
Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia

 , , , , , , , , , and
, , , , , , , , , and 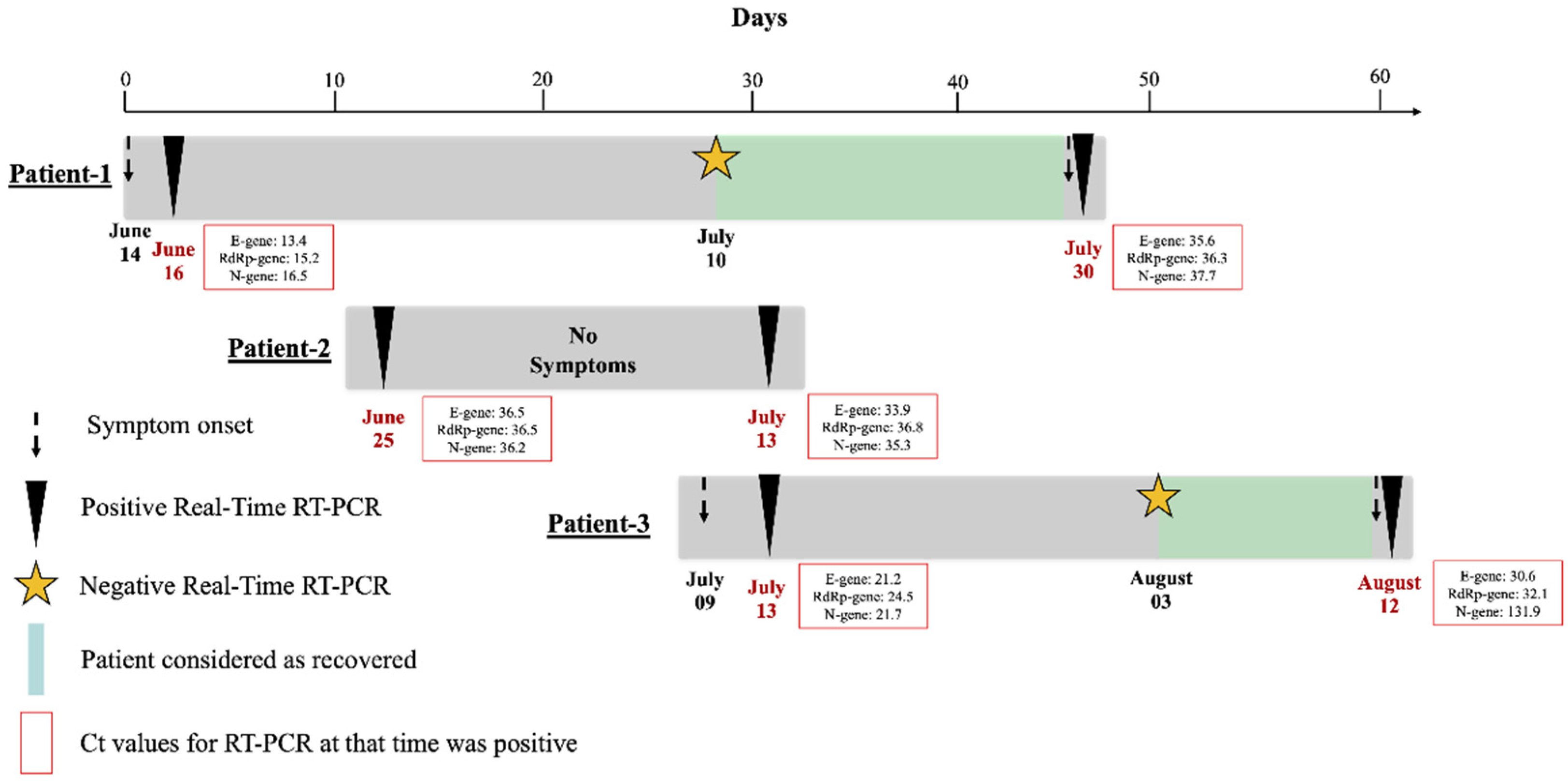{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
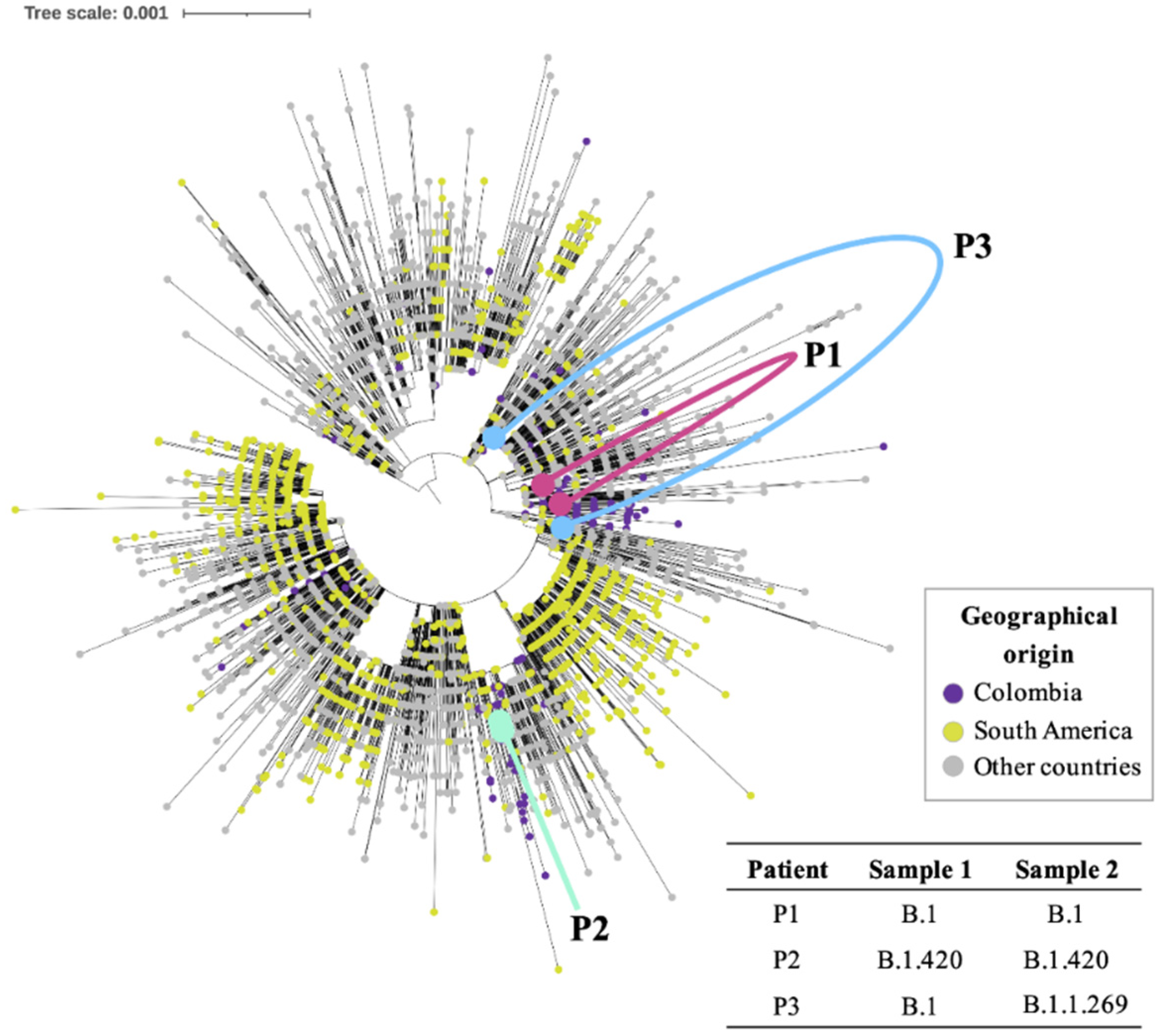
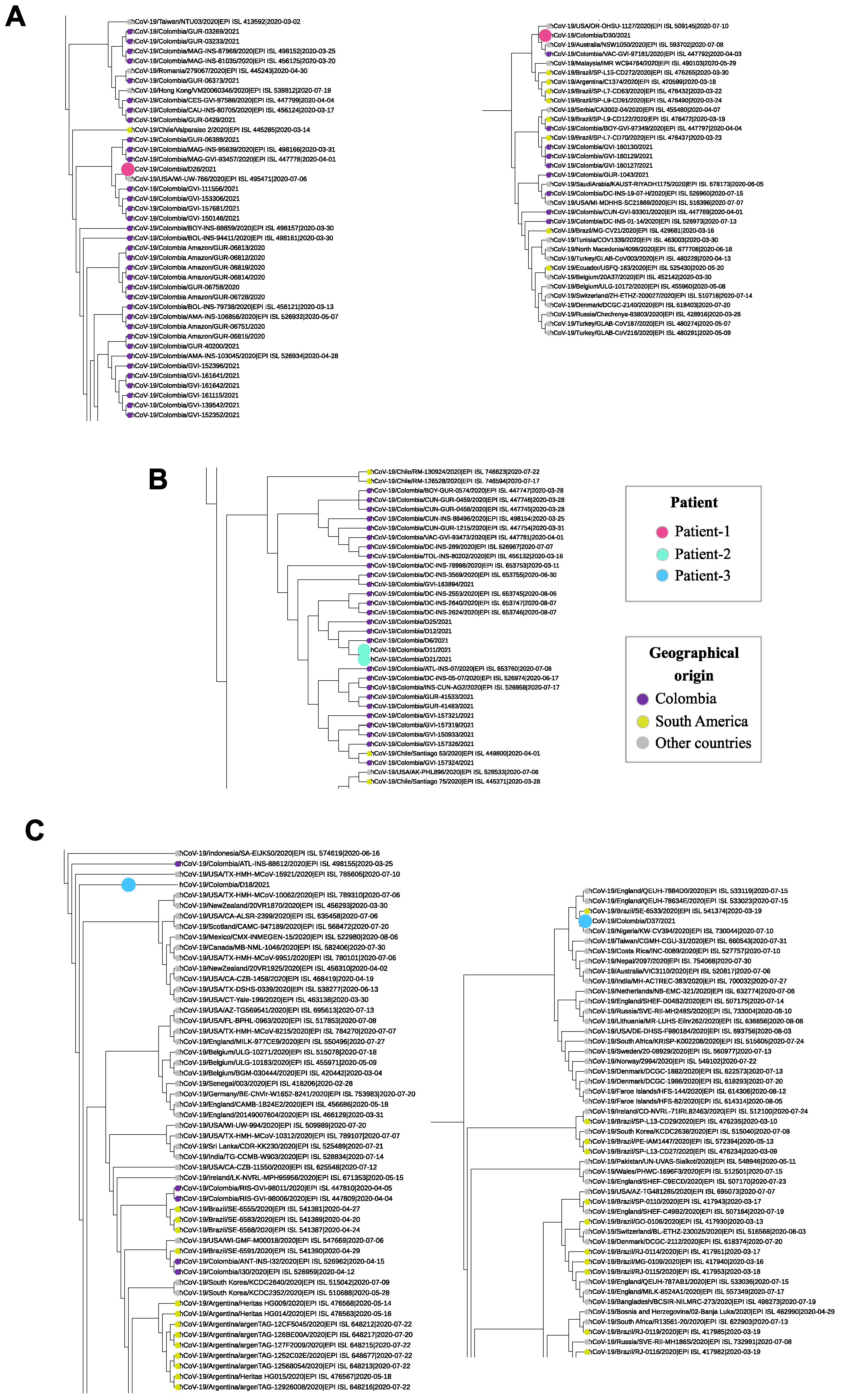
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 evolution and vaccines: Cause for concern? Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.-K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine–elicited human sera. Science 2021, eabg6105. Available online: http://science.sciencemag.org/content/early/2021/01/28/science.abg6105.abstract (accessed on 29 January 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. Available online: http://medrxiv.org/content/early/2020/12/22/2020.12.21.20248640.abstract (accessed on 1 January 2021).
- Faria, N.R.; Claro, I.M.; Candido, D.; Moyses Franco, L.A.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S.; et al. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 3 February 2021).
