G.; Dwyer, D.E. Effects of severe acute respiratory syndrome (SARS) coronavirus infection on peripheral blood lymphocytes and their subsets. Int. J. Infect. Dis. 2005, 9, 323–330. [Google Scholar] [CrossRef]
- Panesar, N.S. What caused lymphopenia in SARS and how reliable is the lymphokine status in glucocorticoid-treated patients? Med. Hypotheses 2008, 71, 298–301. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Neutrophil-to-lymphocyte ratio and lymphocyte-to-C-reactive protein ratio in patients with severe coronavirus disease 2019 (COVID-19): A meta-analysis. J. Med. Virol. 2020, 92, 1733–1734. [Google Scholar] [CrossRef]
- Yang, A.-P.; Liu, J.-P.; Tao, W.-Q.; Li, H.-M. The diagnostic and predictive role of NLR, d-NLR and PLR in COVID-19 patients. Int. Immunopharmacol. 2020, 84, 106504. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Demaria, O.; Carvelli, J.; Batista, L.; Thibult, M.-L.; Morel, A.; André, P.; Morel, Y.; Vély, F.; Vivier, E. Identification of druggable inhibitory immune checkpoints on Natural Killer cells in COVID-19. Cell Mol. Immunol. 2020, 17, 995–997. [Google Scholar] [CrossRef]
- Kim, O.Y.; Monsel, A.; Bertrand, M.; Coriat, P.; Cavaillon, J.M.; Adib-Conquy, M. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit. Care 2010, 14, R61. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.S.; Melnyk, B.M.; Tan, A. Associations Between Stress and Cardiovascular Disease Risk Factors Among Million Hearts Priority Populations. Am. J. Health Promot. 2019, 33, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Brennan, K.J.; Gaughan, J.P.; Ciccolella, D.E.; Kuzma, A.M.; Criner, G.J. African Americans and men with severe COPD have a high prevalence of osteoporosis. COPD 2008, 5, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.; Park, I.W.; Robinson, J.R.; Jones, H.P. The Perfect Storm: COVID-19 Health Disparities in US Blacks. J. Racial Ethn. Health Disparities 2020, 1–8. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.O.; Abate, K.H.; Abd-Allah, F.; Ahmed, M.; Alam, K.; Alam, T.; Alvis-Guzman, N.; Ansari, H.; Arnlov, J.; et al. The Burden of Cardiovascular Diseases Among US States, 1990–2016. JAMA Cardiol. 2018, 3, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Suleyman, G.; Fadel, R.A.; Malette, K.M.; Hammond, C.; Abdulla, H.; Entz, A.; Demertzis, Z.; Hanna, Z.; Failla, A.; Dagher, C.; et al. Clinical Characteristics and Morbidity Associated With Coronavirus Disease 2019 in a Series of Patients in Metropolitan Detroit. JAMA Netw. Open 2020, 3, e2012270. [Google Scholar] [CrossRef]
- Anguiano, L.; Riera, M.; Pascual, J.; Soler, M. Circulating ACE2 in cardiovascular and kidney diseases. Curr. Med. Chem. 2017, 24, 3231–3241. [Google Scholar] [CrossRef]
- Chhabra, K.H.; Xia, H.; Pedersen, K.B.; Speth, R.C.; Lazartigues, E. Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E874–E884. [Google Scholar] [CrossRef] [PubMed]
- Jia, H. Pulmonary angiotensin-converting enzyme 2 (ACE2) and inflammatory lung disease. Shock 2016, 46, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Wurth, R.; Kamilaris, C.D.C.; Eisenhofer, G.; Barrera, F.J.; Hajdenberg, M.; Tonleu, J.; Hall, J.E.; Schiffrin, E.L.; Porter, F.; et al. Endocrine Conditions and COVID-19. Horm. Metab. Res. 2020, 52, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Dalan, R.; Bornstein, S.R.; El-Armouche, A.; Rodionov, R.N.; Markov, A.; Wielockx, B.; Beuschlein, F.; Boehm, B.O. The ACE-2 in COVID-19: Foe or Friend? Horm. Metab. Res. 2020, 52, 257–263. [Google Scholar] [CrossRef]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448–7464. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Y.; Wang, G.Q. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. J. Med. Virol. 2020, 92, 726–730. [Google Scholar] [CrossRef]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Schijns, V.; Lavelle, E.C. Prevention and treatment of COVID-19 disease by controlled modulation of innate immunity. Eur. J. Immunol. 2020, 50, 932–938. [Google Scholar] [CrossRef]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 50, 932–938. [Google Scholar] [CrossRef]
- Grimaud, M.; Starck, J.; Levy, M.; Marais, C.; Chareyre, J.; Khraiche, D.; Leruez-Ville, M.; Quartier, P.; Léger, P.L.; Geslain, G.; et al. Acute myocarditis and multisystem inflammatory emerging disease following SARS-CoV-2 infection in critically ill children. Ann. Intensive Care 2020, 10, 69. [Google Scholar] [CrossRef]
- Dhochak, N.; Singhal, T.; Kabra, S.K.; Lodha, R. Pathophysiology of COVID-19: Why Children Fare Better than Adults? Indian J. Pediatr. 2020, 87, 537–546. [Google Scholar] [CrossRef]
- Shah, M.; Sachdeva, M.; Dodiuk-Gad, R.P. COVID-19 and racial disparities. J. Am. Acad. Dermatol. 2020, 83, e35. [Google Scholar] [CrossRef]
- Brandt, E.B.; Beck, A.F.; Mersha, T.B. Air pollution, racial disparities, and COVID-19 mortality. J. Allergy Clin. Immunol. 2020, 146, 61–63. [Google Scholar] [CrossRef]
- Alcendor, D.J. Racial Disparities-Associated COVID-19 Mortality among Minority Populations in the US. J. Clin. Med. 2020, 9, 2442. [Google Scholar] [CrossRef]
- COVID-19 Racial and Ethnic Health Disparities. Available online: https://www.cdc.gov/coronavirus/2019-ncov/community/health-equity/racial-ethnic-disparities/increased-risk-exposure.html (accessed on 2 June 2021).
