1. Introduction
Coronavirus disease 2019 (COVID-19), caused by the virus SARS-CoV-2, continues to spread globally leading to significant impacts on public health [
1]. SARS-CoV-2, or Severe Acute Respiratory Syndrome coronavirus 2, causes primarily respiratory infections in humans and is related to other coronaviruses like Middle East Respiratory Syndrome Coronavirus (MERS-CoV) and SARS-CoV [
2]. Not only has SARS-CoV-2 caused a public health emergency on a global scale but it continues to have major social, cultural, and economic impacts. Vaccination is the most effective countermeasure for mitigating pandemics and has proven effective against viral pathogens such as smallpox and polio.
The emergence of SARS-CoV-2 in human populations spurred the scientific community to development methods to produce vaccines against COVID-19 [
3]. Many development paths were initiated using methods that have been applied historically to the production of vaccine candidates as well as new approaches aimed at delivering candidates in a rapid and efficient manner [
4]. These methods have included the use of RNA and DNA vaccines, subunit vaccines, attenuated vaccines, as well as vectored vaccines utilizing virus-like particles (VLP), adenovirus or bacterial host constructs. Inactivated vaccines have been a mainstay of vaccinology for decades. Even today, examples of inactivated vaccines include constructs for influenza, cholera, bubonic plague and polio [
5]. Each method of vaccine production has merits and limitations. These limitations range from issues in production at scale, cost to produce, stability and delivery process and the potential for side effects.
The use of these vaccine production methods is predicated on the ability to achieve inactivation of the pathogen’s ability to replicate while preserving the antigenic protein structural potency and integrity [
6]. Methods for producing such candidates require the use of chemicals such as beta-propiolactone, ethylenimine, formalin as well as the use of physical inactivation methods such as gamma irradiation, high energy UV light (UVC) and heat inactivation. These methods work primarily by chemically modifying protein and nucleic acid structures of pathogens through covalent modification, crosslinking, oxidation and structural alteration, rendering the treated agents unable to infect target cells or replicate in vivo. The key to success with such approaches resides in the ability to balance the alterations required for inactivation with the preservation of antigen epitopes similar to the native agent that are required to stimulate immune response when administered to a host. A method which can successfully prevent pathogen replication without inducing alterations to antigen targets is ideal. Candidates produced by such a method would possess the native protein structures that are as close to natural structures in the intact pathogen as possible without the ability to induce disease.
We report in this preliminary evaluation on a novel method (SolaVAX) to produce a candidate vaccine for COVID-19. This method employs the use of a photochemical (riboflavin or vitamin B2) in combination with UV light in the UVA and UVB wavelength regions to carry out specific nucleic acid alterations through electron transfer chemistry-based processes [
7]. The method was originally developed for the treatment of blood products to prevent transfusion transmitted diseases [
8] and has been in routine clinical use for prevention of transfusion transmitted viral, bacterial and parasitic diseases since 2008 [
9]. The process utilizes a well-established and demonstrated capability of riboflavin and UV light to modify nucleic acid structure primarily through modification of guanine bases in a non-oxygen dependent process utilizing the natural electron donor-acceptor chemistry associated with guanine and riboflavin, respectively [
10].
The specificity of the riboflavin photochemistry used in this process avoids the alkylation, crosslinking and covalent modifications that are associated with other chemical and photochemical mechanisms of pathogen inactivation [
11]. The process allows for retention of plasma and cell-bound protein structure post treatment to an extent that such products may still be efficacious in functional utilization for transfusion support of patients [
12,
13]. Unlike the standard chemical agents such as beta-propiolactone and ethyleneimine derivatives that are routinely used for inactivated vaccine production, the photochemical used in this approach (riboflavin) has a well-established safety toxicology profile, is non-mutagenic and non-carcinogenic and poses little to no toxicity or disposal risk to facility personnel or the environment. This safety profile has been documented extensively in pre-clinical and clinical programs in human subjects [
14].
For this work, we hypothesized that the use of these methods for production of an inactivated virus of SARS-CoV-2 could have several advantages. These included the ability to utilize existing equipment, reagents and disposables that are in routine use for treatment of blood products to produce an inactivated vaccine preparation when using purified viral stocks of the target virus, e.g., SARS-CoV-2 in this particular application [
15]. We hypothesized that the selectivity of the chemistry applied in this process would generate a vaccine candidate that was fully attenuated with regard to replication capabilities while maintaining viral protein structural integrity as close to the native virus as possible. We further hypothesized that such a candidate produced by this method would be able to induce a potent immune response with relatively low doses of immunogen and thus provide protective immunity against live virus challenge.
Our approach to evaluate this candidate utilized both in vitro analysis and animal models (Syrian Golden Hamsters), shown to be susceptible to disease in the upper and lower respiratory tract following exposure to live SARS-CoV-2 virus administered intranasally [
16,
17,
18,
19]. Animals exposed in this way were demonstrated to produce and shed virus extensively at days 3–7 post-exposure. Our methods for evaluation of an effective vaccine candidate in this model included measurement of neutralizing antibodies with a Plaque Reduction Neutralization Test (PRNT), assessment of tissue viremia via plaque assays for live virus, flow cytometric analysis of leukocyte subpopulations, single cell mRNA sequencing analysis to assess host transcriptional responses and histopathology of respiratory system post-challenge with live virus via the intranasal route.
As part of these studies, we also utilized adjuvant formulations intended to drive immune response to vaccines predominantly via a Th1 immune pathway [
20,
21]. Our motivation for employing this approach was generated by previous observations of antibody dependent enhancement (ADE) and resulting immunopathology in animal models where Th2 type immune stimulation predominated [
20]. We monitored immune response via cellular and humoral pathways in adjuvanted and non-adjuvanted formulations. We also monitored lung histopathology for evidence indicative of immunopathology induced by the virus or subsequent challenge post-vaccination.