- Mulder, M.; van der Vegt, D.S.J.M.; Oude Munnink, B.B.; GeurtsvanKessel, C.H.; van de Bovenkamp, J.; Sikkema, R.S.; Jacobs, E.M.G.; Koopmans, M.P.G.; Wegdam-Blans, M.C.A. Reinfection of Severe Acute Respiratory Syndrome Coronavirus 2 in an Immunocompromised Patient: A Case Report. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Tillett, R.L.; Sevinsky, J.R.; Hartley, P.D.; Kerwin, H.; Crawford, N.; Gorzalski, A.; Laverdure, C.; Verma, S.C.; Rossetto, C.C.; Jackson, D.; et al. Genomic evidence for reinfection with SARS-CoV-2: A case study. Lancet Infect. Dis. 2021, 21, 52–58. [Google Scholar] [CrossRef]
- Prado-Vivar, B.; Becerra-Wong, M.; Guadalupe, J.J.; Márquez, S.; Gutierrez, B.; Rojas-Silva, P.; Grunauer, M.; Trueba, G.; Barragán, V.; Cárdenas, P. A case of SARS-CoV-2 reinfection in Ecuador. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kim, S.Y.; Kim, T.S.; Hong, K.H.; Ryoo, N.-H.; Lee, J.; Park, J.H.; Cho, S.I.; Kim, M.J.; Kim, Y.-G.; et al. Evidence of Severe Acute Respiratory Syndrome Coronavirus 2 Reinfection After Recovery from Mild Coronavirus Disease 2019. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Instituto Nacional de Salud Distribución Geográfica de los Linajes de SARS-CoV-2 Circulantes en Colombia. 2021. Available online: https://twitter.com/INSColombia/status/1359347653566476288/photo/1 (accessed on 1 March 2021).
- Novoa, W.; Miller, H.; Mattar, S.; Faccini-Martínez, Á.A.; Rivero, R.; Serrano-Coll, H. A first probable case of SARS-CoV-2 reinfection in Colombia. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Ramírez, J.D.; Florez, C.; Muñoz, M.; Hernández, C.; Castillo, A.; Gomez, S.; Rico, A.; Pardo, L.; Barros, E.C.; Castañeda, S.; et al. The arrival and spread of SARS-CoV-2 in Colombia. J. Med. Virol. 2021, 93, 1158–1163. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Muñoz, M.; Patiño, L.H.; Ballesteros, N.; Paniz-Mondolfi, A.; Ramírez, J.D. Characterizing SARS-CoV-2 genome diversity circulating in South American countries: Signatures of potentially emergent lineages? Int. J. Inf. Dis. 2021. [Google Scholar] [CrossRef]
- Stokel-Walker, C. What we know about covid-19 reinfection so far. BMJ 2021, 372, n99. Available online: http://www.bmj.com/content/372/bmj.n99.abstract (accessed on 19 January 2021). [CrossRef]
- Naveca, F.; da Costa, C.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Costa, Á.; Duarte, D.; Silva, G.; Mejía, M.; et al. SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P.1 in Amazonas, Brazil. 2021. Available online: https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596 (accessed on 1 March 2021).
- Zucman, N.; Uhel, F.; Descamps, D.; Roux, D.; Ricard, J.-D. Severe reinfection with South African SARS-CoV-2 variant 501Y.V2: A case report. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- de Souza, W.M.; Amorim, M.R.; Sesti-Costa, R.; Coimbra, L.D.; Toledo-Teixeira, D.A.; Parise, P.L.; Barbosa, P.P.; Bispo-dos-Santos, K.; Mofatto, L.S.; Simeoni, C.L.; et al. Levels of SARS-CoV-2 Lineage P.1 Neutralization by Antibodies Elicited after Natural Infection and Vaccination. Ssrn Electron. J. 2021. [Google Scholar] [CrossRef]
- Truong, T.T.; Ryutov, A.; Pandey, U.; Yee, R.; Goldberg, L.; Bhojwani, D.; Aguayo-Hiraldo, P.; Pinsky, B.A.; Pekosz, A.; Shen, L.; et al. Persistent SARS-CoV-2 infection and increasing viral variants in children and young adults with impaired humoral immunity. medRxiv 2021. Available online: http://medrxiv.org/content/early/2021/03/02/2021.02.27.21252099.abstr (accessed on 1 January 2021).
- Voloch, C.M.; da Silva F, R.; de Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; de C Guimarães, A.P.; de C Leitão, I.; Mariani, D.; Ota, V.A.; et al. Intra-host evolution during SARS-CoV-2 persistent infection. medRxiv 2020. Available online: http://medrxiv.org/content/early/2020/11/16/2020.11.13.20231217.abstract (accessed on 1 January 2021).
