1. Introduction
Infectious diseases are present throughout history, emerging and reemerging as decades pass [
1]. In the most recent years, the world has seen outbreaks of H1N1 influenza, severe acute respiratory syndrome coronavirus (SARS-CoV), human immunodeficiency virus (HIV) [
2] and, notably, SARS-CoV-2 [
3]. Vaccines have been a key player in containing the spread and reducing the mortality of bacterial and viral pathogens, taking part in national and global immunization strategies that have led to eradication of smallpox and near eradication of polio [
4].
Recombinant viral vectors have become an important platform for vaccination, with growing interest in a variety of possible vectors. Viral vector vaccines have been approved against Ebola [
5]—using adenovirus [
6], modified Vaccinia Ankara [
7,
8], and vesicular stomatitis virus [
9] as vectors—and against SARS-CoV-2, using adenovirus as a vector (Johnson & Johnson, Gamaleya, Oxford-Astrazeneca and CanSino) [
1,
10]. There are also examples of approved viral vector vaccines for veterinary use, using vectors such as poxviruses, herpesvirus of turkeys (HVT) [
11] and adenovirus [
12,
13]. This technology fits the concept of platform-based vaccines, in which the viral vector is a backbone that can be modified to express and carry different antigens to quickly adapt the vaccine to target other pathogens, including for emerging outbreaks. By developing a platform-based vaccine and establishing a production process for it, both the product and process can be adapted to other targets with minimal changes. Thus, the time to develop, scale up and, consequently, deliver the vaccine can be greatly reduced, making this a promising approach for pandemic preparedness [
14].
In this context, the Newcastle Disease Virus (NDV) is an avian paramyxovirus which is non-pathogenic in humans [
2]. NDV is classified into three types of strains: velogenic, mesogenic or lentogenic, based on virulence and pathogenicity in avian species [
15]. Lentogenic strains, such as LaSota and B1, are avirulent in both birds and humans, making them the strains of choice for viral vector studies. NDV expresses a fusion protein (F) in its precursor form (F
0), which must be cleaved by host cell proteases for viral entry [
2]. In cell culture, trypsin can be added at the moment of infection to activate the virus [
16], as it is done for production of influenza [
17]. As an avian virus, there is a lack of pre-existing immunity against NDV among humans, as well as remarkable safety, which has been documented in clinical trials for oncolytic treatments using this virus [
2]. Another advantage of using this vector for vaccines includes operation at biosafety level 2, rather than working directly with the target pathogens, which can require costly operation at biosafety level 3 [
18]. Additionally, there are well established methods to generate recombinant NDV constructs bearing protective key antigens from other viral pathogens. These aspects make NDV a promising vaccine vector that has already been explored in vaccine candidates against H1N1 influenza, SARS-CoV, HIV, among others [
2].
Despite having garnered interest as a viral vector for vaccination and for cancer therapy [
15], there are still few studies on the development of production processes for NDV. Typically, this virus is produced in embryonated chicken eggs and collected in the allantoic fluid. Although this is a cost-effective method that can take advantage of existing manufacturing structures for influenza [
18], it also presents disadvantages when compared to cell culture technologies and their potential large-scale production under controlled operational conditions in bioreactors. Virus production in cell culture allows for greater control over several parameters, leading to less variation between batches and the capacity to further optimize each aspect. It may also avoid issues with allergens and the dependency on chicken egg supply [
19,
20]. As such, developing a cell culture-based NDV production process would be highly valuable in the pursuit of establishing a reliable vaccine production platform that allows for quick adaptation to emerging pathogens.
The COVID-19 pandemic highlighted the importance of mass vaccination and the race for fast implementation. Notably, vaccines were developed and approved within months, rather than the usual timespan of years [
3], which was partly due to extensive previous research on the technologies used. These vaccines were key to reduce cases and deaths [
21], as well as to recover economies [
22], in contrast to the 2009 H1N1 pandemic, in which vaccines were only widely available after the main onset of the pandemic [
23]. This further showcases the importance of using vaccine platforms to implement vaccination early on in a pandemic, maximizing the positive contribution of vaccines and avoiding the peak of cases and deaths.
In this work, we set out to develop a novel cell-based production process for two NDV constructs: NDV-GFP and the vaccine candidate NDV-FLS, which expresses the full-length SARS-CoV-2 spike protein. We first adapted both constructs to HEK293 and Vero cells, and then evaluated several infection parameters. In shake flasks, viral production kinetics were compared for different multiplicities of infection (MOI) and a Design of Experiment (DoE) was performed to analyze the effects of temperature, trypsin concentration and trypsin addition. The best conditions were then implemented for batch bioreactor productions of both viruses. For the analytics, we established two assays for viral quantification: median tissue culture infectious dose (TCID50) for infectious viral titer and digital droplet PCR (ddPCR) for genomic/total viral titer. As such, this is an innovative work in exploring and establishing these essential aspects for robust production and quality assessments of NDV in cell culture.
2. Materials and Methods
2.1. Cell Lines and Culture Media
The Vero cell line adapted to suspension was provided by the National Research Council of Canada (NRC), and the adaptation was described in a previous work [
24]. For routine passaging, cells were centrifuged at 800×
g for 5 min and resuspended in fresh media with a seeding density of 3–6 × 10
5 cells/mL. Cell cultures were maintained at 37 °C, 135 rpm and 5% CO
2 in humified Multitron orbital shakers (Infors HT, Bottmingen, Switzerland). Cells were cultured in MDXK medium (Xell AG, Bielefeld, Germany), supplemented with 4 mM GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA), at a working volume of 20 mL, 25 mL, 50 mL or 100–200 mL in polycarbonate shake flasks of volume 125 mL, 250 mL, 500 mL or 1 L (TriForest Enterprises, Irvine, CA, USA), respectively.