2. Materials and Methods
2.1. Study Design
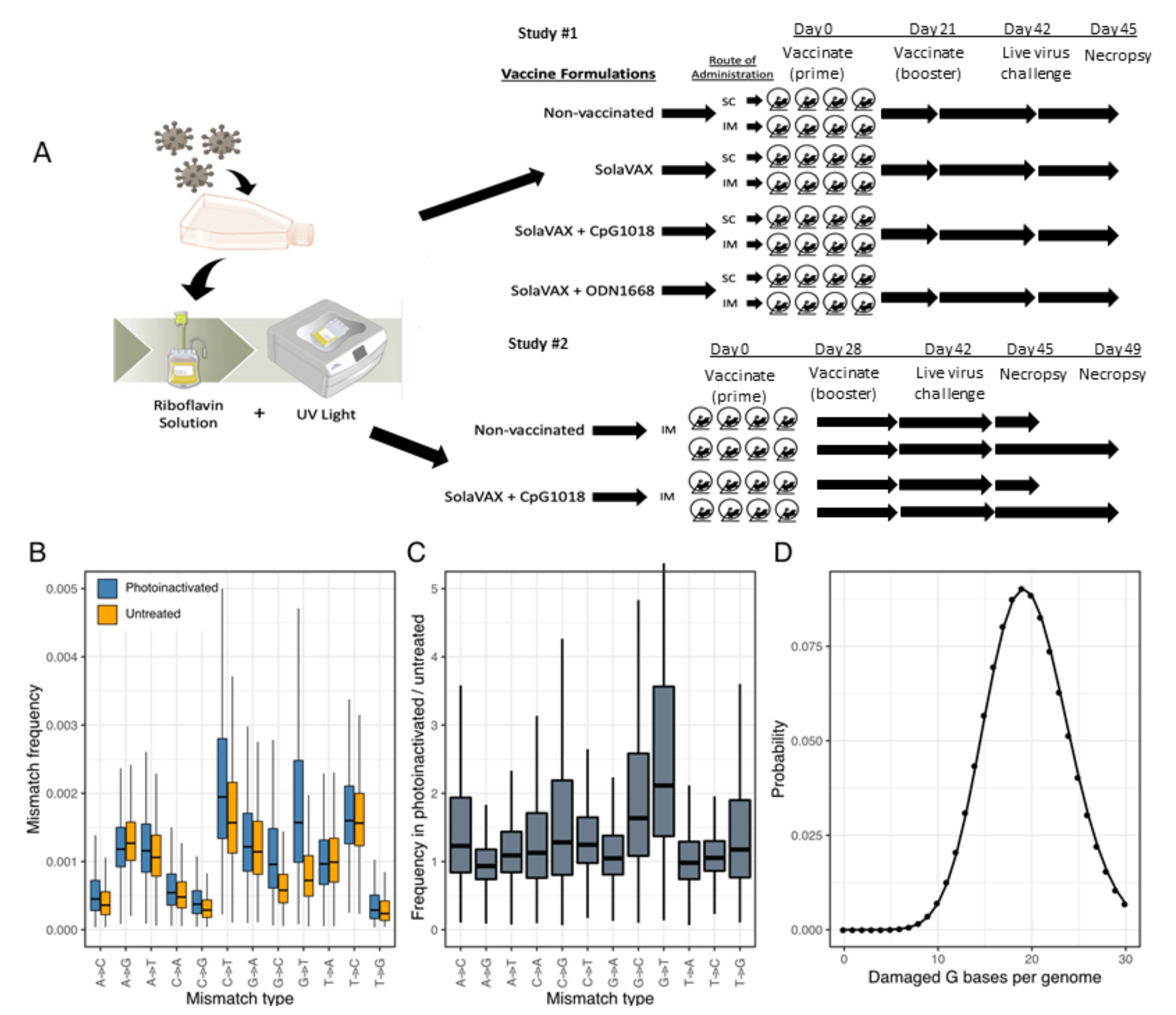
The objective of this preliminary evaluation was to determine and characterize the efficacy of a novel method for creating an inactivated whole virion vaccine (SolaVAX) against SARS-CoV-2 in hamsters. Preliminary data generated from this study will guide additional animal studies. Hamsters were randomly assigned into groups of four hamsters per cohort (vaccine formulation and administration route) to evaluate the performance of the vaccine candidate in response to viral challenge using different routes of administration and in combination with different adjuvants. No animals were excluded from the analyses (1 animal death occurred in the non-adjuvanted group [SvX, SC] prior to administration of the 2nd vaccine dose due to factors not related to the vaccine). Histopathology analyses were performed blinded to the experimental cohort conditions. End points were selected prior to initiation of the study and were selected based on the objective of determining the immune responses to vaccination with the SolaVAX vaccine.
2.2. SARS-CoV-2 Virus
All virus propagation occurred in a BSL-3 laboratory setting. Virus (isolate USA-WA1/2020) was acquired through BEI Resources (product NR-52281) and amplified in Vero C1008 (Vero E6) cell culture. Vero E6 cells (ATCC CRL-1568) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glucose, L-glutamine, sodium pyruvate, 5% fetal bovine serum (FBS) and antibiotics. Inoculation of Vero E6 cells with SARS-CoV-2 was carried out directly in DMEM containing 1% FBS. Medium harvested from infected cells 3-4 days after inoculation was clarified by centrifugation at 800× g, supplemented with FBS to 10% and frozen to −80 °C in aliquots. The virus titer was determined using a standard double overlay plaque assay.
2.3. Viral Inactivation
Viral stock in DMEM with 10% FBS was dispensed into an illumination bag (Mirasol Illumination Bag, Terumo BCT, Lakewood, CO, USA). Riboflavin solution (500 µmol/L) was added, residual air was removed from the product, and the bag was placed into the illu-minator (Mirasol PRT System, Terumo BCT, Lakewood, CO, USA) for treatment with UV light (150 Joules). Upon successful completion of the illumination process, the product was re-moved from the illuminator for vaccine preparation and characterization.
2.4. Sequence Validation of SARS-CoV-2 Isolate
RNA from cell culture supernatant was extracted using the Trizol reagent (Life Technologies) according to the manufacturer’s protocol. Libraries were prepared from total RNA using the Kapa Biosystems RNA HyperPrep kit and sequenced on an Illumina NextSeq instrument to generate single end 150 base reads. Reads were mapped to the USA-WA1 reference sequence (GenBank accession MN985325.1) using bowtie2 [
22]. The position, frequency, and predicted coding impact of variants were tabulated as previously described [
23]. All data related to sequencing for the materials used in this study have been uploaded to the GEO Submission platform (GSE165190) [NCBI tracking system #21610801].
2.5. Assessment of RNA Damage Following Photoinactivation
Libraries were created from RNA of photoinactivated vaccine material and from matching untreated material as above. Reads were mapped to a SARS-CoV-2 reference sequence that corresponded to the consensus sequence of the virus used for vaccine pro-duction using bowtie2. The frequencies of nucleotide substitutions with basecall quality scores ≥ 30 were tabulated and normalized to the number of occurrences of the mutated bases in all reads. Analysis scripts are available at:
https://github.com/stenglein-lab/SolaVAX_sequence_analysis (accessed date 19 February 2021).
2.6. Vaccine Concentration and Preparation
Inactivated virus was concentrated using Amicon Ultra Centrifugal Filter units (Millipore Sigma) at 100k cutoff. Concentrated vaccine material was tested by plaque assay to ensure complete virus inactivation. PCR was performed to determine RNA copies/mL using the Superscript III Platinum One-Step qRT-PCR system (Invitrogen). Standard curves were obtained by using a quantitative PCR (qPCR) extraction control from the original WA1/2020WY96 SARS-COV-2 isolate. Based on the ratio of pfu to virus RNA copy number for the pre-inactivation vaccine, and RNA copy number of the inactivated and concentrated vaccine, we estimated that prime and booster doses of vaccine used in hamsters were equivalent to 2.2 × 106 and 1.8 × 106 PFU-equivalents. We calculated that the amount of virus utilized per dose was on the order of 15 ng (prime) and 13 ng (boost) of virus material, respectively, in each preparation. In the initial study, the viral material used to create the prime and booster doses were derived from the same viral stock. In a second study, the prime and booster doses were created from different batches of viral material, and the concentration of the material was adjusted so that both the prime and booster were equivalent (2e6 PFU-equivalents).
Vaccines were prepared immediately prior to vaccination. For the non-adjuvanted vaccine, the inactivated vaccine material was mixed equally with sterile PBS. Adjuvant CpG 1018 (Dynavax, Lot 1-FIN-3272) was mixed with equal parts of dH20 then mixed with inactivated vaccine at a 1:1 ratio. One mg of adjuvant ODN1668 (Enzo ALX-746-051-M001) powder was reconstituted in 1 mL of dH20 then mixed with inactivated vaccine at 1:1 ratio. Each adjuvant was utilized according to manufacturer’s recommendations for dose. For CpG 1018, 150 μg of adjuvant was used per dose. For ODN1668, 50 µg of adjuvant was used per dose.
2.7. Animals
All hamsters were held at Colorado State University in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International accredited animal facilities. Animal testing and research received ethical approval by the Institutional Animal Care and Use Committee (IACUC) (protocol #18-1234A). In the first study a total of 32 male Golden Syrian hamsters (
Mesocricetus auratus) at 6 weeks of age were acquired from Charles River Laboratories (Wilmington, MA). Hamsters were maintained in a Biosafety Level-2 (BSL-2) animal facility at the Regional Biocontainment Lab at Colorado State University during the vaccination period. The hamsters were group-housed and fed a commercial diet with access to water ad libitum. Each hamster was ear notched for animal identification. As previously described, the hamsters were randomly divided into 4 treatment groups (8 hamsters per group): Control hamsters received no vaccination, a second group (SvX) received inactivated vaccine (SolaVAX) with no adjuvant, a third group (CpG) received inactivated vaccine with CpG 1018 adjuvant, and the final group of hamsters (ODN) received inactivated vaccine with ODN1668 adjuvant. Within each group hamsters were divided in two subgroups (4 hamsters per subgroup) where one subgroup was vaccinated by subcutaneous (SC) route and the second subgroup by intramuscular (IM) route (
Figure 1A).
