
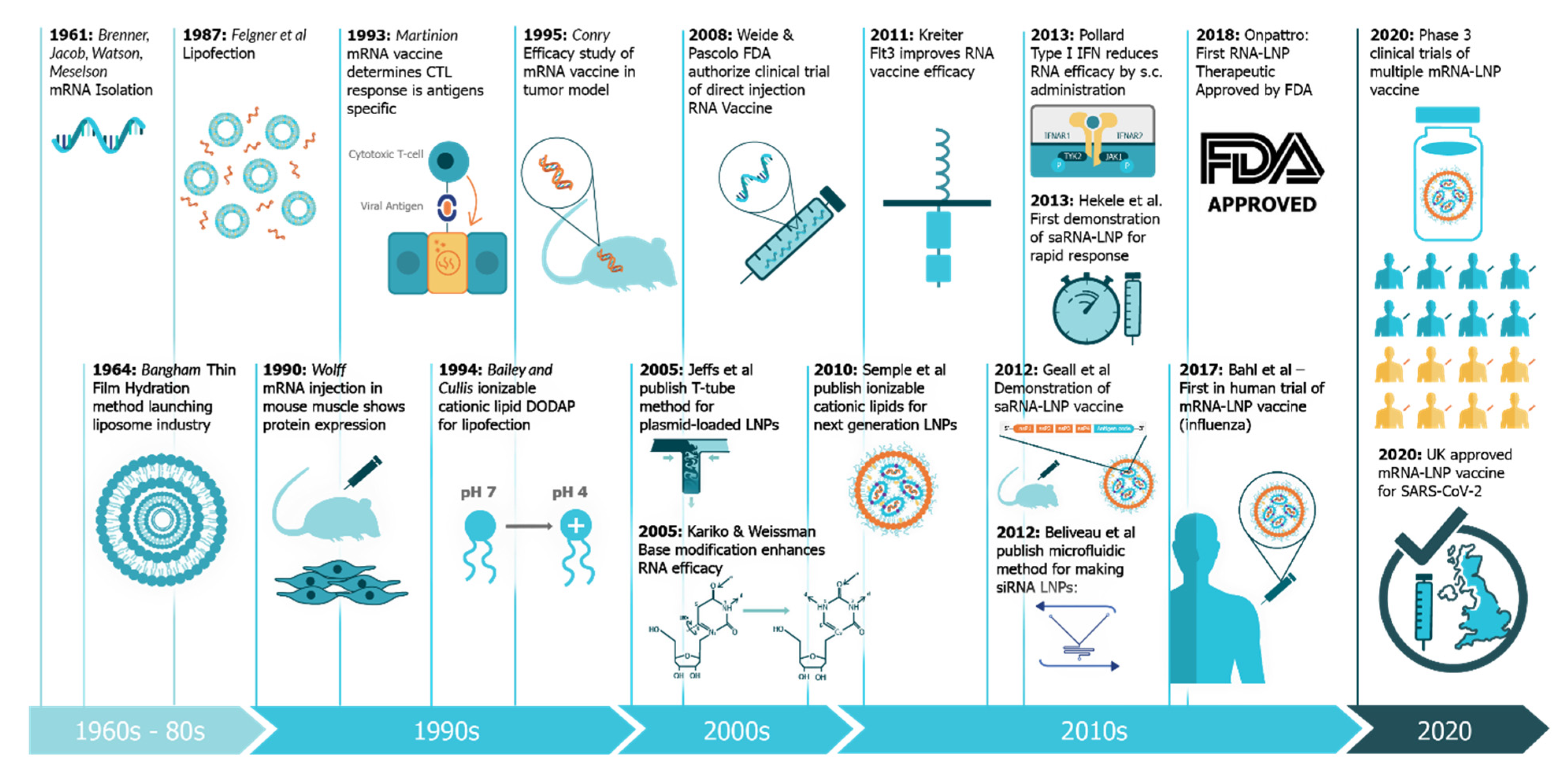
An Update on Self-Amplifying mRNA Vaccine Development


Abstract
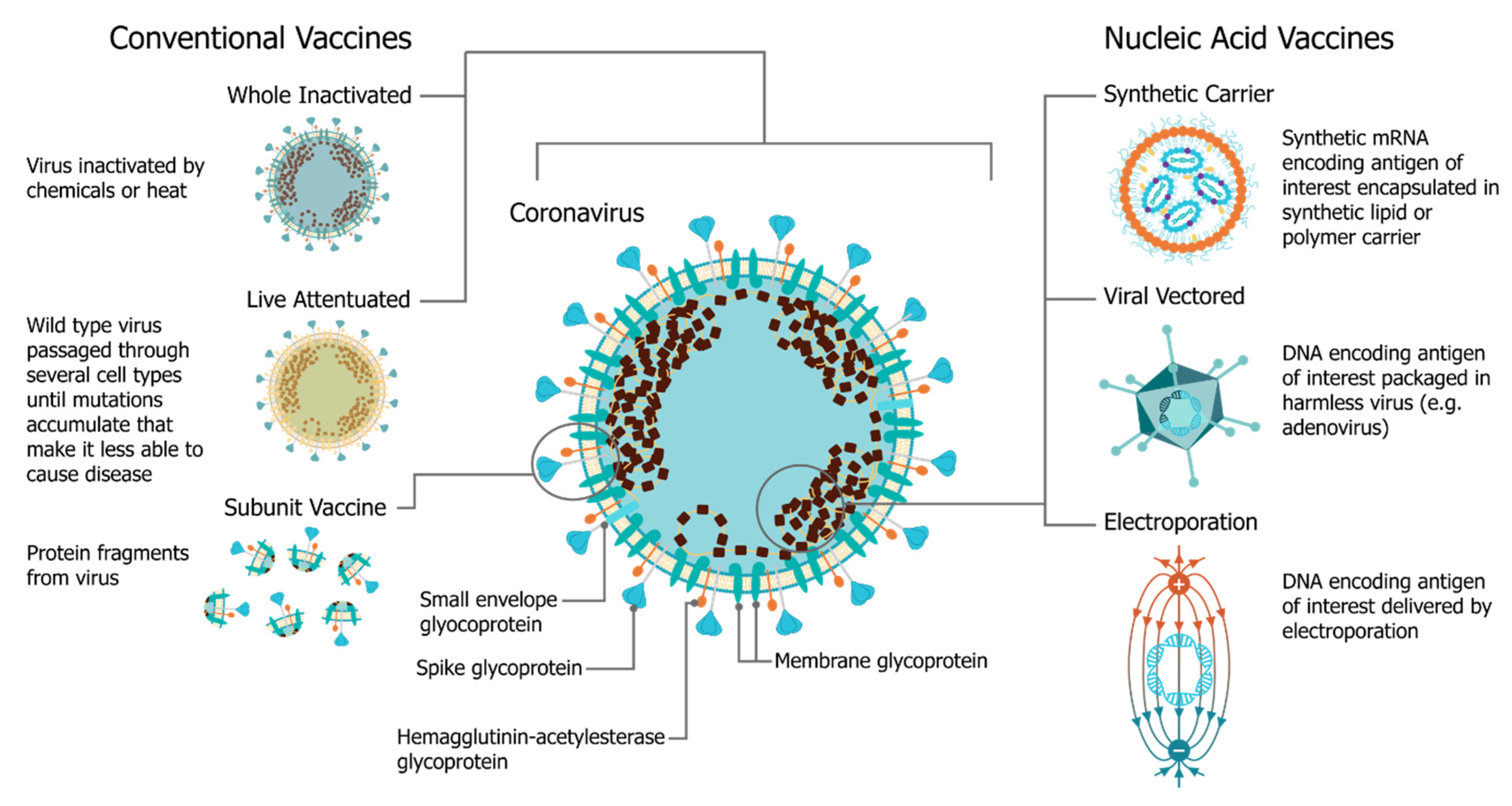
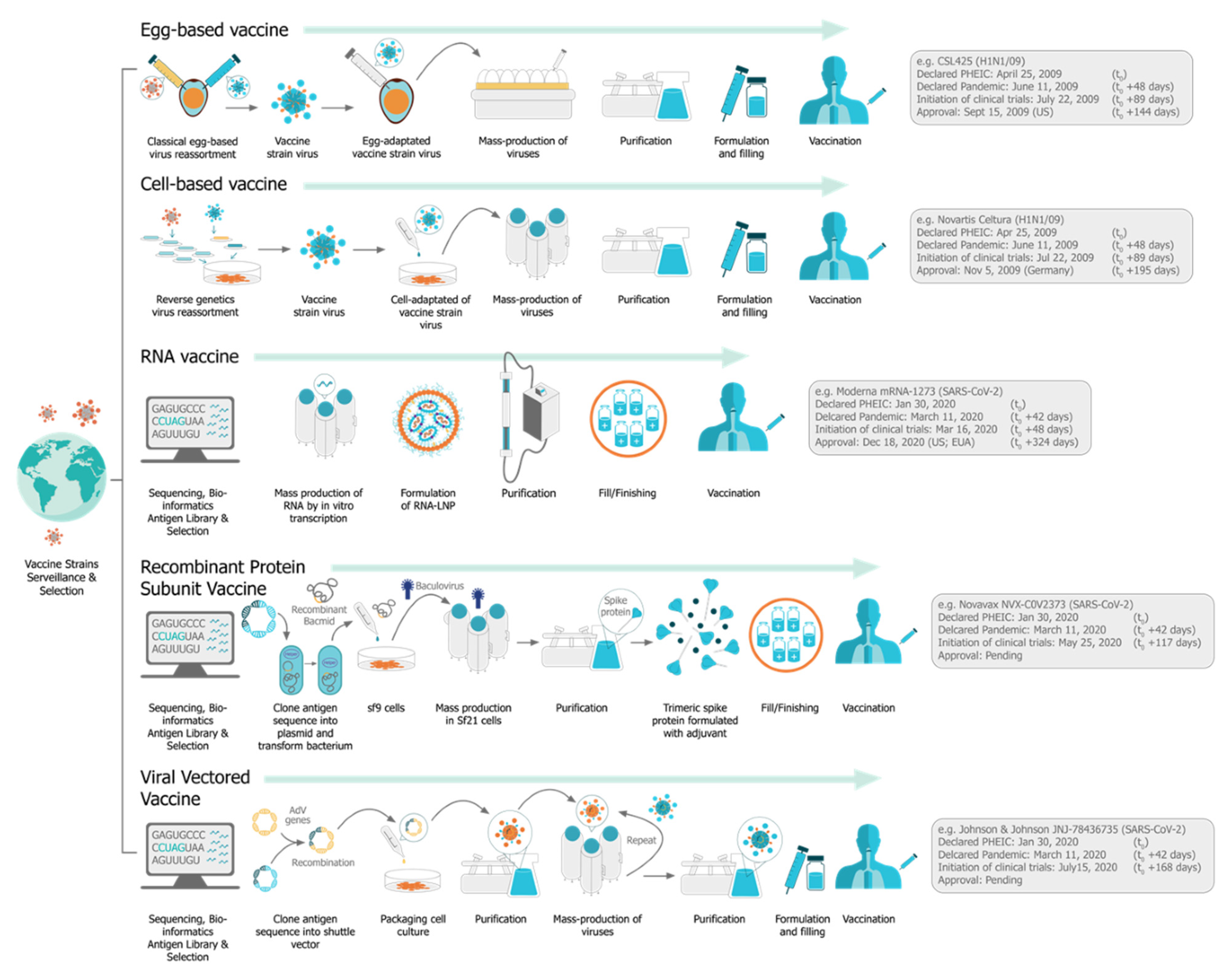
1. Introduction: The Four Pillars of saRNA Vaccines
2. Antigen Design
2.1. Infectious Diseases
2.1.1. Viral Glycoproteins
2.1.2. Bacterial Antigens
2.1.3. Parasitic Antigens
2.1.4. Monoclonal Antibodies for Passive Vaccination
2.2. Cancer
3. Vector Design
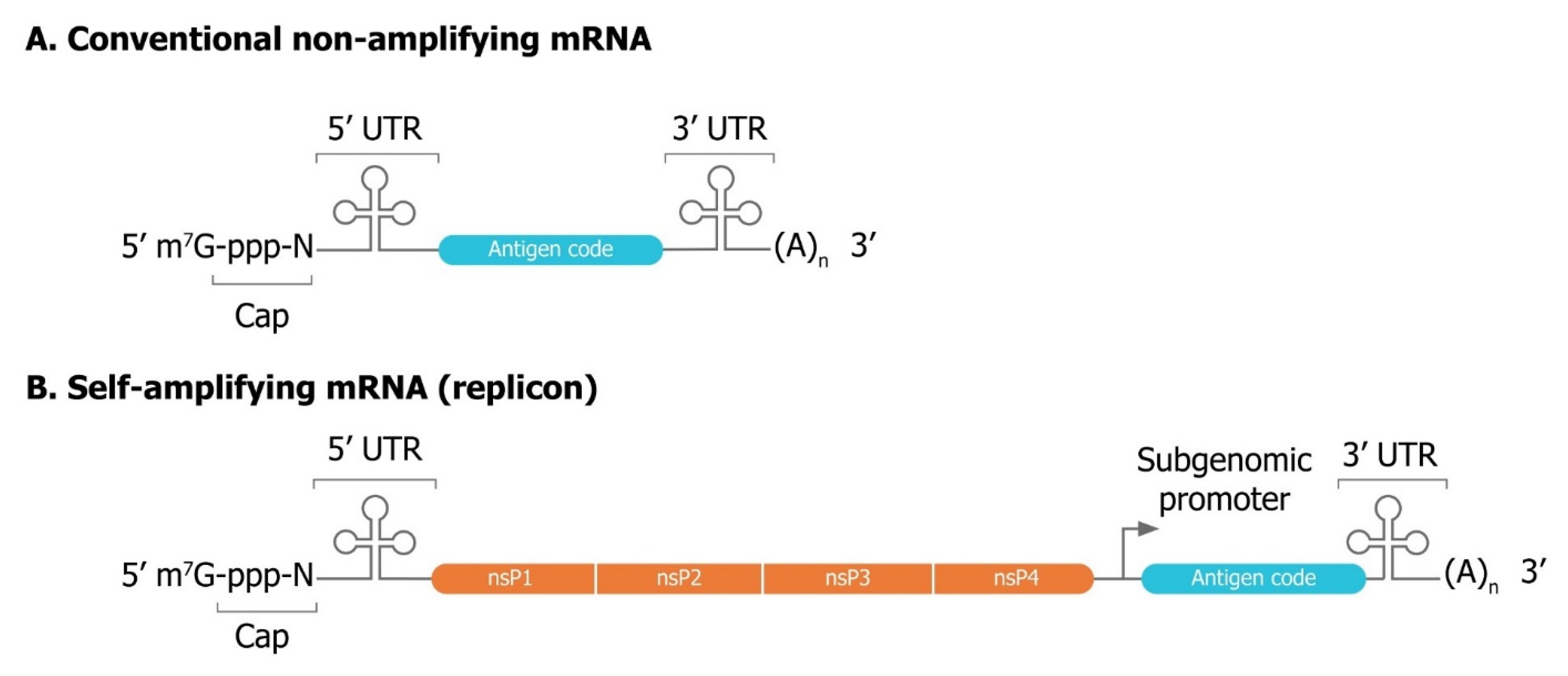
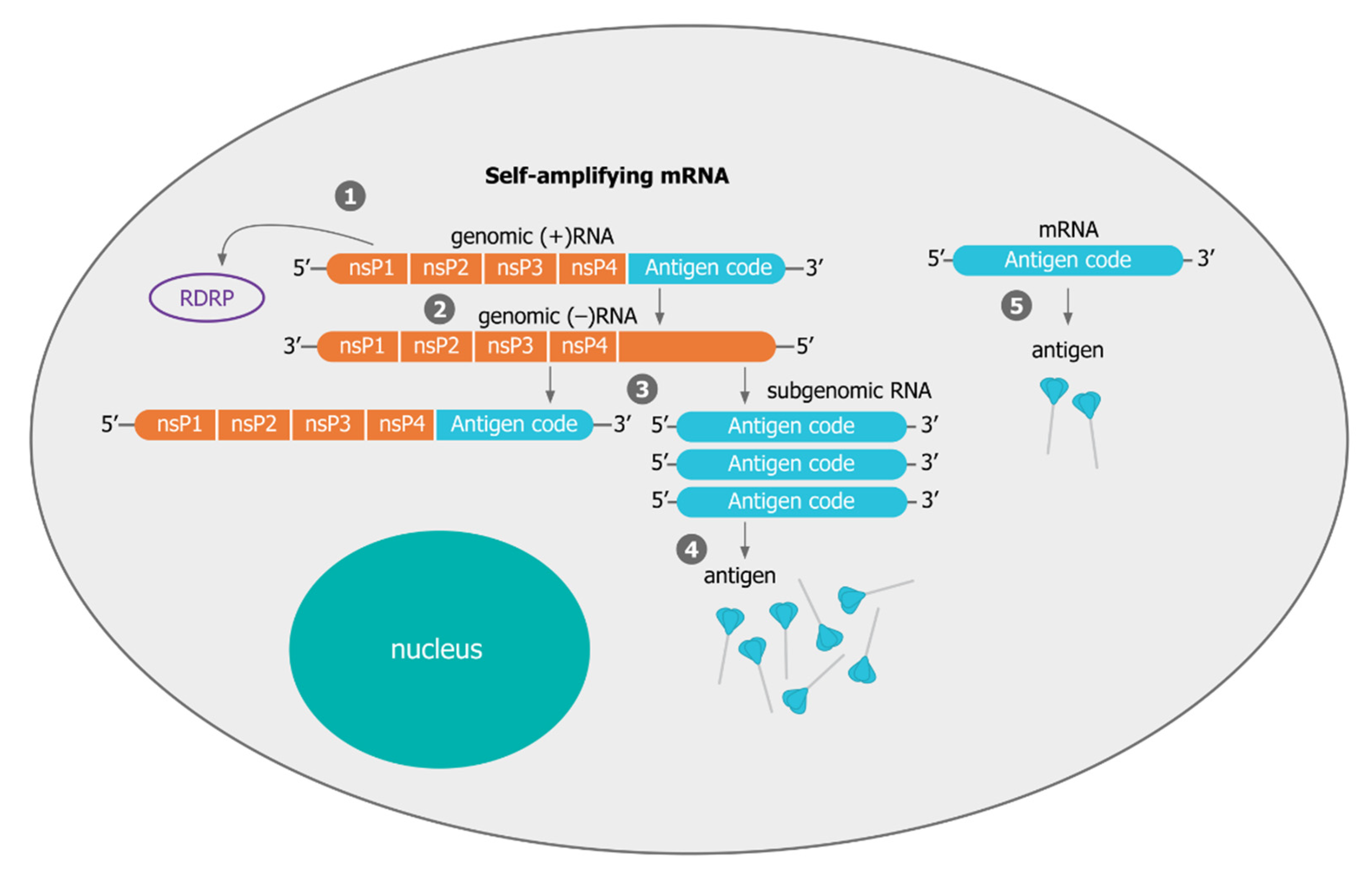
3.1. Mechanisms of Self-Amplification of RNA
3.2. Innovative Self-Amplifying RNA Vector Designs
3.3. Improving Immunogenicity with Molecular Interferon Modulators
4. Delivery Systems
4.1. Naked saRNA
4.2. Polymeric Nanoparticles
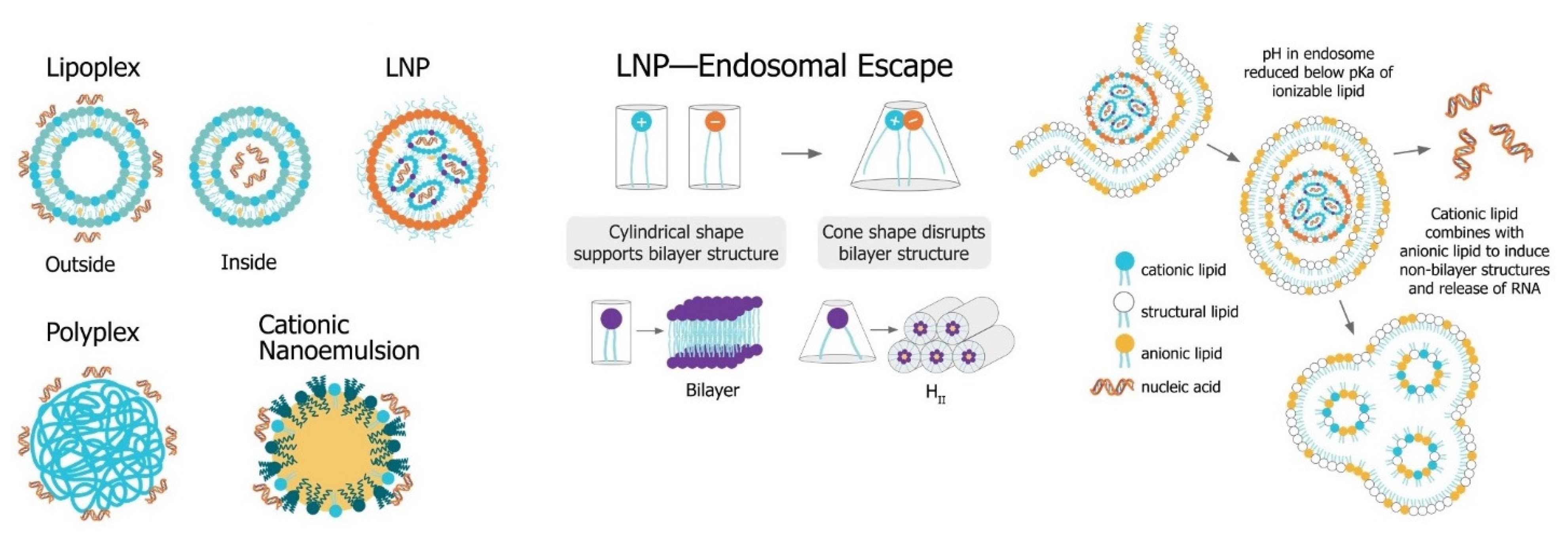
4.3. Lipid Nanoparticles
4.4. Nanoemulsions
4.5. Adjuvanted Delivery Systems
4.6. Delivery Platforms in the Clinic
5. Manufacturing
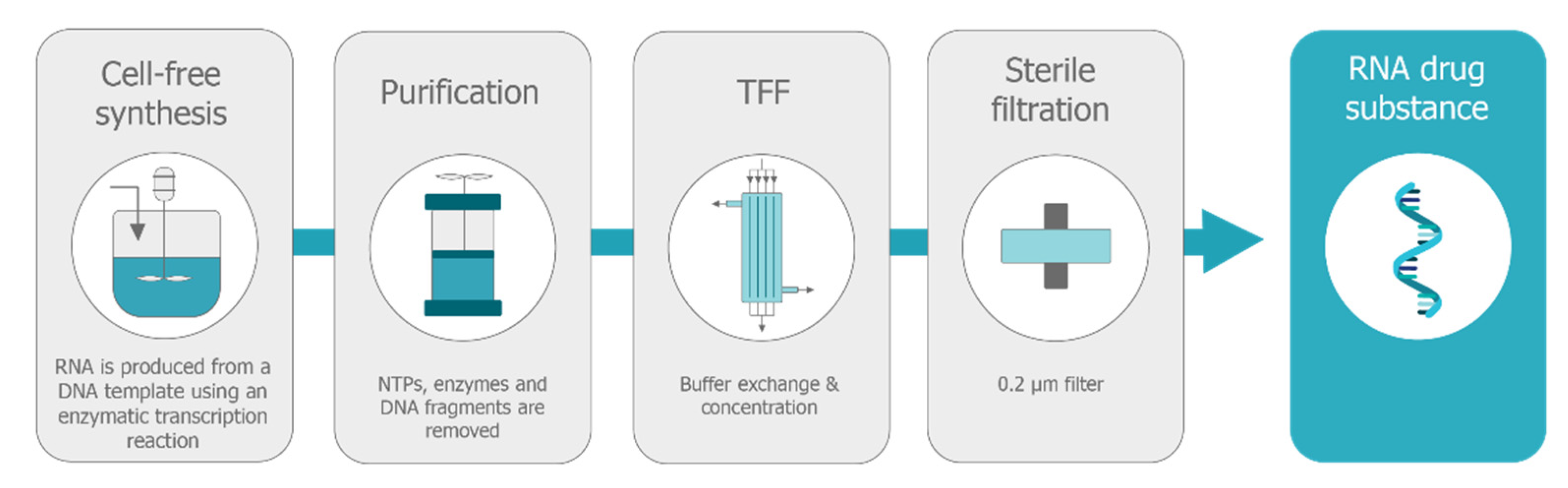
5.1. Production of Self-Amplifying mRNA
5.1.1. Capping Strategies for saRNA
5.1.2. Purification Strategies for saRNA
5.1.3. Immunostimulatory IVT Reaction By-Products
5.1.4. Stability of mRNA
5.2. Manufacturing Considerations for Formulated mRNA Drug Product
5.2.1. Production of Lipid Nanoparticles
5.2.2. Product of Nanoemulsions
6. Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oberfeld, B.; Achanta, A.; Carpenter, K.; Chen, P.; Gilette, N.M.; Langat, P.; Said, J.T.; Schiff, A.E.; Zhou, A.S.; Barczak, A.K.; et al. SnapShot: COVID-19. Cell 2020, 181, 954.e1. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef]
- Koirala, A.; Joo, Y.J.; Khatami, A.; Chiu, C.; Britton, P.N. Vaccines for COVID-19: The current state of play. Paediatr. Respir. Rev. 2020, 35, 43–49. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Funk, C.D.; Laferrière, C.; Ardakani, A. A Snapshot of the Global Race for Vaccines Targeting SARS-CoV-2 and the COVID-19 Pandemic. Front. Pharmacol. 2020, 11, 937. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA Vaccine Development Enabled by Prototype Pathogen Preparedness. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cohen, J. Vaccine designers take first shots at COVID-19. Science 2020, 368, 14. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 3361. [Google Scholar] [CrossRef] [PubMed]
- Laczkó, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castaño, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H., 3rd; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. Concurrent human antibody and Th1 type T-cell responses elicited by a COVID-19 RNA vaccine. medRxiv 2020. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.P.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase 1/2 Study to Describe the Safety and Immunogenicity of a COVID-19 RNA Vaccine Candidate (BNT162b1) in Adults 18 to 55 Years of Age: Interim Report. medRxiv 2020. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- De Alwis, R.; Gan, E.S.; Chen, S.; Leong, Y.S.; Tan, H.C.; Zhang, S.L.; Yau, C.; Matsuda, D.; Allen, E.; Hartman, P.; et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus neutralizing antibodies and mediates protection in rodents. bioRxiv 2020. [Google Scholar] [CrossRef]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kis, Z.; Shattock, R.; Shah, N.; Kontoravdi, C. Emerging Technologies for Low-Cost, Rapid Vaccine Manufacture. Biotechnol. J. 2019, 14, 1800376. [Google Scholar] [CrossRef] [PubMed]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Ulmer, J.B.; Mansoura, M.K.; Geall, A.J. Vaccines ‘on demand’: Science fiction or a future reality. Expert Opin Drug Discov. 2015, 10, 101–106. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Mick Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.-J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Scorza, F.B.; Pardi, N. New Kids on the Block: RNA-Based Influenza Virus Vaccines. Vaccines 2018, 6, 20. [Google Scholar] [CrossRef]
- Lundstrom, K. Self-Amplifying RNA Viruses as RNA Vaccines. Int. J. Mol. Sci. 2020, 21, 5130. [Google Scholar] [CrossRef]
- DeFrancesco, L. The ‘anti-hype’ vaccine. Nat. Biotechnol. 2017, 35, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines 2020, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. How COVID unlocked the power of RNA vaccines. Nature 2021, 589, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Arun Kumar, S.; Jhan, Y.Y.; Bishop, C.J. Engineering DNA vaccines against infectious diseases. Acta Biomater. 2018, 80, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Suschak, J.J.; Williams, J.A.; Schmaljohn, C.S. Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum. Vaccines Immunother. 2017, 13, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Alnylam Pharmaceuticals Press. Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO (Patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults; Alnylam Pharmaceuticals Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Center for Leading Innovation. Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis SARS CoV-2 Infection (COVID-19); Center for Leading Innovation: Rochester, NY, USA, 2020. [Google Scholar]
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef]
- Pfizer. Pfizer and Biontech Achieve First Authorization in the World for a Vaccine to Combat Covid-19. 2020. Available online: https://www.businesswire.com/news/home/20201201006304/en/ (accessed on 26 January 2021).