- Hawkins, D. Differential occupational risk for COVID-19 and other infection exposure according to race and ethnicity. Am. J. Ind. Med. 2020, 63, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Grandi, G.; Facchinetti, F.; Bitzer, J. The gendered impact of coronavirus disease (COVID-19): Do estrogens play a role? Eur. J. Contracept. Reprod. Health Care 2020, 25, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, X.; Shi, J.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Zhou, M.; Xu, S.; Wu, B.; et al. Gendered effects on inflammation reaction and outcome of COVID-19 patients in Wuhan. J. Med. Virol. 2020, 92, 2684–2692. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wenham, C.; Smith, J.; Morgan, R. COVID-19: The gendered impacts of the outbreak. Lancet 2020, 395, 846–848. [Google Scholar] [CrossRef]
- Ghosh, S.; Klein, R.S. Sex Drives Dimorphic Immune Responses to Viral Infections. J. Immunol. 2017, 198, 1782–1790. [Google Scholar] [CrossRef]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost Agents 2020, 34, 339–343. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Horby, P.W.; Pessoa-Amorim, G.; Peto, L.; Brightling, C.E.; Sarkar, R.; Thomas, K.; Jeebun, V.; Ashish, A.; Tully, R.; Chadwick, D.; et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): Preliminary results of a randomised, controlled, open-label, platform trial. MedRxiv 2021. [Google Scholar] [CrossRef]
- Izda, V.; Jeffries, M.A.; Sawalha, A.H. COVID-19: A review of therapeutic strategies and vaccine candidates. Clin. Immunol. 2021, 222, 108634. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Farber, D.L. COVID-19 vaccines: Modes of immune activation and future challenges. Nat. Rev. Immunol. 2021, 21, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Priyadarsini, S.L.; Suresh, M.; Huisingh, D. What can we learn from previous pandemics to reduce the frequency of emerging infectious diseases like COVID-19? Glob. Transit. 2020, 2, 202–220. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Catherina, R.; Frye, H.; Shelley, L. Illicit Wildlife Trade, Wet Markets, and COVID-19: Preventing Future Pandemics. World Med. Health Policy 2020, 12, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Nabi, G.; Wang, Y.; Lü, L.; Jiang, C.; Ahmad, S.; Wu, Y.; Li, D. Bats and birds as viral reservoirs: A physiological and ecological perspective. Sci. Total Environ. 2021, 754, 142372. [Google Scholar] [CrossRef]
- Brook, C.E.; Boots, M.; Chandran, K.; Dobson, A.P.; Drosten, C.; Graham, A.L.; Grenfell, B.T.; Müller, M.A.; Ng, M.; Wang, L.F.; et al. Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. Elife 2020, 9, e48401. [Google Scholar] [CrossRef] [PubMed]
- Donnik, I.M.; Popov, I.V.; Sereda, S.V.; Popov, I.V.; Chikindas, M.L.; Ermakov, A.M. Coronavirus Infections of Animals: Future Risks to Humans. Biol. Bull. 2021, 48, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Carbo, E.C.; Sidorov, I.A.; Zevenhoven-Dobbe, J.C.; Snijder, E.J.; Claas, E.C.; Laros, J.F.J.; Kroes, A.C.M.; de Vries, J.J.C. Coronavirus discovery by metagenomic sequencing: A tool for pandemic preparedness. J. Clin. Virol. 2020, 131, 104594. [Google Scholar] [CrossRef]
- Watsa, M. Rigorous wildlife disease surveillance. Science 2020, 369, 145–147. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanan, N.; Doud, R.L., Jr.; Park, I.-W.; Jones, H.P.; Mathew, S.O. The Many Faces of Innate Immunity in SARS-CoV-2 Infection. Vaccines 2021, 9, 596. https://doi.org/10.3390/vaccines9060596
Hanan N, Doud RL Jr., Park I-W, Jones HP, Mathew SO. The Many Faces of Innate Immunity in SARS-CoV-2 Infection. Vaccines. 2021; 9(6):596. https://doi.org/10.3390/vaccines9060596
Chicago/Turabian StyleHanan, Nicholas, Ronnie L. Doud, Jr., In-Woo Park, Harlan P. Jones, and Stephen O. Mathew. 2021. "The Many Faces of Innate Immunity in SARS-CoV-2 Infection" Vaccines 9, no. 6: 596. https://doi.org/10.3390/vaccines9060596
APA StyleHanan, N., Doud, R. L., Jr., Park, I.-W., Jones, H. P., & Mathew, S. O. (2021). The Many Faces of Innate Immunity in SARS-CoV-2 Infection. Vaccines, 9(6), 596. https://doi.org/10.3390/vaccines9060596