In a follow-up study to this initial evaluation, 16 hamsters were randomly divided into 2 treatment groups (8 hamsters per group): Control hamsters received no vaccination, and a second group received the SolaVAX vaccine with CpG 1018 adjuvant via IM route. Within each group, hamsters were divided into two subgroups (4 hamsters per group), in which one subgroup was sacrificed at 3 days post-challenge with live virus, and the second subgroup at 7 days post-challenge (
Figure 1A).
2.8. Clinical Observations
Body weights were recorded one day before vaccination, at time of prime vaccination and booster vaccination, and then daily after challenge. Hamsters were observed daily post-vaccination for the duration of the study. Clinical evaluation included temperament, ocular discharge, nasal discharge, weight loss, coughing/sneezing, dyspnea, lethargy, anorexia, and moribund.
2.9. Vaccination
Prior to vaccination, blood was collected from all hamsters under anesthesia and sera isolated. Each hamster in the vaccinated groups received 100 µL of vaccine (15 ng). No vaccination was administered to Control hamsters. The hamsters were maintained and monitored for 21 days in the first study, and 28 days in the second study. Prior to the second (booster) vaccination, blood was collected from all hamsters again under anesthesia and sera isolated. A booster vaccination (13 ng) was administered to hamsters as described in the prime vaccination. Hamsters were again maintained and monitored for an additional 21 days.
2.10. Virus Challenge
All hamsters were transferred to a Biosafety Level-3 animal facility at the Regional Biocontainment Lab at Colorado State University prior to live virus challenge. Hamsters were bled under anesthesia and sera collected prior to live virus challenge to determine antibody response post vaccination.
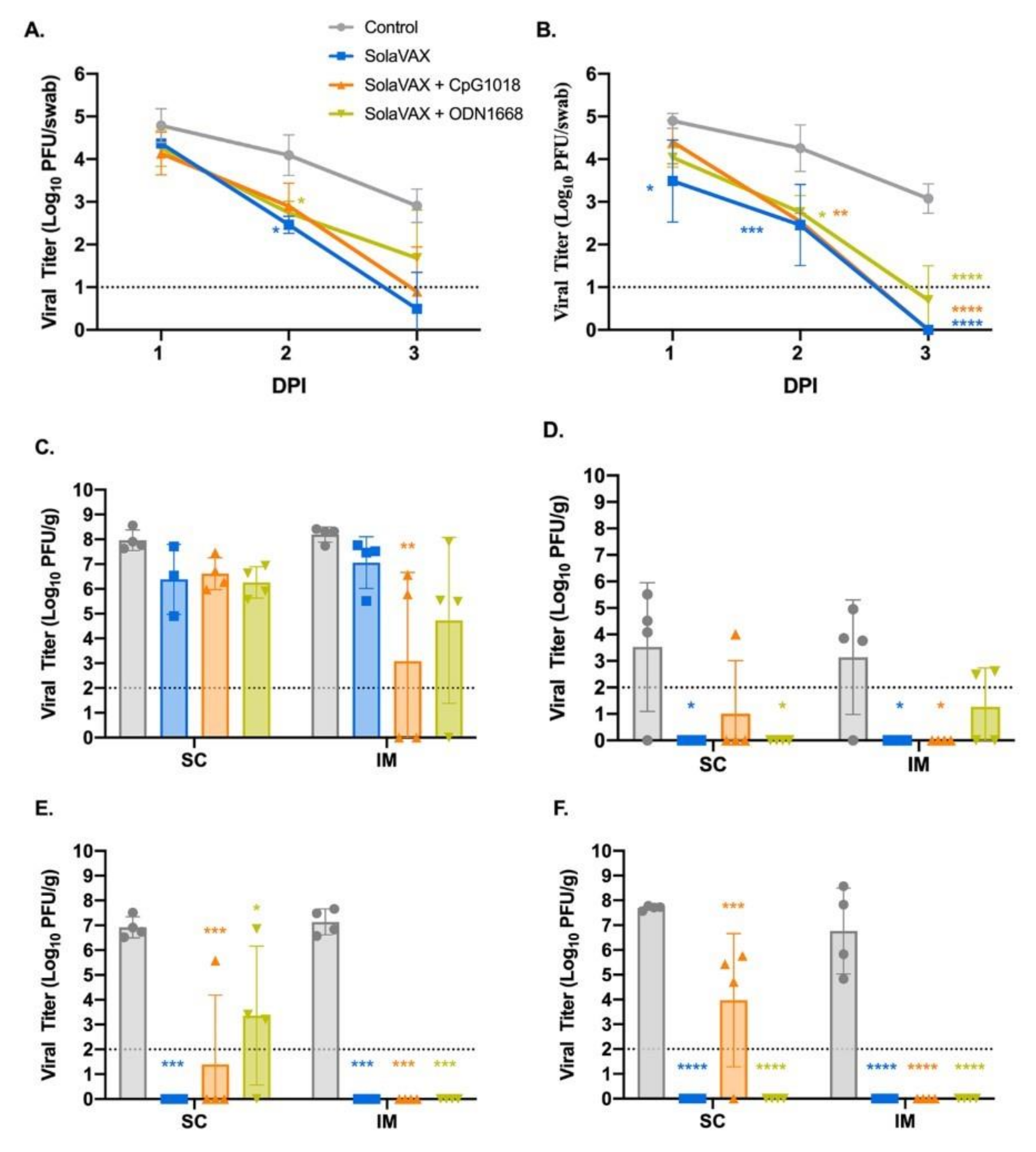
In the initial study, SARS-CoV-2 virus was diluted in phosphate buffered saline (PBS) to 1 × 106 pfu/mL, and to 1 × 105 pfu/mL in the second study. The hamsters were first lightly anesthetized with 10 mg of ketamine hydrochloride and 1 mg of xylazine hydrochloride. Each hamster was administered virus via 200 µL pipette into the nares (50 µL/nare) for a total volume of 100 µL per hamster. Virus back-titration was performed on Vero E6 cells immediately following inoculation. Hamsters were observed until fully recovered from anesthesia. All hamsters were maintained for three days then humanely euthanized and necropsied.
Oropharyngeal swabs were also taken prior to live virus challenge and days 1–3 after challenge to evaluate viral shedding. Swabs were placed in BA-1 medium (Tris-buffered MEM containing 1% BSA) supplemented with antibiotics then stored at −80 °C until further analysis.
2.11. Virus Titration
Plaque assays were used to quantify infectious virus in oropharyngeal swabs and tissues. Briefly, all samples were serially diluted 10-fold in BA-1 media supplemented with antibiotics. Confluent Vero E6 cell monolayers were grown in 6-well tissue culture plates. The growth media was removed from the cell monolayers and washed with PBS immediately prior to inoculation. Each well was inoculated with 0.1 mL of the appropriate diluted sample. The plates were rocked every 10–15 min for 45 min and then overlaid with 0.5% agarose in media with 7.5% bicarbonate and incubated for 1 day at 37 °C, 5% CO
2. A second overlay with neutral red dye was added at 24 h and plaques were counted at 48–72 h post-plating. Viral titers are reported as the log
10 pfu per swab or gram (g). Samples were considered negative for infectious virus if viral titers were below the limit of detection (LOD). The theoretical limit of detection was calculated using the following equation:
where N is the number of replicates per sample at the lowest dilution tested; V is the volume used for viral enumeration (volume inoculated/well in mL). For oropharyngeal swabs the LOD was 10 pfu/swab or 1.0 log
10 pfu/swab. For tissues the LOD was 100 pfu/g or 2.0 log
10 pfu/g. For testing of the inactivated virus preparations, the LOD was < 0.1 pfu/mL or < −1.0 log
10 pfu/mL.