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660-IN610. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A Scalable, Extrusion-Free Method for Efficient Liposomal Encapsulation of Plasmid DNA. Pharm. Res. 2005, 22, 362–372. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Diken, M.; Selmi, A.; Diekmann, J.; Attig, S.; Hüsemann, Y.; Koslowski, M.; Huber, C.; Türeci, Ö.; Sahin, U. FLT3 Ligand Enhances the Cancer Therapeutic Potency of Naked RNA Vaccines. Cancer Res. 2011, 71, 6132. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Cullis, P.R. Modulation of Membrane Fusion by Asymmetric Transbilayer Distributions of Amino Lipids. Biochemistry 1994, 33, 12573–12580. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; McKay, P.F.; Christensen, D.; Yus, B.I.; Aldon, Y.; Follmann, F.; Shattock, R.J. Effects of cationic adjuvant formulation particle type, fluidity and immunomodulators on delivery and immunogenicity of saRNA. J. Control. Release 2019, 304, 65–74. [Google Scholar] [CrossRef]
- Maruggi, G.; Chiarot, E.; Giovani, C.; Buccato, S.; Bonacci, S.; Frigimelica, E.; Margarit, I.; Geall, A.; Bensi, G.; Maione, D. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 2017, 35, 361–368. [Google Scholar] [CrossRef]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef]
- Luo, F.; Zheng, L.; Hu, Y.; Liu, S.; Wang, Y.; Xiong, Z.; Hu, X.; Tan, F. Induction of Protective Immunity against Toxoplasma gondii in Mice by Nucleoside Triphosphate Hydrolase-II (NTPase-II) Self-amplifying RNA Vaccine Encapsulated in Lipid Nanoparticle (LNP). Front. Microbiol. 2017, 8, 605. [Google Scholar] [CrossRef]
- Li, Y.; Su, Z.; Zhao, W.; Zhang, X.; Momin, N.; Zhang, C.; Wittrup, K.D.; Dong, Y.; Irvine, D.J.; Weiss, R. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 2020, 1, 882–893. [Google Scholar] [CrossRef]
- Li, Y.; Teague, B.; Zhang, Y.; Su, Z.; Porter, E.; Dobosh, B.; Wagner, T.; Irvine, D.J.; Weiss, R. In vitro evolution of enhanced RNA replicons for immunotherapy. Sci. Rep. 2019, 9, 6932. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, Q.; Durnell, L.A.; et al. Intramuscular Delivery of Replicon RNA Encoding ZIKV-117 Human Monoclonal Antibody Protects against Zika Virus Infection. Mol. Ther. Methods Clin. Dev. 2020, 18, 402–414. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Bouton, C.R.; Hu, K.; Samnuan, K.; Shattock, R.J. Innate Inhibiting Protiens Enhance Expression and Immunogenicity of Self-Amplifying RNA. Mol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Samsa, M.M.; Dupuy, L.C.; Beard, C.W.; Six, C.M.; Schmaljohn, C.S.; Mason, P.W.; Geall, A.J.; Ulmer, J.B.; Yu, D. Self-Amplifying RNA Vaccines for Venezuelan Equine Encephalitis Virus Induce Robust Protective Immunogenicity in Mice. Mol. Ther. 2019, 27, 850–865. [Google Scholar] [CrossRef] [PubMed]
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; De Gregorio, E.; Geall, A.J.; Brazzoli, M.; Bertholet, S. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLoS ONE 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Chapter Seven—Self-Amplifying mRNA Vaccines. In Advances in Genetics; Huang, L., Liu, D., Wagner, E., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 89, pp. 179–233. [Google Scholar]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Moyo, N.; Vogel, A.B.; Buus, S.; Erbar, S.; Wee, E.G.; Sahin, U.; Hanke, T. Efficient Induction of T Cells against Conserved HIV-1 Regions by Mosaic Vaccines Delivered as Self-Amplifying mRNA. Mol. Ther. Methods Clin. Dev. 2019, 12, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.; Porter, E.; Zhang, Y.; Silva, M.; Li, N.; Dobosh, B.; Liguori, A.; Skog, P.; Landais, E.; Menis, S.; et al. Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol. Ther. 2019, 27, 2080–2090. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef]
- Ajbani, S.P.; Velhal, S.M.; Kadam, R.B.; Patel, V.V.; Bandivdekar, A.H. Immunogenicity of Semliki Forest virus based self-amplifying RNA expressing Indian HIV-1C genes in mice. Int. J. Biol. Macromol. 2015, 81, 794–802. [Google Scholar] [CrossRef]
- Baeza Garcia, A.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B.; et al. Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nat. Commun. 2018, 9, 2714. [Google Scholar] [CrossRef]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M.; et al. Induction of Broad-Based Immunity and Protective Efficacy by Self-amplifying mRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2015, 90, 332–344. [Google Scholar] [CrossRef]
- Lazzaro, S.; Giovani, C.; Mangiavacchi, S.; Magini, D.; Maione, D.; Baudner, B.; Geall, A.J.; De Gregorio, E.; D’Oro, U.; Buonsanti, C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 2015, 146, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Démoulins, T.; Ebensen, T.; Schulze, K.; Englezou, P.C.; Pelliccia, M.; Guzmán, C.A.; Ruggli, N.; McCullough, K.C. Self-replicating RNA vaccine functionality modulated by fine-tuning of polyplex delivery vehicle structure. J. Control. Release 2017, 266, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Englezou, P.C.; Sapet, C.; Démoulins, T.; Milona, P.; Ebensen, T.; Schulze, K.; Guzman, C.-A.; Poulhes, F.; Zelphati, O.; Ruggli, N.; et al. Self-Amplifying Replicon RNA Delivery to Dendritic Cells by Cationic Lipids. Mol. Ther. Nucleic Acids 2018, 12, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Démoulins, T.; Milona, P.; Englezou, P.C.; Ebensen, T.; Schulze, K.; Suter, R.; Pichon, C.; Midoux, P.; Guzmán, C.A.; Ruggli, N.; et al. Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 711–722. [Google Scholar] [CrossRef]
- Perche, F.; Clemençon, R.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Pichon, C. Neutral Lipopolyplexes for In Vivo Delivery of Conventional and Replicative RNA Vaccine. Mol. Ther. Nucleic Acids 2019, 17, 767–775. [Google Scholar] [CrossRef]
- Goswami, R.; Chatzikleanthous, D.; Lou, G.; Giusti, F.; Bonci, A.; Taccone, M.; Brazzoli, M.; Gallorini, S.; Ferlenghi, I.; Berti, F.; et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect. Dis. 2019, 5, 1546–1558. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef]
- Blakney, A.K.; Zhu, Y.; McKay, P.F.; Bouton, C.R.; Yeow, J.; Tang, J.; Hu, K.; Samnuan, K.; Grigsby, C.L.; Shattock, R.J.; et al. Big Is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711–5727. [Google Scholar] [CrossRef]
- Manara, C.; Brazzoli, M.; Piccioli, D.; Taccone, M.; D’Oro, U.; Maione, D.; Frigimelica, E. Co-administration of GM-CSF expressing RNA is a powerful tool to enhance potency of SAM-based vaccines. Vaccine 2019, 37, 4204–4213. [Google Scholar] [CrossRef]