HEK293 suspension cells are originated from HEK293SF (clone 293SF-3F6) cells, which derive from a GMP-grade master cell bank [
25]. The cells were cultured in HEK GM medium (Xell AG, Bielefeld, Germany), supplemented with 4 mM GlutaMAX. Routine passaging and incubation in shakers was the same as for suspension Vero cells.
Adherent Vero cells (ATCC CCL-81.5) were routinely passaged by washing with PBS without calcium and magnesium (WISENT Inc., Saint-Jean-Baptiste, QC, Canada), detaching with TrypLE™ Express Enzyme (Gibco, Gaithersburg, MD, USA) and adding VP Serum-Free Medium (VP-SFM) (Gibco, Gaithersburg, MD, USA) with 4 mM GlutaMAX and 1% Penicillin-Streptomycin solution (WISENT Inc., Saint-Jean-Baptiste, QC, Canada) to collect. Once collected, cells are pelleted at 300× g for 5 min and resuspended in VP-SFM to remove TrypLE. Cells are plated onto T-175 flasks or 150 mm plates, at 5–10 × 106 cells and are passaged every 2–3 days.
Adherent HEK293 (HEK293A, ATCC CRL-1573 [
26]) cells were routinely passaged in the same way as adherent Vero cells, but using Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) with 10% Fetal Bovine Serum (FBS) (Gibco, Gaithersburg, MD, USA) and 1% Penicillin-Streptomycin solution instead of VP-SFM.
2.2. Virus Adaptation
The engineering and rescue of the Newcastle Disease Virus constructs NDV-GFP and NDV-FLS were described in another publication [
27]. Briefly, the gene of interest (encoding green fluorescent protein or human codon-optimized full-length spike from SARS-CoV-2, respectively) was inserted between the P and M genes of the NDV (LaSota strain) genome. These viruses were initially produced in allantoic fluid and passaged for adaptation to Vero and HEK293 cells. Passages consisted of infecting cells, harvesting the virus produced and using it to reinfect cells for the next passage.
In Vero cells, for passages 1 and 2, adherent Vero cell cultures in T-25 flasks with VP-SFM media and 4 mM GlutaMAX were infected at a confluency of 80–90% and an MOI of 0.5 with TPCK-treated trypsin (MilliporeSigma, Oakville, ON, Canada) to a final concentration of 1 μg/mL. The supernatant was collected at 24 hpi. From passage 3 onwards, suspension Vero cells were seeded at 1 × 106 cells/mL in 25 mL MDXK media with 4 mM GlutaMAX in 250 mL shake flasks. The cells were immediately infected at an MOI of 0.01 with 1 μg/mL trypsin. At 36 hpi, the culture was centrifuged at 800× g for 5 min to collect the supernatant, which was stored at −80 °C.
In HEK293 cells, for all passages, suspension cells were seeded at 1 × 106 cells/mL in 25 mL Xell HEK GM media with 4 mM GlutaMAX in 250 mL shake flasks. The cells were immediately infected at an MOI of 0.01 with 1 μg/mL trypsin. At 36 hpi, the culture was centrifuged at 800× g for 5 min to collect the supernatant, which was stored at −80 °C.
2.3. Median Tissue Culture Infectious Dose (TCID50)