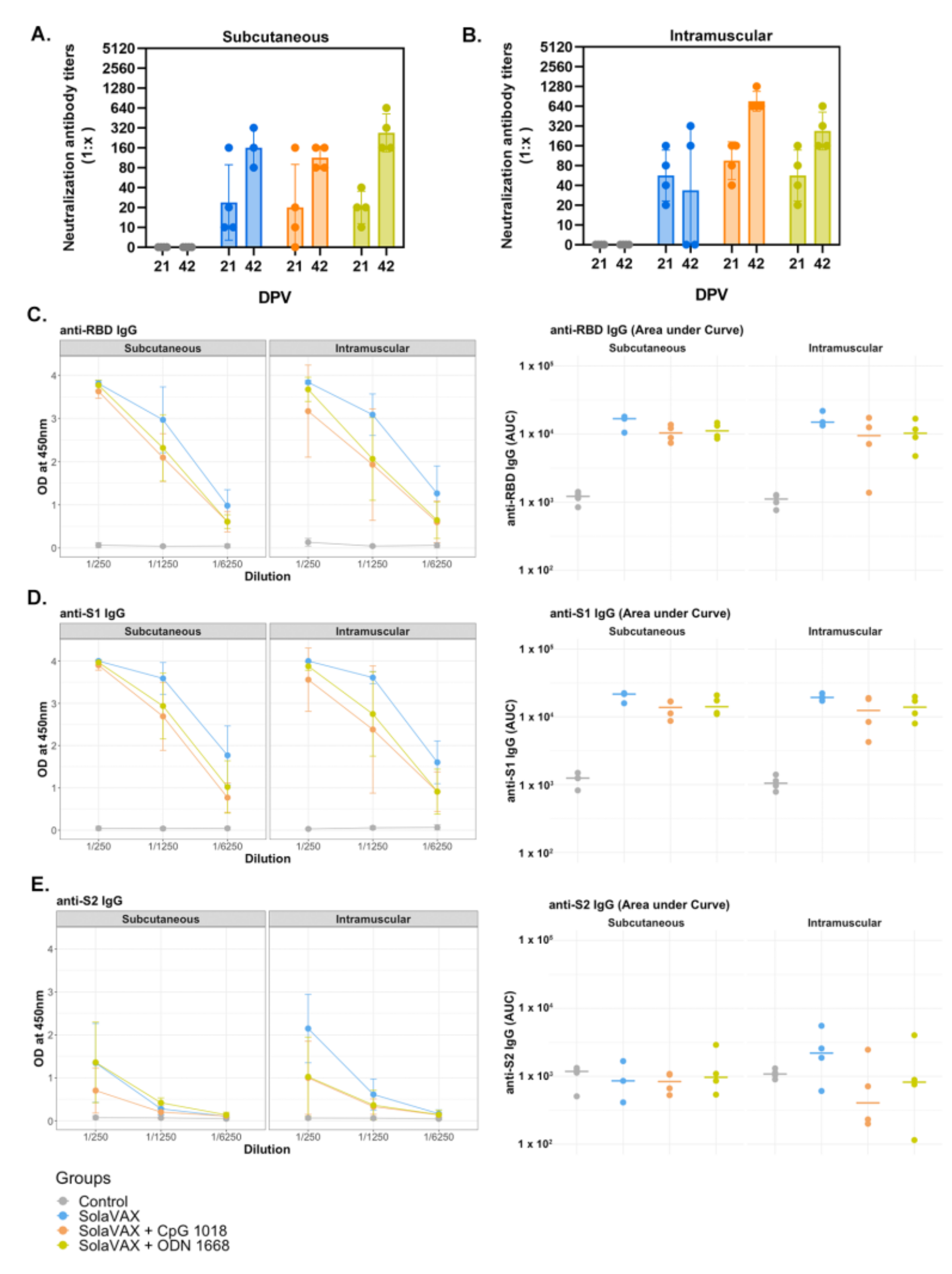
2.12. Plaque Reduction Neutralization Test
The production of neutralizing antibodies was determined by plaque reduction neutralization test. Briefly, serum was first heat-inactivated for 30 min at 56 °C in a waterbath. Then, serum samples were diluted two-fold in BA-1 media starting at a 1:5 dilution on a 96-well plate. An equal volume of SARS-CoV-2 virus (isolate USA-WA1/2020) was added to the serum dilutions and the sample-virus mixture was gently mixed. The plates were incubated for 1-h at 37 °C. Following incubation, serum-virus mixtures were plated onto Vero E6 plates as described for virus plaque assays. Antibody titers were recorded as the reciprocal of the highest dilution in which >90% of virus was neutralized. All hamsters were tested for the presence of antibodies against SARS-CoV-2 prior to vaccination.
2.13. ELISA for Anti S1, S2 and RBD Antibodies
ELISA was performed to evaluate antibody binding to SARS-CoV-2 spike protein region S1 (16-685 amino acids), S2 (686-1213 amino acids), and RBD (319-541 amino acids) (all recombinant proteins from SinoBiological, Wayne, PA, USA). The procedure was adapted from Robbiani et al. [
24], with few modifications. Briefly, high binding 96-well plates (Corning, St. Louis, MO, USA) were coated with 50 ng of S1, S2, and RBD protein prepared in PBS and incubated overnight at 4 °C. Plates were washed 5 times with PBS + 0.05% Tween 20 (Sigma, St. Louis, MO) and incubated with blocking buffer (PBS + 2% BSA + 2% normal goat serum + 0.05% Tween 20) for 2 h at room temperature (RT). Serial dilutions (1/250, 1/1250, and 1/6250) of serum obtained from naïve, non-vaccinated and vaccinated hamsters were prepared in blocking buffer and added to the plates for 1 h. After washing, 1:10,000 dilution of HRP conjugated anti-hamster IgG (H+L) secondary antibody (Jackson Immuno Research, 107-035-142) prepared in blocking buffer was added and incubated for 1 h. Plates were washed, TMB substrate (ThermoFisher, Waltham, MA, USA) added, and the reaction was stopped after 10 min by adding 1M H
2SO
4. Absorbance was measured at 450 nm using a Biotek Synergy 2 plate reader (Winnoski, VT, USA).
2.14. Tissue Collection
Necropsies were performed at three or seven days after live virus challenge to note gross pathological changes. Tissues were collected for virus quantification, histopathology, flow cytometry, and single cell sequencing. For virus quantitation, portions of right cranial lung lobe, right caudal lung lobe, trachea, and nasal turbinate specimens from each hamster were weighed (100 mg per specimen) and homogenized in BA-1 media with antibiotics then frozen to −80 °C until the time of analysis. The tissue homogenates were briefly centrifuged and virus titers in the clarified fluid was determined by plaque assay. Viral titers of tissue homogenates are expressed as pfu/g (log10). For histopathology, portions of the right cranial lung lobe, right caudal lung lobe, trachea, nasal turbinates, and spleen were collected from each hamster. The tissues were placed in 10% neutral buffered formalin for seven days then paraffin embedded and stained with hematoxylin and eosin using routine methods for histological examination. For flow cytometry and single cell sequencing, a portion of the left cranial lung lobe and spleen from each hamster was placed in PBS and immediately processed for analysis.