- Stokes, A.; Pion, J.; Binazon, O.; Laffont, B.; Bigras, M.; Dubois, G.; Blouin, K.; Young, J.K.; Ringenberg, M.A.; Ben Abdeljelil, N.; et al. Nonclinical safety assessment of repeated administration and biodistribution of a novel rabies self-amplifying mRNA vaccine in rats. Regul. Toxicol. Pharmacol. 2020, 113, 104648. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Lou, G.; Gallorini, S.; Brazzoli, M.; Johnson, R.; O’Hagan, D.T.; Baudner, B.C.; Perrie, Y. Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines. Vaccines 2020, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Anderluzzi, G.; Schmidt, S.T.; Woods, S.; Gallorini, S.; Brazzoli, M.; Giusti, F.; Ferlenghi, I.; Johnson, R.N.; Roberts, C.W.; et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: The impact of cationic lipid selection. J. Control. Release 2020, 325, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 252. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef]
- Zhong, Z.; Portela Catani, J.P.; Mc Cafferty, S.; Couck, L.; Van Den Broeck, W.; Gorlé, N.; Vandenbroucke, R.E.; Devriendt, B.; Ulbert, S.; Cnops, L.; et al. Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine. Vaccines 2019, 7, 96. [Google Scholar] [CrossRef]
- Luisi, K.; Morabito, K.M.; Burgomaster, K.E.; Sharma, M.; Kong, W.-P.; Foreman, B.M.; Patel, S.; Fisher, B.; Aleshnick, M.A.; Laliberte, J.; et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci. Adv. 2020, 6, eaba5068. [Google Scholar] [CrossRef]
- Elong Ngono, A.; Syed, T.; Nguyen, A.-V.; Regla-Nava, J.A.; Susantono, M.; Spasova, D.; Aguilar, A.; West, M.; Sparks, J.; Gonzalez, A.; et al. CD8+ T cells mediate protection against Zika virus induced by an NS3-based vaccine. Sci. Adv. 2020, 6, eabb2154. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef] [PubMed]
- Diken, M.; Kranz, L.M.; Kreiter, S.; Sahin, U. mRNA: A Versatile Molecule for Cancer Vaccines. Curr. Issues Mol. Biol. 2017, 22, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, K.; Lazzaro, S.; Lutz, J.; Rauch, S.; Heidenreich, R. mRNA Cancer Vaccines. Recent Results Cancer Res. 2016, 209, 61–85. [Google Scholar] [CrossRef]
- Lundstrom, K. Alphavirus-Based Antigen Preparation. In Vaccine Delivery Technology: Methods and Protocols; Pfeifer, B.A., Hill, A., Eds.; Springer US: New York, NY, USA, 2021; pp. 63–81. [Google Scholar] [CrossRef]
- Tews, B.A.; Meyers, G. Self-Replicating RNA. Methods Mol. Biol. 2017, 1499, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, K.; Liljeström, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Götte, B.; Liu, L.; McInerney, G.M. The Enigmatic Alphavirus Non-Structural Protein 3 (nsP3) Revealing Its Secrets at Last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Hellström, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5′ and 3′ ends of alphavirus RNAs—Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef]
- Carrasco, L.; Sanz, M.A.; González-Almela, E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Lello, L.S.; Utt, A.; Bartholomeeusen, K.; Wang, S.; Rausalu, K.; Kendall, C.; Coppens, S.; Fragkoudis, R.; Tuplin, A.; Alphey, L.; et al. Cross-utilisation of template RNAs by alphavirus replicases. PLoS Pathog. 2020, 16, e1008825. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, C.; O’Hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Shattock, R.J. Structural Components for Amplification of Positive and Negative Strand VEEV Splitzicons. Front. Mol. Biosci. 2018, 5, 71. [Google Scholar] [CrossRef]
- Kallen, K.J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive(®) vaccines. Hum. Vaccin Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef]
- De Haro, C.; Méndez, R.; Santoyo, J. The eIF-2α kinases and the control of protein synthesis1. FASEB J. 1996, 10, 1378–1387. [Google Scholar] [CrossRef]
- Liang, S.L.; Quirk, D.; Zhou, A. RNase L: Its biological roles and regulation. IUBMB Life 2006, 58, 508–514. [Google Scholar] [CrossRef]
- Beissert, T.; Koste, L.; Perkovic, M.; Walzer, K.C.; Erbar, S.; Selmi, A.; Diken, M.; Kreiter, S.; Türeci, Ö.; Sahin, U. Improvement of In Vivo Expression of Genes Delivered by Self-Amplifying RNA Using Vaccinia Virus Immune Evasion Proteins. Hum. Gene Ther. 2017, 28, 1138–1146. [Google Scholar] [CrossRef]
- Liu, Y.; Chin, J.M.; Choo, E.L.; Phua, K.K.L. Messenger RNA translation enhancement by immune evasion proteins: A comparative study between EKB (vaccinia virus) and NS1 (influenza A virus). Sci. Rep. 2019, 9, 11972. [Google Scholar] [CrossRef]
- Yoshioka, N.; Gros, E.; Li, H.R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.D.; Yu, B.D.; et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Huysmans, H.; Zhong, Z.; De Temmerman, J.; Mui, B.L.; Tam, Y.K.; Mc Cafferty, S.; Gitsels, A.; Vanrompay, D.; Sanders, N.N. Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying mRNA in the Skin. Mol. Ther. Nucleic Acids 2019, 17, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Kunath, K.; von Harpe, A.; Fischer, D.; Petersen, H.; Bickel, U.; Voigt, K.; Kissel, T. Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: Comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. J. Control. Release 2003, 89, 113–125. [Google Scholar] [CrossRef]
- Blakney, A.K.; Yilmaz, G.; McKay, P.F.; Becer, C.R.; Shattock, R.J. One Size Does Not Fit All: The Effect of Chain Length and Charge Density of Poly(ethylene imine) Based Copolymers on Delivery of pDNA, mRNA, and RepRNA Polyplexes. Biomacromolecules 2018, 19, 2870–2879. [Google Scholar] [CrossRef]
- Gurnani, P.; Blakney, A.K.; Terracciano, R.; Petch, J.E.; Blok, A.J.; Bouton, C.R.; McKay, P.F.; Shattock, R.J.; Alexander, C. The In Vitro, Ex Vivo, and In Vivo Effect of Polymer Hydrophobicity on Charge-Reversible Vectors for Self-Amplifying RNA. Biomacromolecules 2020, 21, 3242–3253. [Google Scholar] [CrossRef]
- Blakney, A.K.; Abdouni, Y.; Yilmaz, G.; Liu, R.; McKay, P.F.; Bouton, C.R.; Shattock, R.J.; Becer, C.R. Mannosylated Poly(ethylene imine) Copolymers Enhance saRNA Uptake and Expression in Human Skin Explants. Biomacromolecules 2020, 21, 2482–2492. [Google Scholar] [CrossRef]
- Saviano, F.; Lovato, T.; Russo, A.; Russo, G.; Bouton, C.R.; Shattock, R.J.; Alexander, C.; Quaglia, F.; Blakney, A.K.; Gurnani, P.; et al. Ornithine-derived oligomers and dendrimers for in vitro delivery of DNA and ex vivo transfection of skin cells via saRNA. J. Mater. Chem. B 2020, 8, 4940–4949. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef]
- Ansaldi, F.; Canepa, P.; Parodi, V.; Bacilieri, S.; Orsi, A.; Compagnino, F.; Icardi, G.; Durando, P. Adjuvanted seasonal influenza vaccines and perpetual viral metamorphosis: The importance of cross-protection. Vaccine 2009, 27, 3345–3348. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Asahara, H.; Tzertzinis, G.; Roy, B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 2020, 26, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Control, Center for Disease. H1N1/09 Swine Flu Pandemic Timeline. Available online: https://www.cdc.gov/flu/pandemic-resources/2009-pandemic-timeline.html (accessed on 26 January 2021).
- Control, Center for Disease. H1N1/09 PHEIC Declaration. Available online: https://wwwnc.cdc.gov/eid/article/15/8/09-0665_article (accessed on 26 January 2021).
- Control, Center for Disease. H1N1/09 Pandemic Declaration. Available online: https://www.cdc.gov/h1n1flu/who/ (accessed on 26 January 2021).