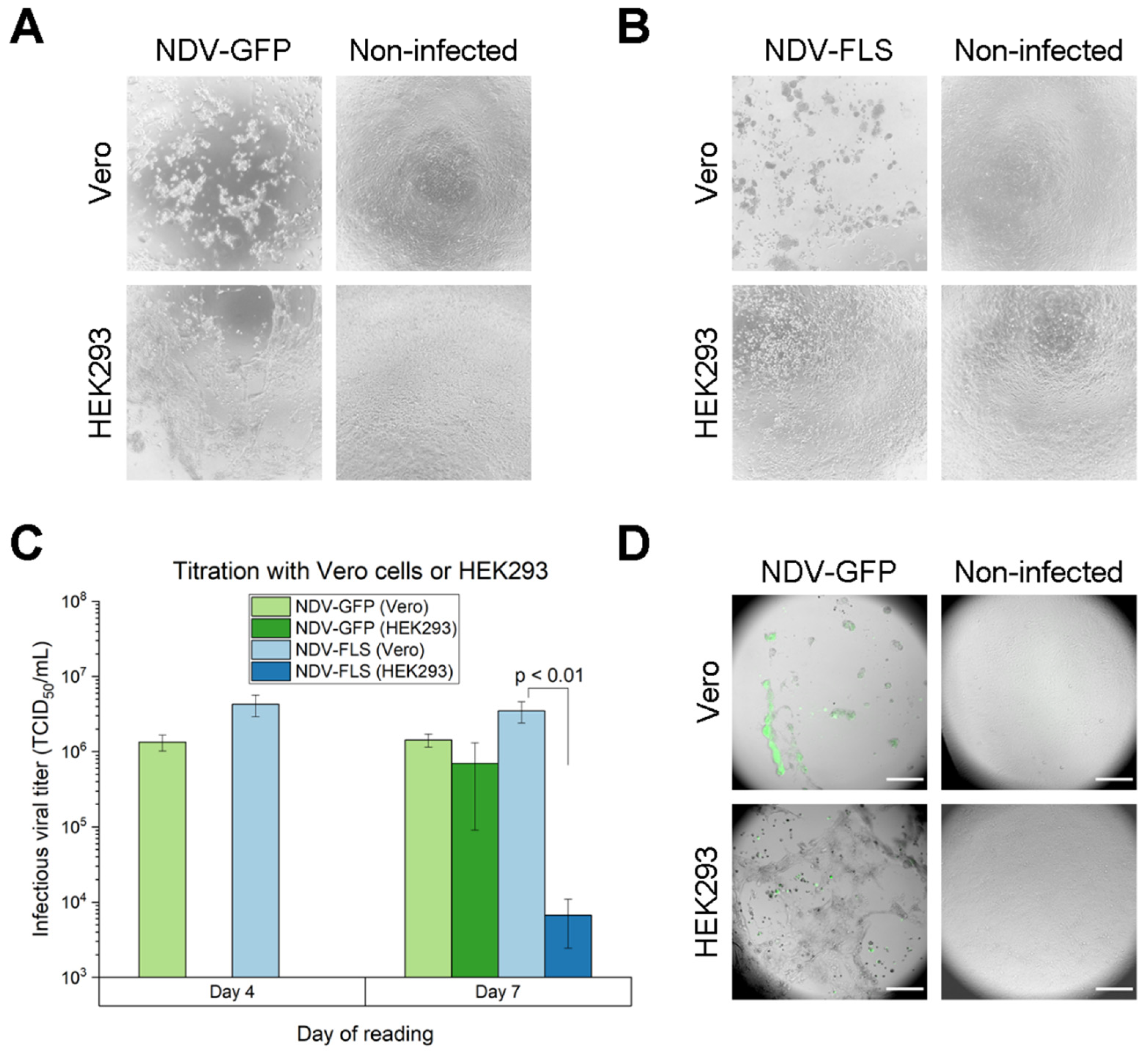
For routine quantification, adherent Vero cells were seeded on 96-well plates with 15,000 cells in 100 μL of media (VP-SFM) per well. For media and cell line comparison during TCID
50 development, adherent HEK293 cells were used with DMEM. When using DMEM, BSA 2.5 μg/mL was added instead of FBS, to avoid trypsin activity inhibition. The following day, the media was aspirated and replaced by 100 μL of media containing 1 μg/mL trypsin and a serial dilution of the virus (1:5 or 1:10). After 4 and 7 days of incubation at 37 °C with 5% CO
2, wells were analyzed on a standard light microscope for cytopathic effect (CPE), consisting of rounded cells, a disrupted monolayer and/or clumps. The number of CPE-positive wells in each column was used to quantify the experiment by the Spearman and Kärber algorithm [
28,
29,
30].
The assay with 1:5 dilutions (Coefficient of Variation: 34.57%) was chosen for all the TCID50 development and for samples which were below the range of detection of the 1:10 dilutions (<3.16 × 102 TCID50/mL). The assay with 1:10 dilutions (Coefficient of Variation: 34.69%) was chosen for all samples from shake flask experiments and bioreactors.
For comparison of CPE readings and Alamar blue readings, CPE was read first on the microscope before addition of the dye. The cell viability reagent Alamar blue (Invitrogen, Waltham, MA, USA) was diluted 1:10 in PBS without calcium and magnesium, and 100 µL of the dilution was added to each well, as described previously [
31]. Plates were incubated at 37 °C with 5% CO
2 and the absorbance was analyzed after 4 h. The absorbances at 570 nm and 600 nm were measured, and the absorbance at 600 nm was subtracted from the absorbance at 570 nm (ABS
570nm − ABS
600nm) to obtain the normalized value. Cut-off values were determined in a way that none of the wells in the (non-infected) negative control would be considered infected.
For comparison with fluorescence readings, a triplicate of an NDV-GFP sample was used for TCID50 and plates were read both by CPE, using a standard light microscope, and by fluorescence, using a plate reader with the excitation at 485/20 nm and emission at 528/20 nm.
After classifying the wells as positive through the cell viability (Alamar blue) or the fluorescence, the viral titer was determined by the Spearman and Kärber algorithm [
28,
29,
30], in the same way as when reading CPE.
For fluorescent microscope imaging, the TCID50 plates infected with NDV-GFP samples were observed on day 7 on Olympus IX-83 microscope using a 10× objective lens. Images were processed on ImageJ to merge bright-field images with green fluorescence channel images.
One-way ANOVA with the Tukey method was performed to determine statistical significance when comparing titration between different cell lines and different reading methods.