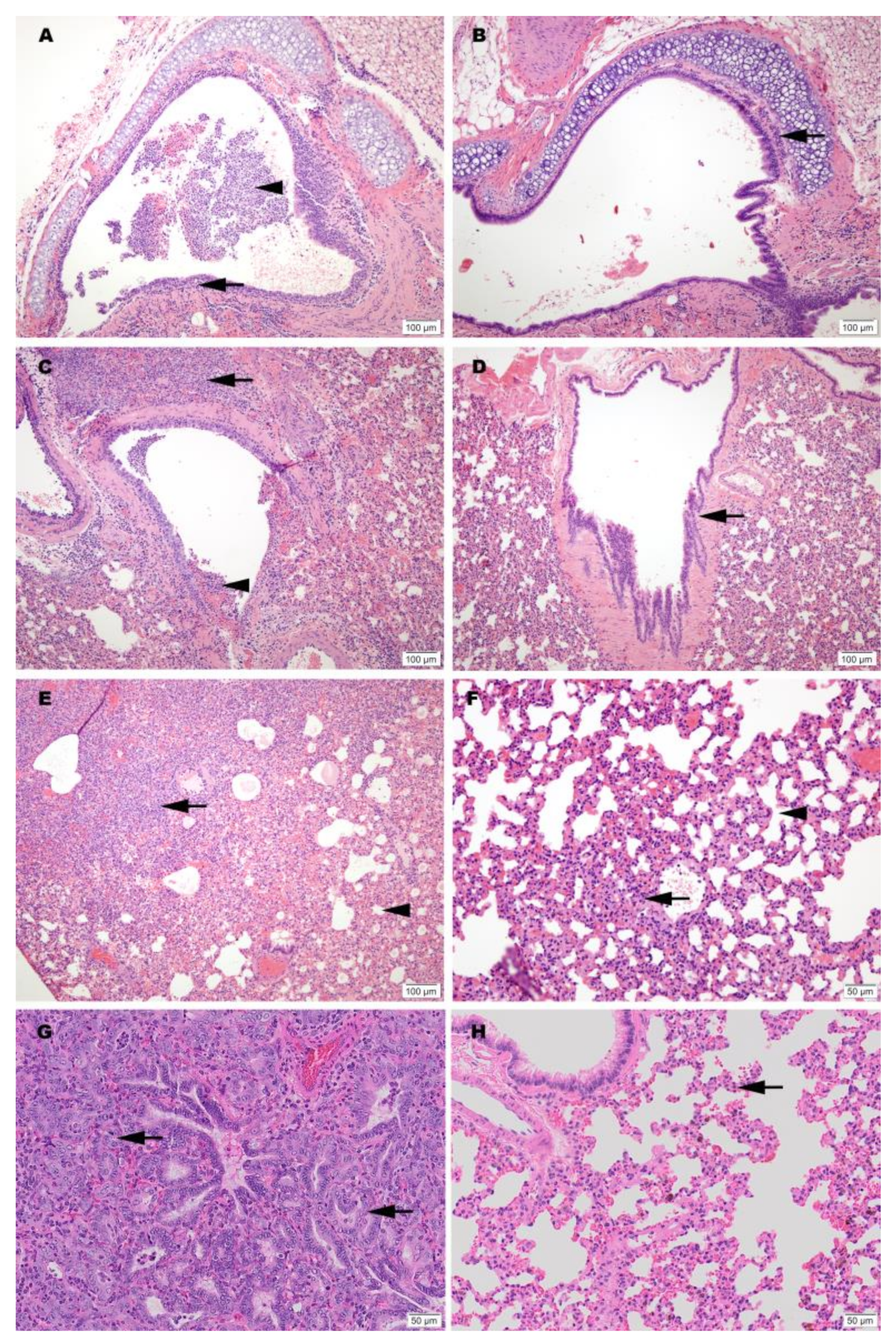
2.15. Histopathology
Histopathology was blindly interpreted by a board-certified veterinary pathologist (Podell BK) at Colorado State University. The H&E slides were evaluated for morphological evidence of inflammatory-mediated pathology in lung, trachea, heart and spleen, and reduction or absence of pathological features used as an indicator of vaccine-associated protection. Each hamster was assigned a score of 0–3 based on absent, mild, moderate, or severe manifestation, respectively, for each manifestation of pulmonary pathology including mural bronchial inflammation, neutrophilic bronchitis, consolidating pneumonia, and interstitial alveolar thickening, then the sum of all scores provided for each hamster.
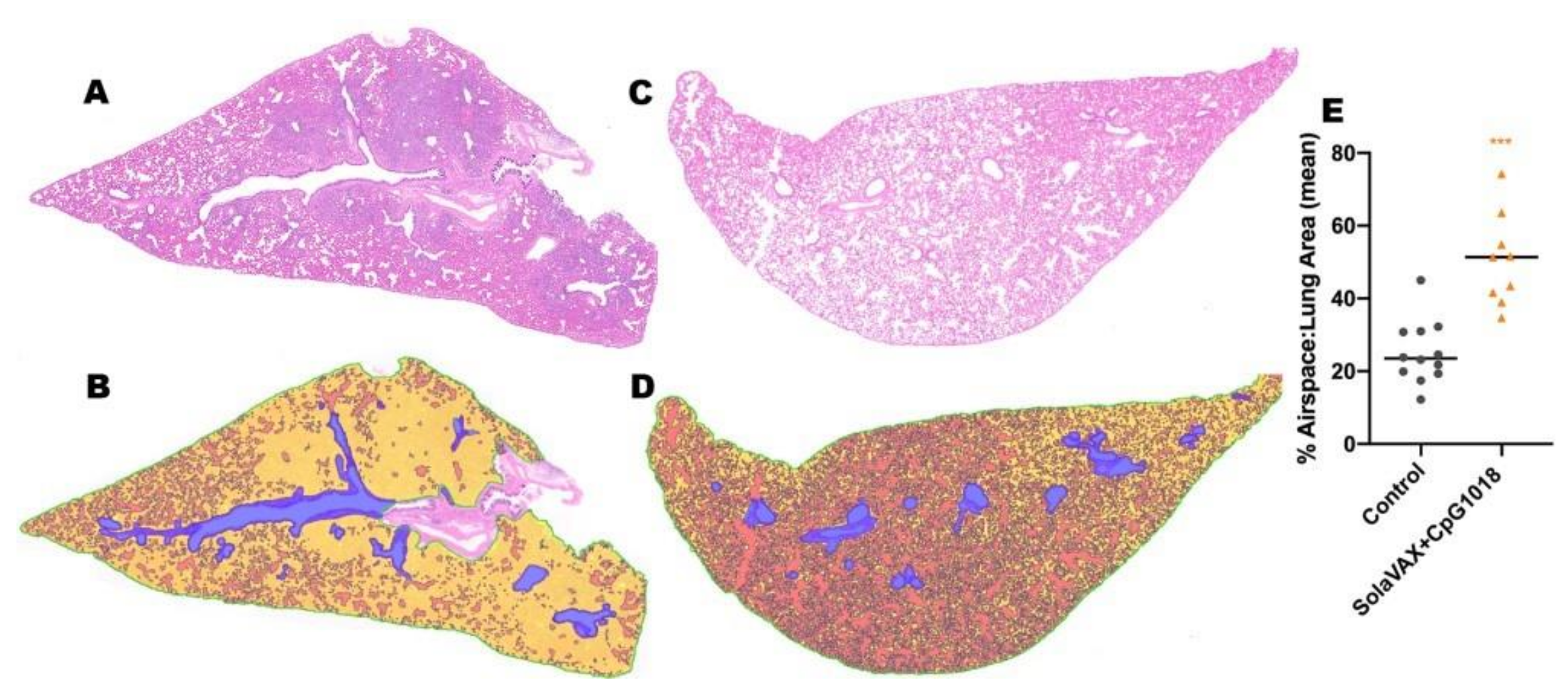
H&E-stained lung tissue slides were scanned at 20× magnification using an Olympus VS120 microscope, Hamamatsu ORCA-R2 camera, and Olympus VS-ASW 2.9 software through the Experimental Pathology Facility at Colorado State University. Visiopharm software was used for image analysis to detect and quantify alveolar airspace. For each tissue section, a region of interest (ROI) identification algorithm was generated at a low magnification with custom decision forest training and classification to differentiate tissue versus background based on color and area. Alveolar airspace was identified within tissue ROIs using a formulated K-means clustering algorithm based on staining intensity, area, and morphological features. Varying tissue areas were normalized by identifying and excluding major airways and vessels from the final analysis. Alveolar airspace identification and quantification were then reviewed and edited by a pathologist (Podell BK) and data expressed as a percent ratio of airspace to solid tissue.