- Martin, D.; Weaver, S.; Tegally, H.; San, E.J.; Wilkinson, E.; Giandhari, J.; Naidoo, S.; Pillay, Y.; Singh, L.; Lessells, R.J.; et al. The Emergence and Ongoing Convergent Evolution of the N501Y Lineages Coincided with a Major Global Shift in the SARS-Cov-2 Selective Landscape. Available online: https://virological.org/t/the-emergence-and-ongoing-convergent-evolution-of-the-n501y-lineages-coincided-with-a-major-global-shift-in-the-sars-cov-2-selective-landscape/618 (accessed on 1 March 2021).
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine 2020, 61. [Google Scholar] [CrossRef]
- Arons, M.M.; Hatfield, K.M.; Reddy, S.C.; Kimball, A.; James, A.; Jacobs, J.R.; Taylor, J.; Spicer, K.; Bardossy, A.C.; Oakley, L.P.; et al. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N. Engl. J. Med. 2020, 382, 2081–2090. [Google Scholar] [CrossRef]
- Vibholm, L.K.; Nielsen, S.S.F.; Pahus, M.H.; Frattari, G.S.; Olesen, R.; Andersen, R.; Monrad, I.; Andersen, A.H.F.; Thomsen, M.M.; Konrad, C.V.; et al. SARS-CoV-2 persistence is associated with antigen-specific CD8 T-cell responses. EBioMedicine 2021, 64. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Song, S.; Ma, L.; Zou, D.; Tian, D.; Li, C.; Zhu, J.; Chen, M.; Wang, A.; Ma, Y.; Li, M.; et al. The Global Landscape of SARS-CoV-2 Genomes, Variants, and Haplotypes in 2019nCoVR. Genomics. Proteom. Bioinform. 2020. Available online: https://www.sciencedirect.com/science/article/pii/S1672022920301315 (accessed on 1 March 2021). [CrossRef]
- Wang, L.; Qu, L.; Yang, L.; Wang, Y.; Zhu, H. NanoReviser: An Error-Correction Tool for Nanopore Sequencing Based on a Deep Learning Algorithm. Fron. Genet. 2020, 11, 900. Available online: https://www.frontiersin.org/article/10.3389/fgene.2t020.00900 (accessed on 1 March 2021). [CrossRef]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and epidemic spread of SARS-Cov-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.S.; Au, C.H.; Lam, H.Y.; Wang, C.L.N.; Ho, D.N.-Y.; Lam, Y.M.; Chu, D.K.W.; Poon, L.L.M.; Chan, T.L.; Zee, J.S.-T.; et al. Evaluation on the use of Nanopore sequencing for direct characterization of coronaviruses from respiratory specimens, and a study on emerging missense mutations in partial RdRP gene of SARS-CoV-2. Virol. J. 2020, 17, 183. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health. 2021. Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 1 March 2021).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, J.D.; Muñoz, M.; Ballesteros, N.; Patiño, L.H.; Castañeda, S.; Rincón, C.A.; Mendez, C.; Oliveros, C.; Perez, J.; Márquez, E.K.; et al. Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia. Vaccines 2021, 9, 282. https://doi.org/10.3390/vaccines9030282
Ramírez JD, Muñoz M, Ballesteros N, Patiño LH, Castañeda S, Rincón CA, Mendez C, Oliveros C, Perez J, Márquez EK, et al. Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia. Vaccines. 2021; 9(3):282. https://doi.org/10.3390/vaccines9030282
Chicago/Turabian StyleRamírez, Juan David, Marina Muñoz, Nathalia Ballesteros, Luz H. Patiño, Sergio Castañeda, Carlos A. Rincón, Claudia Mendez, Carolina Oliveros, Julie Perez, Elizabeth K. Márquez, and et al. 2021. "Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia" Vaccines 9, no. 3: 282. https://doi.org/10.3390/vaccines9030282
APA StyleRamírez, J. D., Muñoz, M., Ballesteros, N., Patiño, L. H., Castañeda, S., Rincón, C. A., Mendez, C., Oliveros, C., Perez, J., Márquez, E. K., Ortiz, F. d. l. S., Correa-Cárdenas, C. A., Duque, M. C., & Paniz-Mondolfi, A. (2021). Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia. Vaccines, 9(3), 282. https://doi.org/10.3390/vaccines9030282