2.4. Polymerase Chain Reaction (PCR)
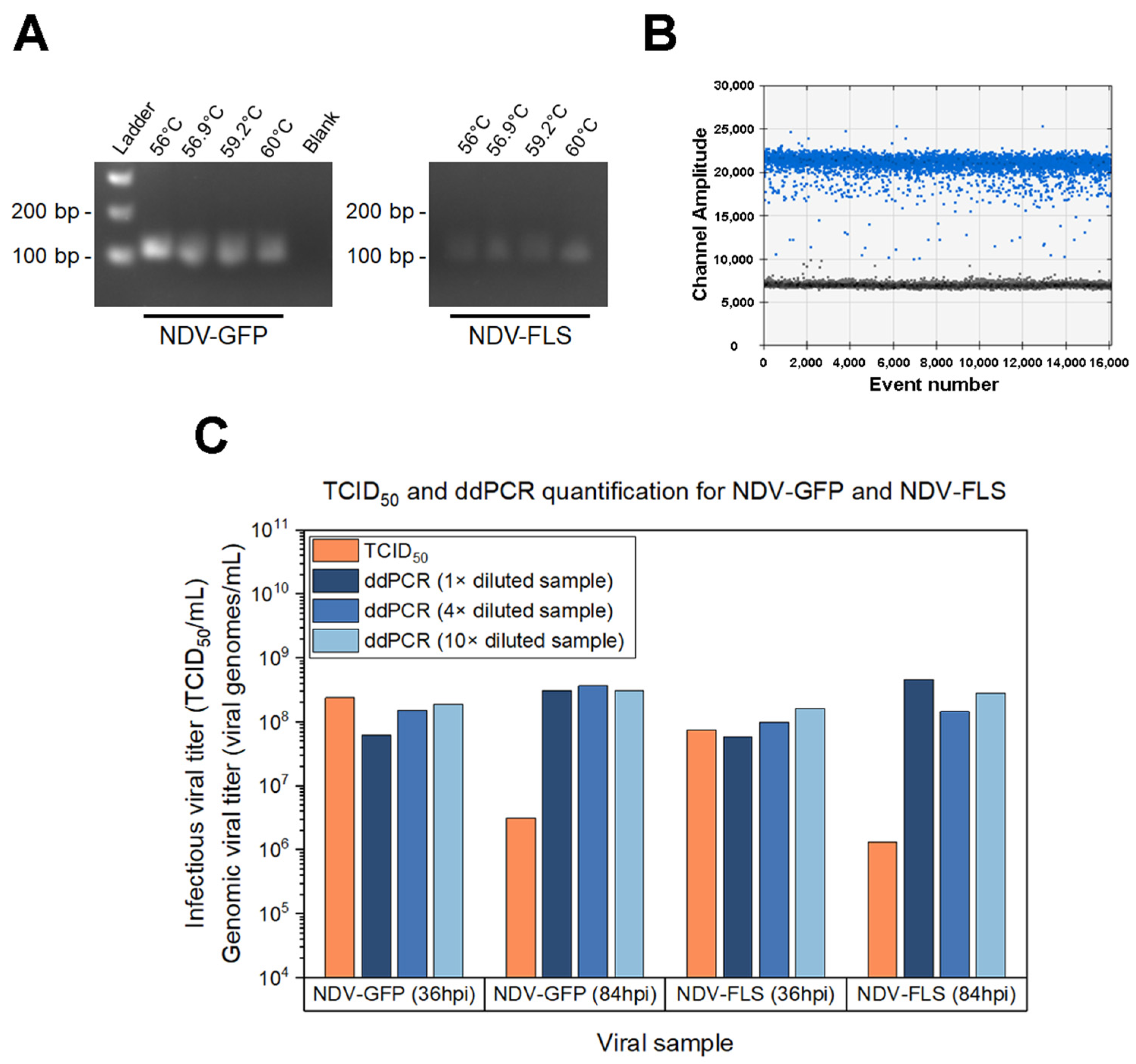
The Q5 High Fidelity Polymerase (New England Biolabs, Ipswich, MA, USA) was used with primers targeting the L gene (polymerase) of NDV: NDV-L F [5′-ATATGTTCTGACTCCTGCCC-3′] and NDV-L R [5′-TCTAGTCGCTTGATCTCTGC-3′]. PCR was performed according to the manufacturer’s instructions, with the following thermocycler program: initial denaturation (1 min at 98 °C), followed by 30 cycles of the steps: 10 s at 98 °C, 30 s at the annealing temperature, 30 s at 72 °C. Next, the final elongation step happens for 2 min at 72 °C. The same NDV-GFP and NDV-FLS cDNA samples were used for PCR with different annealing temperatures: 56 °C, 57.6 °C, 59.2 °C and 60 °C. The amplified bands were visualized in a 2.5% agarose gel with SYBR Safe DNA gel stain (Thermofisher, Waltham, MA, USA).
2.5. Digital Droplet Polymerase Chain Reaction (ddPCR)
For routine quantification, RNA extraction was done for 20 µL of supernatant samples diluted with 180 µL PBS (without calcium and magnesium) using the High Pure Viral Nucleic Acid kit (Roche, Basel, Switzerland). During assay development, different dilutions of the sample were also tested: 1× (no dilution—200 µL sample), 4× (50 µL sample with 150 µL PBS) and 10× (20 µL sample with 180 µL PBS). Next, 2 µL of the extracted RNA was used with the iScript Select cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) to generate cDNA using random RT-PCR primers. Then, the cDNA was diluted (between 1:10 to 1:10,000) to target the linear range of ddPCR. 5 µL of the template dilution was used with the QX200 ddPCR kit (Bio-Rad Laboratories, Hercules, CA, USA), using the EvaGreen master mix and the same primers listed for PCR. The manufacturer’s instructions were followed to prepare the reaction and generate droplets. As for the thermocycler program: after initial denaturation (5 min at 95 °C), 34 cycles of the following steps were repeated: 30 s at 95 °C, 1 min at 59 °C, 30 s at 72 °C. Then, the final elongation step happened for 5 min at 72 °C.
Droplets are analyzed individually in the droplet reader and the copies/µL of each sample is given. This output is corrected for the dilution and volumes used to determine the viral genomes/mL of the original sample with the following calculation:
in which: I = Copies/µL (ddPCR output); J = volume of the ddPCR reaction; K = dilution of the cDNA template; L = volume of RT-PCR reaction; M = volume of the cDNA dilution added in the ddPCR reaction; N = volume of RNA added in the RT-PCR reaction; O = elution volume for RNA extraction; P = initial sample volume used for the RNA extraction; Q = dilution of the sample in RNA extraction.