2.16. Processing of Lungs, Spleen and Blood
Lungs and spleens were processed as described by Fox et al. [
25]. Briefly, a portion of the left cranial lung lobe and spleen from each hamster were aseptically transferred from PBS into DMEM then teased apart. Lungs were treated with a solution of DNase IV (500 units/mL) and Liberase (0.5 mg/mL) for 30 min at 37 °C to dissociate and digest collagen. Both lung and spleen cells were homogenized using a syringe plunger and passed through a 70 µm filter to prepare single cell suspension. Erythrocytes were lysed using Gey’s RBC lysis buffer (0.15 M NH
4Cl, 10 mM HCO
3) and cells were resuspended in 1 mL of complete media.
For blood, buffy coat was harvested by adding equal volume of PBS + 2% FBS to the blood and centrifuging at 800× g for 10 min at 25 °C with brakes off. The buffy coat was collected and washed, and erythrocytes were lysed using 1× Miltenyi RBC lysis buffer (Miltenyi, CA, USA). Cells were washed and resuspended in 1 mL complete media. After adding absolute counting beads (Invitrogen), total cell numbers of lung, spleen and blood were determined by flow cytometry analysis using an LSR-II (BD).
2.17. Flow Cytometry Staining
Flow cytometry staining was performed as mentioned by Fox et al. [
25]. Briefly, 2 × 10
6 cells were added into each well of a 96-well v-bottom plate and incubated with 1× Brefeldin A at 37 °C for 4 h. Cells were washed and stained with Zombie NIR live/dead stain, washed and further stained with predetermined optimal titrations of specific surface antibodies (
Table S1) and fluorescence minus one (FMOs). For intracellular staining, cells were further incubated with 1× Foxp3 Perm/Fix buffer (eBiosciences, San Diego, CA, USA) for 1 h at 37 °C, washed with 1× permeabilization buffer (eBiosciences, San Diego, CA, USA) twice and stained with intracellular antibodies cocktail (prepared in 1× permeabilization buffer) and respective FMOs overnight at 4 °C. The next day, cells were washed twice and resuspended in 300 μL of 1× Permeabilization buffer. Samples were acquired using a Cytek Aurora spectral flow cytometer where 100,000 events were recorded.
2.18. Single Cell RNA Sequencing
Lungs cells were prepared as described above, filtered, washed, and resuspended in PBS + 0.4% BSA. Cells were counted using a hemocytometer, and ~12,000 cells were added to the 10× Genomics chromium Next GEM Chip for a target recovery of 8000 cells. GEMs were placed in a thermal cycler and cDNA purified using Dynabeads. cDNA amplification was done using 10× Genomics single cell v3′ chemistry as per the manufacturer’s recommendations. The amplification PCR was set at 11 cycles and to eliminate any traces of primer-dimers, the PCR amplified cDNA product was purified using 0.6× SPRI beads (Beckman Coulter) before using the DNA for sequencing library preparation. Quality and quantity of cDNA was determined via Agilent TapeStation analysis using a HS-D5000 screen tape (
Figure S1). Twenty-five percent (25%) of the total cDNA amount was carried forward to generate barcoded sequencing libraries with 10 cycles of Sample Index PCR in 35-mL reaction volume (
Figure S2). Libraries were then pooled at equal molar concentration (
Figure S3) and sequenced on an Illumina NextSeq 500 sequencer to obtain a total of 941M read pairs (Illumina). An average of 78M read pairs per sample were generated with a standard deviation of 10.7M read pairs. Low-quality cells with <200 genes/cell and cells that express mitochondrial genes in >15% of their total gene expression were excluded. Gene expression in each group was normalized based on the total read count and log transformed.
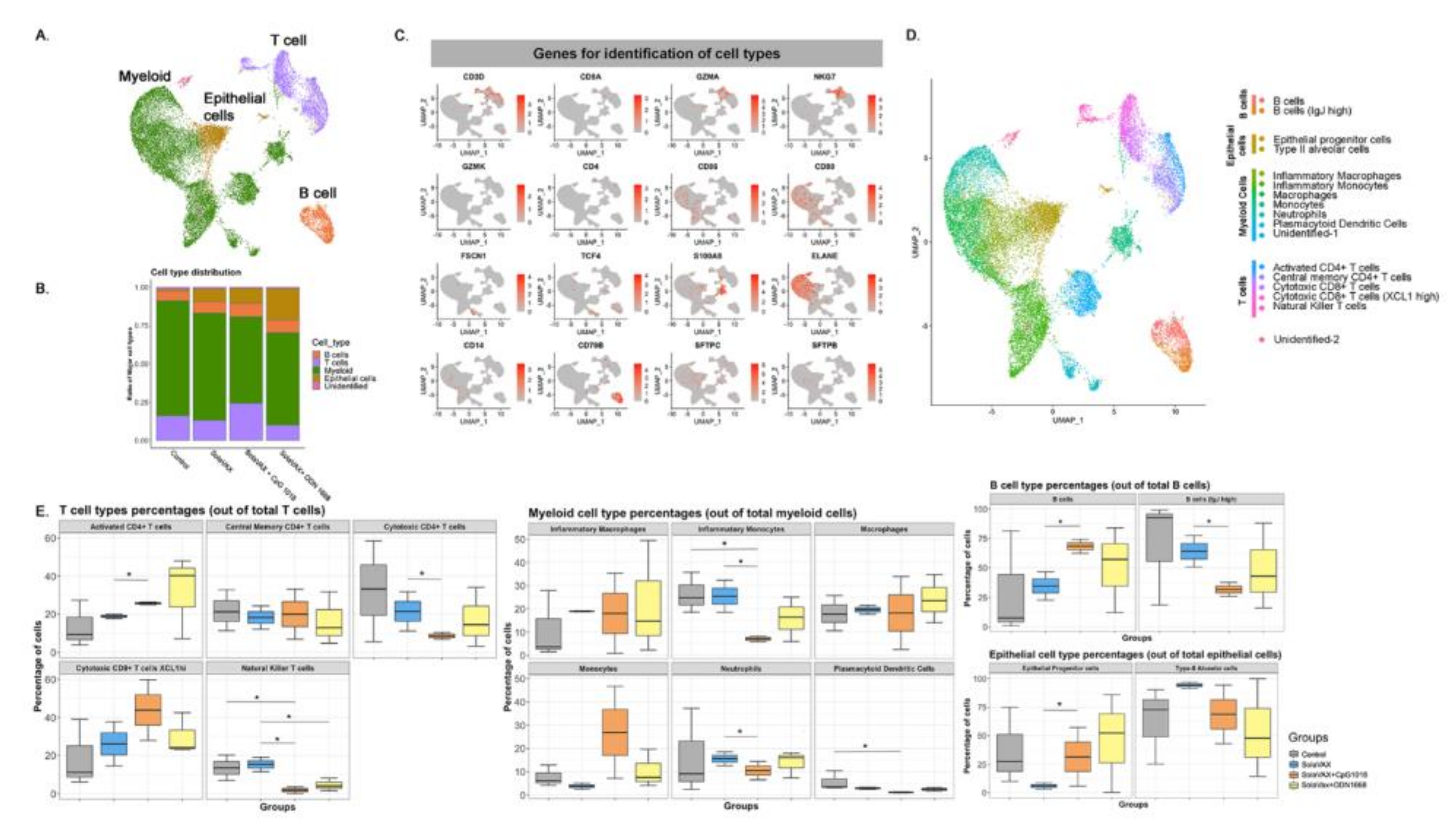
Sequenced samples were de-multiplexed using Cell Ranger mkfastq (Cell Ranger 10× Genomics, v3.0.2) to generate fastq files and aligned to the Mesocricetus auratus (accession GCA_000349665) and SARS-CoV-2 (reference genome MN985325) reference genomes using CellRanger count pipeline. Filtered barcode matrices were analyzed by Seurat package Version 3.0. Low quality cells, defined as expressing <200 genes/cell or those in which mitochondrial genes corresponded to >15% of their total gene expression, were excluded. Samples within groups were merged and downsampled to the 5466 cells per group. Thereafter, gene expression for each group was normalized based on total read counts and log transformed. All groups were integrated using Seurat integration strategy [
26], aligned samples scaled, and cells analyzed by unsupervised clustering (0.5 resolution), after principal components analysis (PCA). The top 15 principal components were visualized using UMAP. Differentially up-regulated genes in each cluster were selected with >0.25 log fold change and an adjusted
p < 0.05. Cell types were assigned by manually inspecting the top 20 upregulated genes, in addition to identifying previously published specific markers such as FSCN1 and GZMA for dendritic cells and CD8+ effector T cells, respectively [
27]. Differentially expressed genes (DEGs) between non-vaccinated group and vaccinated groups were identified using DESeq2 algorithm, with a Bonferroni-adjusted
p < 0.05 and a log
2 fold change > 1.