- ClinicalTrials.gov. CSL Vaccine A/H1N1. Available online: https://clinicaltrials.gov/ct2/show/NCT00938639?term=CSL425&draw=2&rank=4 (accessed on 26 January 2021).
- FDA. FDA Approves H1N1 Vaccines. Available online: https://www.cidrap.umn.edu/news-perspective/2009/09/fda-approves-four-companies-h1n1-vaccines (accessed on 26 January 2021).
- ClinicalTrials.gov. Novartis Cell Based A/H1N1 Vaccine. Available online: https://clinicaltrials.gov/ct2/show/NCT00943358 (accessed on 26 January 2021).
- Reuters. Novartis A/H1N1 Vaccine Approval. 2021. Available online: https://www.reuters.com/article/novartis-idUKZAT01056620091105?edition-redirect=ca (accessed on 26 January 2021).
- FDA. H1N1 Swine Flu EUA. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization-archived-information#H1N1 (accessed on 26 January 2021).
- World Health Organization. COVID-19 Declared PHEIC. 2021. Available online: https://www.who.int/publications/m/item/covid-19-public-health-emergency-of-international-concern-(pheic)-global-research-and-innovation-forum (accessed on 26 January 2021).
- World Health Organization. COVID-19 Declard Pandemic. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 26 January 2021).
- ClinicalTrials.gov. Novavax Clinical Trial Start Date. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04368988 (accessed on 26 January 2021).
- ClinicalTrials.gov. Moderna Clinical Trial Start Date. 2021. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04283461 (accessed on 26 January 2021).
- FDA. Moderna COVID-19 Vaccine Approval. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/moderna-covid-19-vaccine (accessed on 26 January 2021).
- ClinicalTrials.gov. J&J Clinical Trial Start Date. 2021. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04436276 (accessed on 26 January 2021).
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Samnuan, K.; Blakney, A.K.; McKay, P.F.; Shattock, R.J. Design-of-Experiments In Vitro Transcription Yield Optimization of Self-Amplifying RNA. bioRxiv 2021. [Google Scholar] [CrossRef]
- Davis, R.H. Large-scale oligoribonucleotide production. Curr. Opin. Biotechnol. 1995, 6, 213–217. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Gingras, A.-C.; Sonenberg, N.; Burley, S.K. Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-methyl-GDP. Cell 1997, 89, 951–961. [Google Scholar] [CrossRef]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef]
- Yisraeli, J.K.; Melton, D.A. Synthesis of long, capped transcripts in Vitro by SP6 and T7 RNA polymerases. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1989; Volume 180, pp. 42–50. [Google Scholar]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef]
- Peng, Z.-H.; Sharma, V.; Singleton, S.F.; Gershon, P.D. Synthesis and Application of a Chain-Terminating Dinucleotide mRNA Cap Analog. Org. Lett. 2002, 4, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.E.; Lorsch, J. Chapter Nineteen—RNA Purification—Precipitation Methods. In Methods in Enzymology; Lorsch, J., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 530, pp. 337–343. [Google Scholar]
- Baronti, L.; Karlsson, H.; Marušič, M.; Petzold, K. A guide to large-scale RNA sample preparation. Anal. Bioanal. Chem. 2018, 410, 3239–3252. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.T. Advances in methods for native expression and purification of RNA for structural studies. Curr. Opin. Struct. Biol. 2014, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Queiroz, J.A.; Sousa, F. Ribonucleic acid purification. J. Chromatogr. A 2014, 1355, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deering, R.P.; Kommareddy, S.; Ulmer, J.B.; Brito, L.A.; Geall, A.J. Nucleic acid vaccines: Prospects for non-viral delivery of mRNA vaccines. Expert Opin Drug Deliv 2014, 11, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Gholamalipour, Y.; Karunanayake Mudiyanselage, A.; Martin, C.T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef]
- Triana-Alonso, F.J.; Dabrowski, M.; Wadzack, J.; Nierhaus, K.H. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J. Biol. Chem. 1995, 270, 6298–6307. [Google Scholar] [CrossRef]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Moon, S.L.; Wilusz, J. In Vitro Transcription of Modified RNAs. In Recombinant and In Vitro RNA Synthesis: Methods and Protocols; Conn, G.L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 171–180. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. The Many Pathways of RNA Degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Wayment-Steele, H.K.; Kim, D.S.; Choe, C.A.; Nicol, J.J.; Wellington-Oguri, R.; Sperberg, R.A.P.; Huang, P.-S.; Das, R. Theoretical basis for stabilizing messenger RNA through secondary structure design. biorxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Brunelle, J.L.; Green, R. Chapter Five—In Vitro Transcription from Plasmid or PCR-amplified DNA. In Methods in Enzymology; Lorsch, J., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 530, pp. 101–114. [Google Scholar]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Viger-Gravel, J.; Schantz, A.; Pinon, A.C.; Rossini, A.J.; Schantz, S.; Emsley, L. Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B 2018, 122, 2073–2081. [Google Scholar] [CrossRef]
- Yaghi, N.K.; Wei, J.; Hashimoto, Y.; Kong, L.Y.; Gabrusiewicz, K.; Nduom, E.K.; Ling, X.; Huang, N.; Zhou, S.; Kerrigan, B.C.; et al. Immune modulatory nanoparticle therapeutics for intracerebral glioma. Neuro Oncol. 2017, 19, 372–382. [Google Scholar] [CrossRef]
- Richner, J.M.; Jagger, B.W.; Shan, C.; Fontes, C.R.; Dowd, K.A.; Cao, B.; Himansu, S.; Caine, E.A.; Nunes, B.T.D.; Medeiros, D.B.A.; et al. Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell 2017, 170, 273–283. [Google Scholar] [CrossRef]
- Kim, J.; Jozic, A.; Sahay, G. Naturally Derived Membrane Lipids Impact Nanoparticle-Based Messenger RNA Delivery. Cell. Mol. Bioeng. 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Vemuri, S.; Yu, C.-D.; Wangsatorntanakun, V.; Roosdorp, N. Large-Scale Production of Liposomes by A Microfluidizer. Drug Dev. Ind. Pharm. 1990, 16, 2243–2256. [Google Scholar] [CrossRef]
- Jenning, V.; Lippacher, A.; Gohla, S.H. Medium scale production of solid lipid nanoparticles (SLN) by high pressure homogenization. J. Microencapsul. 2002, 19, 1–10. [Google Scholar] [CrossRef]
- Muchow, M.; Maincent, P.; Muller, R.H. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev. Ind. Pharm. 2008, 34, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Shegokar, R.; Singh, K.K.; Müller, R.H. Production & stability of stavudine solid lipid nanoparticles—From lab to industrial scale. Int. J. Pharm. 2011, 416, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Sorgi, F.L.; Huang, L. Large scale production of DC-Chol cationic liposomes by microfluidization. Int. J. Pharm. 1996, 144, 131–139. [Google Scholar] [CrossRef]
- Liu, M.A.; Ulmer, J.B. Human clinical trials of plasmid DNA vaccines. Adv. Genet. 2005, 55, 25–40. [Google Scholar] [CrossRef]
- Scheuber, A. Imperial Social Enterprise to Accelerate Low-Cost COVID-19 Vaccine. Available online: https://www.imperial.ac.uk/news/198053/imperial-social-enterprise-accelerate-lowcost-covid19/ (accessed on 26 January 2021).