2.6. Design of Experiment (DoE) for Infection Parameters
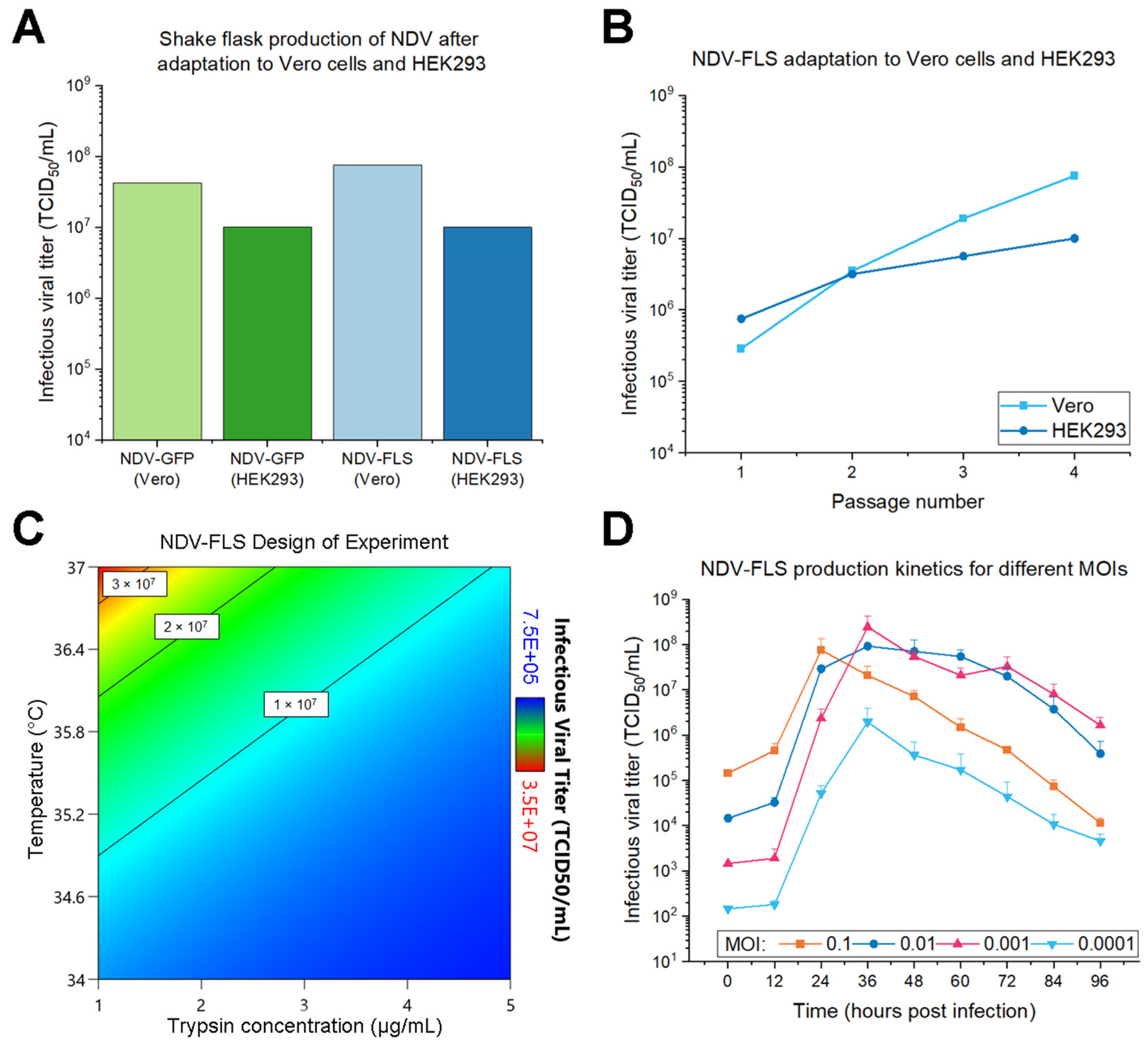
A two-level full factorial design was done with triplicates of each condition to screen 3 parameters at infection: trypsin concentration (from 1 to 5 µg/mL), trypsin addition (no repeated addition or addition at 24 h) and temperature (from 34 to 37 °C). To start the experiment, cultures of suspension Vero cells were centrifuged at 800× g for 5 min and seeded at 1 × 106 cells/mL in 30 mL MDXK media with 4 mM GlutaMAX in 250 mL shake flasks. The flasks were immediately infected with NDV-FLS at an MOI of 0.01 using the chosen DoE parameters. For trypsin addition at 24 h, trypsin was added to a final concentration of 1 or 5 µg/mL, according to the initial trypsin concentration assigned to each flask. Viral samples were taken at 30 hpi by centrifuging at 800× g for 5 min and aliquoting the supernatant (storage at −80 °C). Samples were quantified by TCID50 and analyzed with the Design Expert 13 software (Stat-Ease Inc., Minneapolis, MN, USA) using base 10 log-transformed values. Statistical significance was determined through ANOVA, followed by several residual analyses and diagnostics to confirm the quality of the model.
2.7. Multiplicity of Infection (MOI) Optimization
4 different multiplicities of infection (MOI) were evaluated: 0.1, 0.01, 0.001 and 0.0001 IVP/cell. Cultures of suspension Vero cells were centrifuged at 800× g for 5 min and seeded at 1 × 106 cells/mL in 25 mL MDXK media with 4 mM GlutaMAX in 250 mL shake flasks. Immediately, cells were infected with NDV-FLS at the corresponding MOIs with 1 µg/mL trypsin, in triplicates. Shake flasks were incubated at 37 °C, 135 rpm and 5% CO2 and samples were taken every 12 h after infection. For sampling, 0.8 mL culture of each flask was taken and spun down at 800× g for 5 min to aliquot the supernatant (storage at −80 °C). Viral samples were quantified by TCID50 and triplicates were averaged to plot the viral production kinetics of each MOI. The peak values of viral production for each MOI were compared in a one-way ANOVA with the Tukey method to investigate statistical significance.