2.19. Statistical Analyses
The sample size for this preliminary evaluation will allow differentiation of proportions of 0.9 vs. 0.1 at power of 80% (Fisher’s exact test). Group mean (
n = 4) viral titers were analyzed using a two-way ANOVA analysis followed by a post hoc test to analyze differences between the Control group and the vaccinated groups. In the case where samples reached the LOD, values were entered as 0 for statistical analysis. Data were considered significant if
p < 0.05. Analysis was performed using GraphPad Prism software (version 8.4.2) (GraphPad Software, Inc, La Joia, CA). Mean (
n = 4) subjective pathology scores were compared between groups using the Kruskall-Wallis test for non-parametric data with an alpha of 0.05. The flow cytometry results were analyzed using FlowJo as well as a newly published methodology [
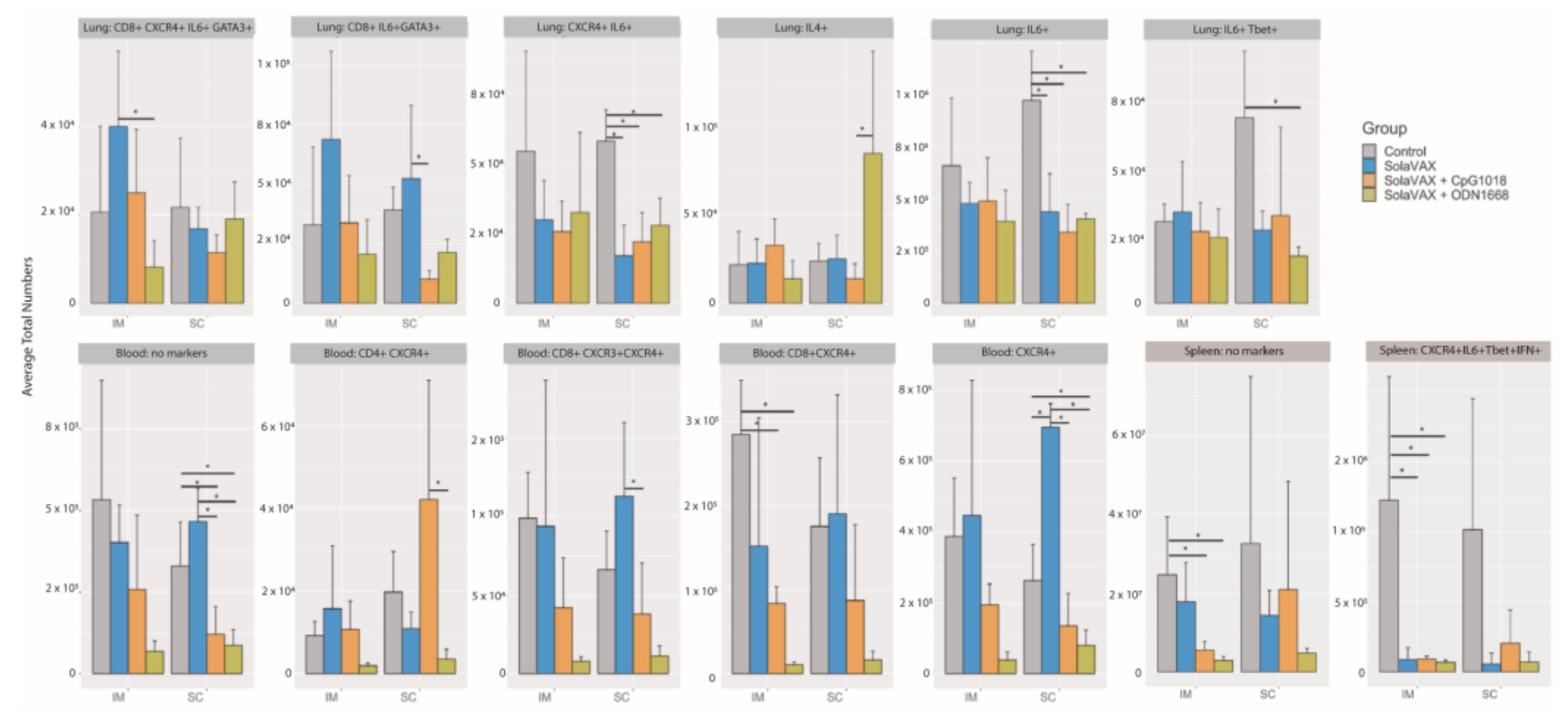
28]. Events were filtered to cell populations that constituted greater than 2% of the live leukocytes of at least one sample, where a cell population is defined by the combination of positive and negative markers. Statistical significance among A and among B groups was determined using Anova and Tukey Honest Significant Difference.
4. Discussion
Many routes to the preparation of vaccine candidates exist. All possess potential benefits and drawbacks. In this preliminary evaluation, we evaluated the ability of a photochemical process for inactivation of pathogens in blood products to use in production of an inactivated SARS-CoV-2 whole virion for prevention of COVID-19 infection.
Limitations of this work include small sample numbers and limited evaluation of the vaccine dose used in the study needed to induce an effect. Despite these limitations, clear differences due to route of administration and formulation of the vaccine (adjuvanted and non-adjuvanted) were observed. A clear difference between vaccinated and non-vaccinated animals was also evident in both humoral and cellular immune responses and as evidenced by protection from disease induced pathologies in lung tissues. Since the current study was conducted as an early evaluation of feasibility, additional replicates will be tested in future studies to further evaluate the immune response as a function of immunogen dose and composition.
Our motivation for studying this approach was based on the hypothesis that the ability to inactivate virus replication without inducing damage to protein epitopes, could result in the generation of a potent vaccine candidate with intact protein antigen targets comparable to native, live-type virus. Such a candidate was hypothesized to have several advantages including the use of chemical reagents with extensive safety toxicology and general handling benefits, equipment and disposables that are currently in routine use for producing human transfusion products and a cost profile in production that could be favorable for mass production and provide for both global affordability and availability.