- Kaiser, J. Temperature concerns could slow the rollout of new coronavirus vaccines. Science 2020. [Google Scholar] [CrossRef]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sponsor | Type of mRNA | Delivery System | Preclinical Data | Clinical Data |
|---|---|---|---|---|
| Moderna | bmRNA | LNP | [9,12] | [7,8] |
| BioNTech/Pfizer | bmRNA | LNP | [13] | [11,14,15,16,17] |
| ICL | saRNA | LNP | [18] | |
| Arcturus | saRNA | LNP | [19] | |
| CureVac | mRNA | LNP | [20] |
| Disease Category | Disease Target | Replicon Backbone | Antigen | Delivery Platform | Preclinical Animal Model | Ref. |
|---|---|---|---|---|---|---|
| Infectious Disease | Chlamydia trachomatis | VEEV | MOMP | CAF, PEI | Mice | [57] |
| Ebola | VEEV | Glycoprotein (EBOV) | Dendrimer | Mice | [59] | |
| Group A Streptococci | VEE-SINV | GAS SLOdm | CNE | Mice | [58] | |
| Group B Streptococci | VEE-SINV | GBS BP-2a | CNE | Mice | [58] | |
| HCV | VEEV | E1-E2 | CNE | Mice | [67] | |
| HCMV | VEEV | gH/gL | LNP | Mice | [67] | |
| HIV-1 | VEE-SINV | TV1 Env gp140 | CNE | NHP | [68] | |
| SFV | Gag/Pol Mosaic | PEI | Mice | [69] | ||
| VEEV | eOD-GT8 gp120 | LNP | Mice | [70] | ||
| VEEV | Env gp140 | Lipoplex | Mice | [71] | ||
| SFV | HIV-1C Env, Gag, PolRT | Naked | Mice | [72] | ||
| Malaria | VEE-SINV | PMIF | CNE | Mice | [73] | |
| Influenza | VEEV | HA (H1N1, A/WSN/33) | Dendrimer | Mice | [59] | |
| VEE-SINV | HA (H1N1, A/Cal/7/09) | CNE | Mice, Ferrets | [74] | ||
| VEE-SINV | NP (H1N1, A/PR/34/07) | LNP | Mice | [75] | ||
| VEE-SINV | NP, M1 or NP+M1 (H1N1, A/PR/8/34) | LNP | Mice | [66] | ||
| CSFV | HA, NP (H5N1/Yamaguchi/2004) | PEI with CPP | Mice, Pigs | [76] | ||
| CSFV | NP (H3N2, Brisbane 2007) | Cationic lipid | Mice | [77] | ||
| n.s. | HA (H1N1, A/PR/8, A/Cal/7/09) | PEI | Mice | [78] | ||
| CSFV | HA, NP (H5N1/Yamaguchi/2004) | PEI | Mice | [79] | ||
| VEEV | HA (A/PR/8/34) | LPPs | Mice | [80] | ||
| n.s. | HA (H1N1, A/Cal/7/09) | MLNPs | Mice | [81] | ||
| SFV taRNA | HA (H1N1, A/Cal/7/09) | Naked | Mice | [82] | ||
| VEEV | HA (H1N1, A/Cal/7/09) | pABOL | Mice | [83] | ||
| VEE-SINV | NP, GMCSF | CNE | Mice | [84] | ||
| Rabies | VEE-SINV | Glycoprotein G | CNE | Rats | [85] | |
| VEE-SINV | Glycoprotein G | PNPs, Liposomes, CNE | Mice | [86] | ||
| VEE-SINV | Glycoprotein G | LNP, CNE | Mice | [87] | ||
| VEEV | Glycoprotein G | LNP, CNE | Mice | [67] | ||
| Respiratory syncytial virus | VEE-SINV | Glycoprotein F | LNP | Mice | [88] | |
| SARS-CoV-2 | VEEV | Pre-fusion stabilized spike protein | LNP | Mice | [18] | |
| VEEV | Spike protein | LION emulsion | Mice, NHP | [89] | ||
| VEEV | Pre-fusion spike protein | LNP | Mice | [62] | ||
| VEEV | Spike protein | LNP | Mice | [19] | ||
| Toxoplasma gondii | VEEV | GRA6, ROP2a, ROP18, SAG1, SAG2A, AMA1 | Dendrimer | Mice | [59] | |
| SFV | NTPase-II | LNP | Mice | [60] | ||
| VEEV | VEEV | E3-E2-6K-E1 | CNE | Mice | [65] | |
| Zika | VEEV | prM-E | Dendrimer | Mice | [90] | |
| VEEV | prM-E | NLC | Mice, Guinea pigs | [91] | ||
| VEEV | prM-E | Naked | Mice | [92] | ||
| VEEV | ZIKV-117 Ab | NLC | Mice | [63] | ||
| n.s. | prM-E | CNE | Mice, NHPs | [93] | ||
| VEEV | NS3, prM-E | LNP | Mice | [94] | ||
| Cancer | Melanoma | VEEV | IL-12 | LNP | Mice | [61] |
| VEEV | IL-2 | LNP | Mice | [62] | ||
| Colon carcinoma | VEEV | IL-12 | LNP | Mice | [61] |
| Disease Target | Institution | Vaccine Components (Route of Administration) | Target | Clinical Trial Number (Phase) | Status |
|---|---|---|---|---|---|
| Rabies | GlaxoSmithKline | VEE-SINV saRNA with CNE (IM) | Rabies glycoprotein G | NCT04062669 (I) | Ongoing, recruiting |
| SARS-CoV-2 | Arcturus Therapeutics | STARR™ (VEEV) saRNA with LUNAR® LNP (IM) | Pre-fusion stabilized spike protein of SARS-CoV-2 | NCT04480957 (I) | Ongoing, recruiting |
| HDT Bio Corp. | VEEV saRNA with LION emulsion (IM) | Spike protein of SARS-CoV-2 | - | Pre-recruiting | |
| Imperial College London | VEEV saRNA with LNPs (IM) | Pre-fusion stabilized spike protein of SARS-CoV-2 | ISRCTN17072692 (II) | Ongoing, recruiting | |
| Imperial College London, University of Oxford | VEEV saRNA with LNPs OR ChAdOx (IN) | Pre-fusion stabilized spike protein of SARS-CoV-2 | - | Pre-recruiting | |
| Non-Small Cell Lung Cancer, Colorectal Cancer, Gastroesophageal Adenocarcinoma, Urothelial Carcinoma | Gritstone Oncology, Inc. | GRT-C901, GRT-R902 | Personalized neoantigens | NCT03639714 (I/II) | Recruiting |
| Non-Small Cell Lung Cancer, Colorectal Cancer, Pancreatic Cancer, Solid Tumor, Shared Neoantigen-Positive Solid Tumors | Gritstone Oncology, Inc. | GRT-C903 GRT-R904 | Personalized neoantigens | NCT03953235 (I/II) | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. https://doi.org/10.3390/vaccines9020097
Blakney AK, Ip S, Geall AJ. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines. 2021; 9(2):97. https://doi.org/10.3390/vaccines9020097
Chicago/Turabian StyleBlakney, Anna K., Shell Ip, and Andrew J. Geall. 2021. "An Update on Self-Amplifying mRNA Vaccine Development" Vaccines 9, no. 2: 97. https://doi.org/10.3390/vaccines9020097
APA StyleBlakney, A. K., Ip, S., & Geall, A. J. (2021). An Update on Self-Amplifying mRNA Vaccine Development. Vaccines, 9(2), 97. https://doi.org/10.3390/vaccines9020097