2.8. Bioreactors
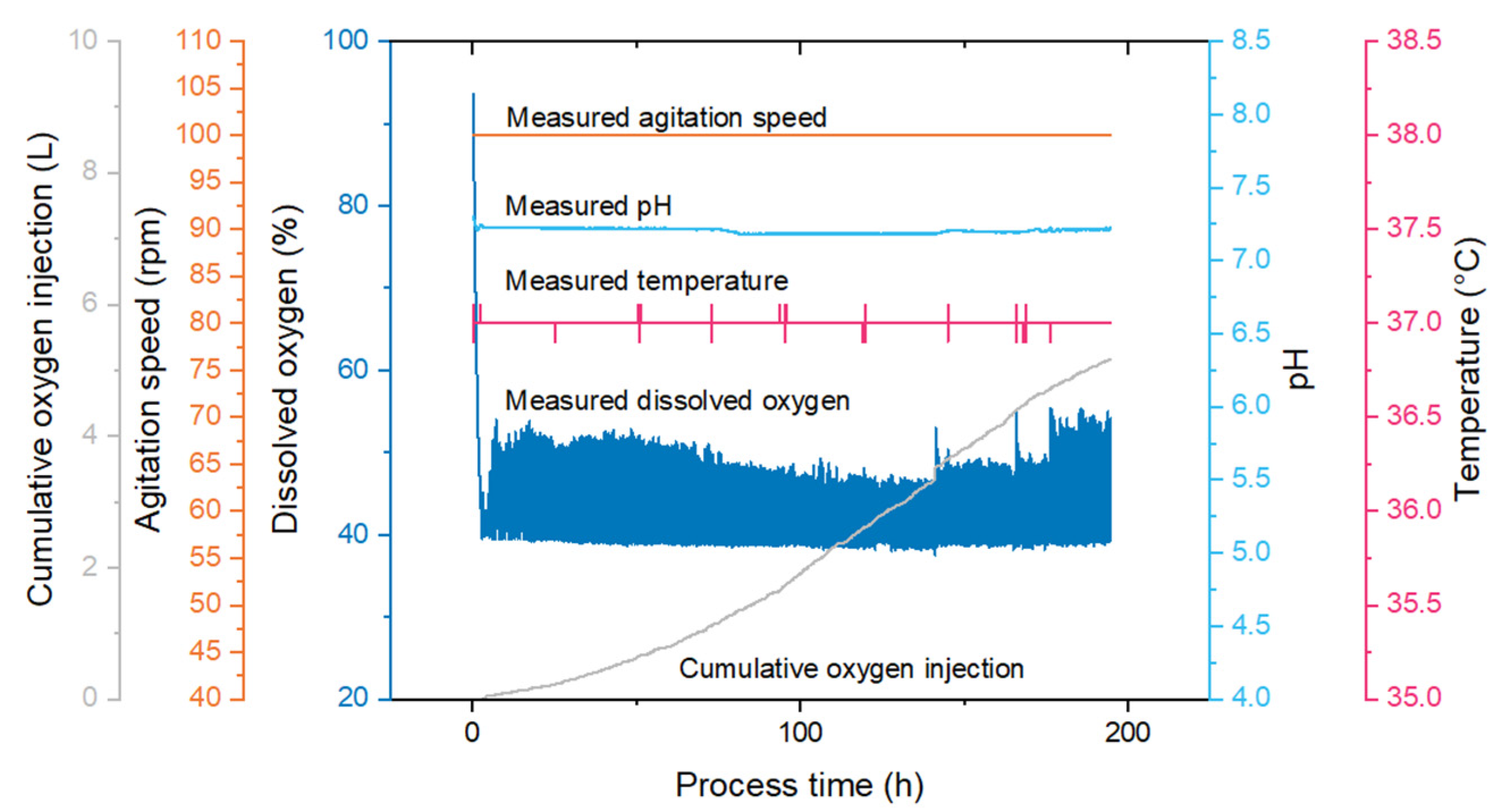
For 1 L bioreactors, a culture of 650 to 850 mL was seeded at 2.5 × 105 cells/mL in MDXK with 4 mM glutamine. The bioreactors (Applikon Biotechnology, Delft, The Netherlands) were assembled with a marine impeller for stirring and sensors for dissolved oxygen (DO) concentration, temperature and pH. The following parameters were controlled in the system: pH at 7.2, temperature at 37 °C, DO at 40–50% and stirring at 100 rpm. DO was maintained by a constant airflow in the headspace of 10 mL/min, along with pure oxygen sparging when necessary. The pH was regulated by addition of CO2 in the headspace or injection of NaHCO3 (90 g/L) (Sigma, USA).
Samples were taken every 24 h to monitor cell growth using the Vi-CELL XR Cell Viability Analyzer (Beckman Coulter Life Sciences, Brea, CA, USA). Glutamine was injected at 1 mM daily. For virus production, cells were infected at an MOI of 0.01 IVP/cell with 1 µg/mL trypsin on day 4 or 5 of culture, when cell density was approximately 8 × 105 cells/mL. After infection, samples were taken every 12 h and the supernatant was obtained by centrifuging at 800× g for 5 min (storage at −80 °C).
4. Discussion
NDV is a promising viral vector for vaccine development that has been studied for its potential application against several human diseases, and it is still commonly produced in embryonated chicken eggs [
2]. In this study, we set out to provide an alternative for NDV production by developing the foundation for a cell culture-based production process. Bioprocesses for vaccine manufacturing are composed of an upstream phase, a downstream phase, and the analytics used throughout the entire process to quantify the production and optimizations. Here, we developed analytical assays and evaluated upstream process parameters by testing cell lines for production, adapting the virus to suspension cell cultures and comparing several infection conditions. After this evaluation, we applied the selected parameters to produce NDV in 1 L scale bioreactors.