This preliminary evaluation demonstrates the potential of this process to inactivate SARS-CoV-2 virus via a specific, targeted guanine base modification. This work has also demonstrated the potential for a product made via this method to induce a potent immune response to vaccination. This response triggered both Th1 and Th2 type immune pathways, leading to generation of neutralizing antibodies and cellular responses capable of protecting vaccinated animals against intranasal challenge with 105 pfu SARS-CoV-2. The use of adjuvants was found to boost the levels of neutralizing antibody titers. Interestingly, the non-adjuvanted formulation still provided sufficient protection to prevent viral production and shedding in challenged animals. Robbianni et al. [
24] have observed varying overall neutralizing antibody levels in plasma of convalescent COVID-19 patients with consistent levels of specific sub-types against RBD epitopes. They have speculated that a subclass of antibody against receptor binding domain (RBD) epitopes may be critical in conferring therapeutic benefit in those products. The extent to which this may play a role in vaccine efficacy is unknown.
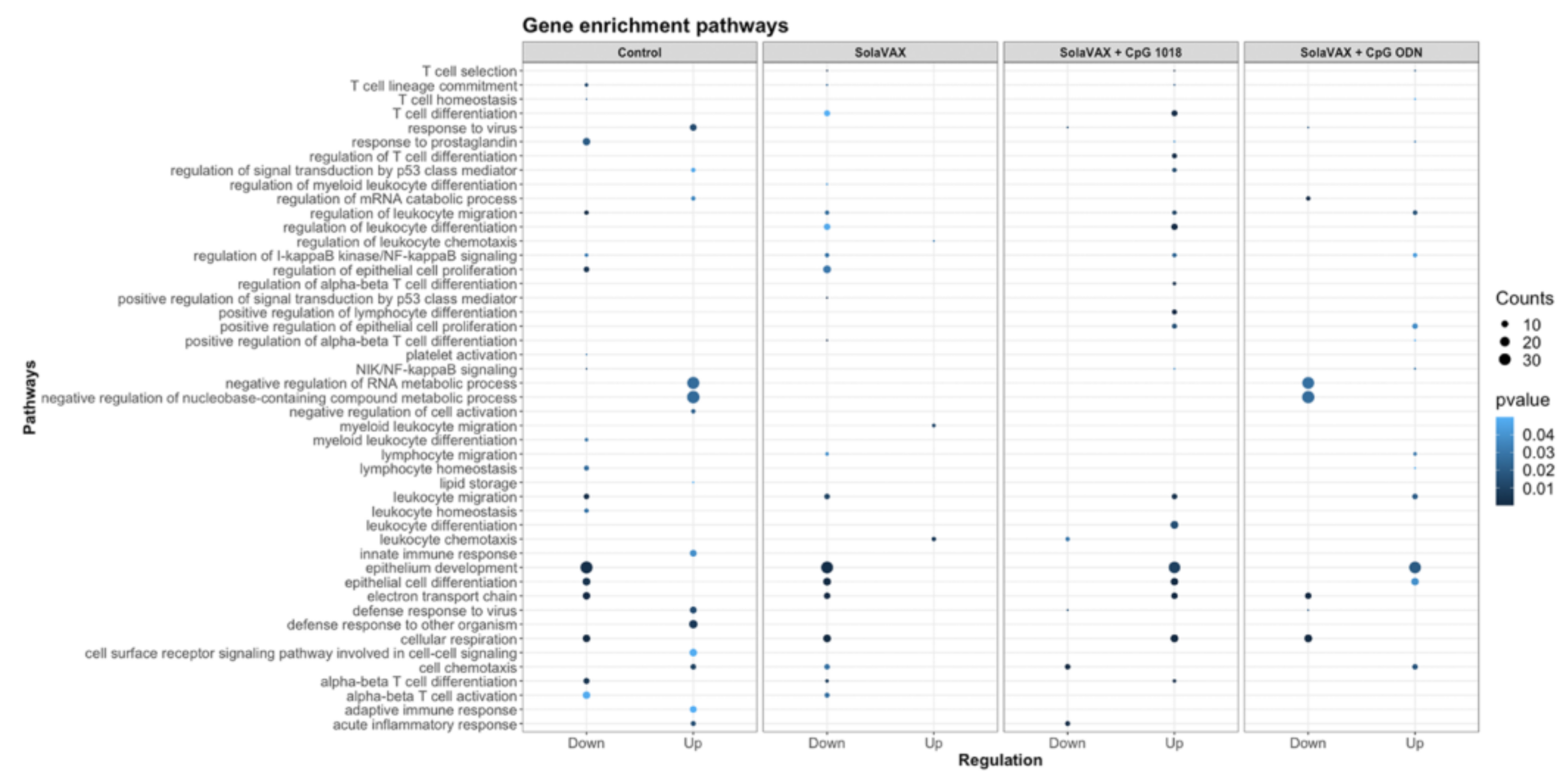
The immune system is a critical target of vaccine development, and to characterize various immune responses and how these responses relate to vaccine protection is of utmost importance. We applied single-cell RNA sequencing (scRNA-seq) to characterize the immune cell populations, and transcriptional changes in the lungs of hamsters between non-vaccinated and SvX vaccinated groups (with and without adjuvant) infected with SARS-CoV-2. These experiments reveal several aspects of the COVID-19 pathogenesis and the vaccine efficiency in terms of immune correlates. Our results demonstrate that compared to non-vaccinated hamsters, SvX vaccinated groups showed significant differences in the distribution of leukocyte populations infiltrating the lungs. Notably, inflammatory genes and cells related to inflammation (inflammatory monocytes, neutrophils, and Natural killer T cells), along with stress response genes, were significantly decreased in the CpG group along with the SARS-CoV-2 viral reads.
On the other hand, epithelial cell development genes, T cell differentiation genes, helper T cells, and B cells are upregulated in the CpG group. These results indicate that SvX if given in combination with CpG, can reduce the inflammation caused by SARS-CoV-2, decreasing the probability of “cytokine storm,” one of the leading causes of death COVID-19. We know that vaccines work by targeting and preparing the immune system to rapidly respond to similar antigens. However, in the case of coronavirus infections like SARS-CoV-2, vaccines may also result in vaccine-induced immunopathology [
20]. Thus, a better understanding of vaccine-elicited protective vs. deleterious immunological responses is necessary.
Adjuvanted formulations, particularly CpG 1018 demonstrated the lowest levels of viral shedding, preservation of normal lung morphology and airway passage integrity and reduced numbers of infiltrates in the trachea and lung tissue. This difference may be due to a difference in the concentrations that were used for the two adjuvants evaluated. The selection of adjuvant dose was based on recommendations from the manufacturer’s which was the best available information available for these formulations at the time of this study. Both adjuvants used in this study are known to promote Th1 immune pathway responses. The two adjuvant formulations selected for this study are commercially available and have been approved by the FDA for use in other human vaccine products (CpG 1018) or were available in sterile, endotoxin free formulations (ODN1668). They thus seemed very suitable for use in this setting. Prior work with vaccine candidates suggested that ADE leading to lung immunopathology might be avoided by using Th1 promoting adjuvants [
33]. Results observed here are consistent with those observations. Further studies with Th2 promoting adjuvant formulations such as alum are required, however, to further elucidate the significance of these findings.
This preliminary evaluation demonstrates that a COVID-19 vaccine candidate produced by this method has the potential to be effective in providing protection against challenge infection in a sensitive hamster model. Importantly, we believe that this initial work demonstrates the potential that such a production method could be applied to vaccine candidates targeting other viral, bacterial, and parasitic pathogens. We have already applied such an approach to the generation of solid tumor vaccines and evaluated their use in both murine and canine disease models [
34,
35]. Results from studies involving vaccine, boost and challenge material with different levels and types of spike variants demonstrate the broad responsiveness that a vaccine candidate prepared via this method may have relative to emerging variants. This suggests that further evaluation of this vaccine against current variant forms in the clinical setting are warranted.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}