MDCK and Vero cells are well established systems for viral vaccine production, but a range of other continuous cell lines (CCLs) have also been studied for this purpose, including HEK293. While HEK293 shows promise as a cell line that has been adapted for suspension and grows to high densities in serum-free media [
32], manufacturers tend to prefer processes using established cells for faster licensing [
33]. Vero cells have a long history of proven safety, being the first CCL approved for viral vaccine production for human use. From a process perspective, these cells are commonly used with adherent cell culture technologies, such as microcarriers or fixed bed bioreactors, which are labor intensive and limited by surface area, resulting in a difficult scale up [
20]. However, recent advances in adapting Vero cells to suspension have been successful [
24], as these suspension Vero cells have been shown to work for virus production using stirred tank bioreactors in batch [
34] and perfusion [
24] modes. These cells have also been adapted to grow in the serum-free commercially available MDXK medium, after screening with several other media [
34]. Recently, this cell line’s genome has been sequenced through de novo assembly and annotated, facilitating future genome editing approaches [
35]. Furthermore, Vero cells are interferon-deficient [
35], making them susceptible to a wide range of viruses that have achieved high productivity when produced in Vero cells [
20].
In this study, suspension Vero cells showed the additional ability of yielding higher viral titers for both NDV-GFP and NDV-FLS constructs, which was in line with the more evident CPE and intensity of fluorescence observed in adherent Vero cells when compared to HEK293. Serial passaging of NDV in Vero cells led to an increase in titer after four passages, similar to what has been shown for other strains of NDV [
36], in which the number of passages required for such an increase varied for each strain. This increase is expected, as the viruses were originally collected in allantoic fluid, and viral adaptation to cell culture may select for viruses with more efficient replication in the new host cell. Further characterization of the viruses adapted to these cell lines could be important to evaluate if there were changes to safety, efficacy and abundance of recombinant protein on the viral surface when compared to the virus produced in eggs.
After defining suspension Vero as the cell line of choice for NDV production, a DoE revealed that the highest NDV-FLS titers were obtained when infecting at 37 °C with 1 µg/mL trypsin, and that repeated trypsin addition had no significant effect. VSV titers are influenced by the temperature in the production phase, and each construct has an optimal temperature [
34]. As the LaSota strain of NDV is not thermostable [
37], similarly to VSV, a lower temperature could have resulted in higher viral titers. However, a production temperature of 37 °C led to significantly higher titers than 34 °C, ruling out the use of low temperatures for these NDV constructs. This may be in line with the 37 °C incubation step that is typically implemented when producing NDV in embryonated eggs [
18,
38]. As for trypsin, the concentrations tested were 1 and 5 µg/mL, which are values reported in the literature for NDV experiments [
37,
39]. In our study, the highest NDV titers were achieved with the lowest trypsin concentration, which is similar to what has been observed for influenza virus [
17]. Vero cells are known to produce trypsin inhibitors [
40], and multiple additions of trypsin have been described as having a positive effect [
41] or no effect [
40] on the multi-cycle production of influenza in this cell line. For NDV, we found that repeated trypsin addition had no apparent effect on the viral titer produced, which prompted us to add trypsin only at the moment of infection.
A range of MOIs (0.1–0.0001) that encompasses the MOIs used for NDV in previous works [
37,
39,
42] was also evaluated. With the exception of the lowest one tested, all MOIs reached a similar peak of approximately 1 × 10
8 TCID
50/mL. The viral production peak was 24 hpi for the highest MOI (0.1), and shifted to a later time point (36 hpi) with lower MOIs. However, this higher MOI showed a greater and earlier loss of infectivity than the next two MOIs assayed (0.01 and 0.001). For the 0.01 MOI, the titer remained relatively constant until 60 hpi, and was still higher than the 0.1 MOI by the end of the experiment at 96 hpi. Such stability is important for a robust process, as it is more likely to result in an adequate yield even if production kinetics shift due to variations in the process. The 0.01 MOI was chosen for the process, since an MOI 10 times lower still yielded similar results, and thus possible volume errors when adding the virus at 0.01 MOI would still lead to a reliable production. Overall, from the first passage in Vero cells to the last shake flask optimization experiment, the produced NDV-FLS titers increased by almost 320-fold, from 2.87 × 10
5 TCID
50/mL to 9.17 × 10
7 TCID
50/mL, indicating that the selection of culture and infection parameters was adequate.
Aside from the cell lines and infection parameters used, analytics are also an essential part of the production process that should not be overlooked. The virus being produced must be characterized and quantified throughout several steps of manufacturing to generate crucial data for process development and for regulatory approval [
43]. As a replicative viral vector [
2], NDV can be quantified regaring the replication-competent particles—also known as functional or infectious titer—and regarding the total particles, which may or may not be functional. The ratio between these two titers is indicative of quality and can be used to assess different time points or conditions of the process [
44]. As such, when developing a process, it is important to establish reliable and scalable analytical methods to increase feasibility of implementing this process in large scale, consequently improving the chances of quickly achieving mass vaccination for a new pathogen of concern.
In this study, not only have we developed assays for each type of quantification, but we have also established methods of reading the TCID
50 assay amenable to automation. NDV-GFP was quantified by reading fluorescence on a plate reader, while other constructs, such as NDV-FLS, can also be quantified on a plate reader when paired with a reagent that detects viability. Alamar blue is a blue dye based on resazurin, which changes to a red color when reduced to resorufin in metabolically active cells, indicating cell health [
45]. Both fluorescence and viability were shown to be comparable to CPE quantification, resulting in valid methods of reading TCID
50. As these methods rely on plate readers, and not visual inspection, they are non-subjective and can be automated for use in industry or for standardization across collaborating institutions and facilities. Therefore, the availability of these tools makes the assay more feasible for high throughput processes and industrial application. Antibody-based assays, such as an immunofluorescence assay (IFA) [
18,
46], could also be of interest, as they can be targeted to quantify only viruses that contain the protein required for immunization, which could be important in vaccine manufacturing. This specificity, however, means having to adapt the assay with a different antibody for each new construct, which could slow down the development of new vaccines using the platform. Therefore, TCID
50 and ddPCR assays were chosen, as they can be used for any NDV construct.
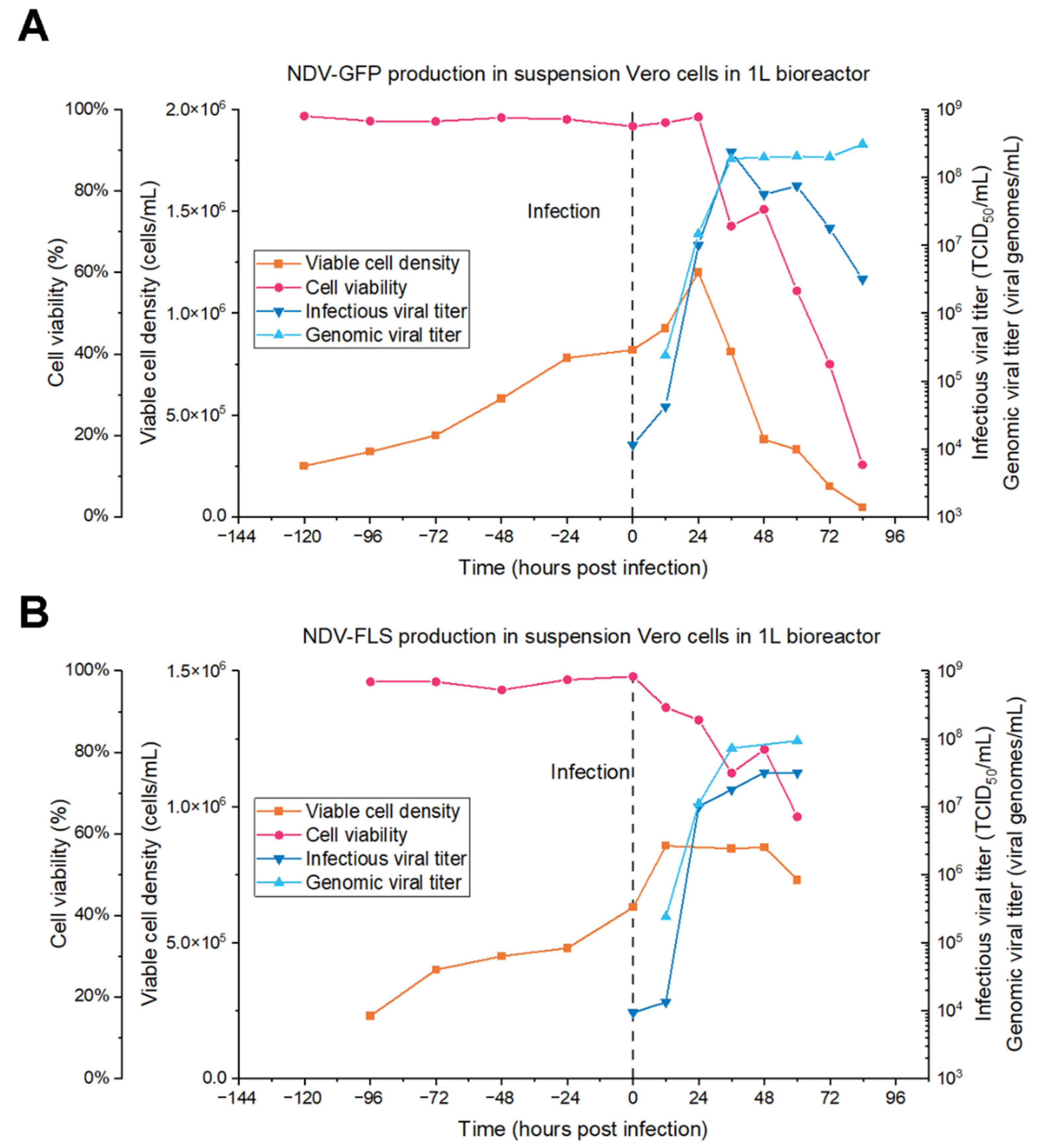
After establishing the infection parameters at small scale and the analytical assays, we set out to produce NDV in batch mode in 1 L stirred tank bioreactors. For NDV-GFP, the peak titer produced was 2.37 ± 0.82 × 10
8 TCID
50/mL at 36 hpi, which is similar to the highest titer observed in shake flasks (1.07 ± 0.37 × 10
8 TCID
50/mL). As for NDV-FLS, the peak production was 3.16 ± 1.09 × 10
7 TCID
50/mL at 48 hpi, which is similar to the value at 36 hpi (1.78 ± 0.62 × 10
7 TCID
50/mL) when considering the analytical error. This is lower than the highest values achieved with this MOI in shake flasks (9.17 ± 1.44 × 10
7 TCID
50/mL), which can occur when scaling up to bioreactors because of differences in stirring and many other factors. Both productions are comparable to the titers produced in embryonated eggs, which is in the order of 10
8 FFU/mL [
18] and 10
7 PFU/mL [
47], indicating that the bioreactor-based process developed in this study is a valuable substitute for existing egg-based productions.
Furthermore, process intensification could increase the quantity of infective particles harvested, using technologies such as fed-batch or perfusion [
48]. The lower infectious titers observed at later time points in bioreactors and shake flasks suggest that NDV could be a good candidate for production in perfusion mode, as the viruses could be continuously harvested before suffering a loss in infectivity due to temperature and shear stress in the bioreactor.
Therefore, we have successfully developed the upstream process and analytical methods for suspension Vero cell-based production of NDV, using the constructs NDV-GFP and NDV-FLS as models. Future steps include establishing a scalable purification protocol and testing different bioreactor production modes, such as fed-batch and perfusion, to move towards a complete process based on continuous manufacturing.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}