


Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation

 ,
,  , , , , ,
, , , , , 

Abstract
:1. Introduction
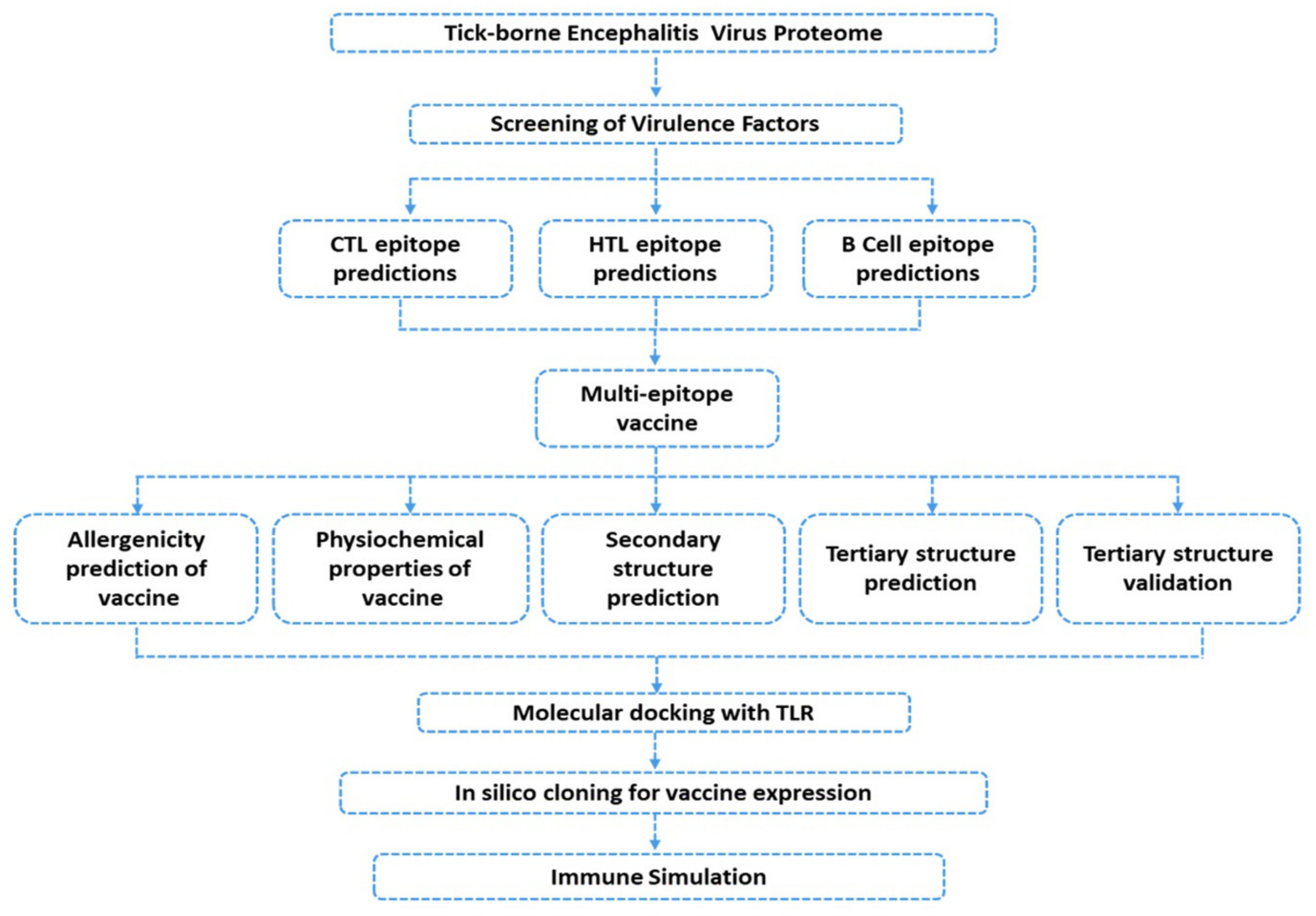
2. Materials and Methods
2.1. Data Retrieval
2.2. Data Processing
2.2.1. Prediction of Immunogenic Epitopes
2.2.2. Multiple-Epitope Vaccine Designing and Evaluation
2.2.3. Prediction of Vaccine Antigenicity
2.2.4. Multiple-Epitope Vaccine 3D Structure Prediction and Validation
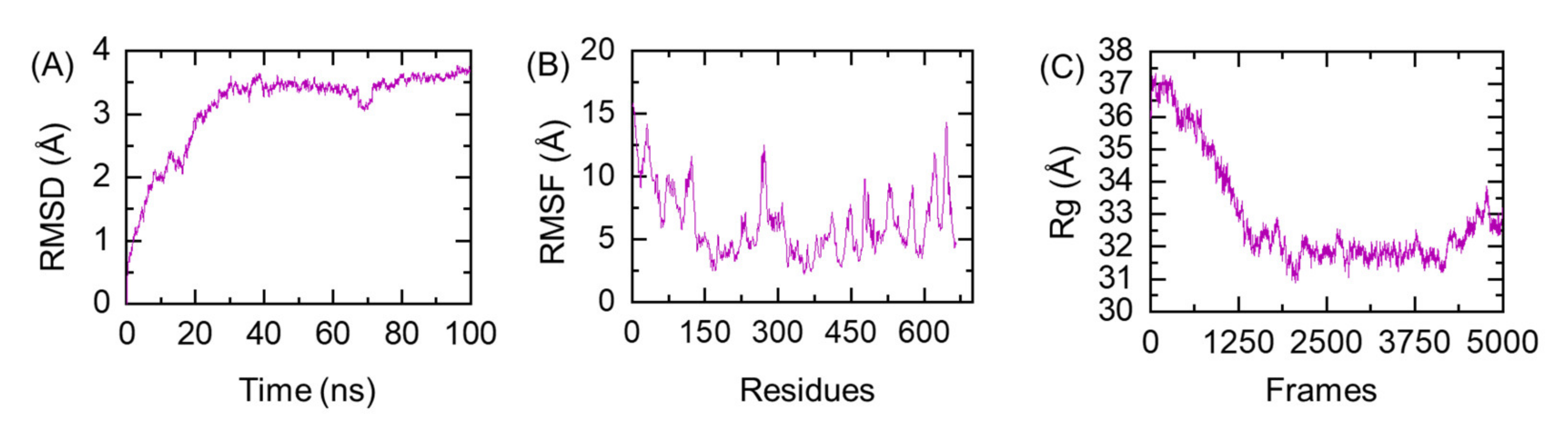
2.2.5. Ensemble Vaccine Folding Evaluation through MD Simulation
2.3. Evaluation of the Immune Response Triggering Properties of MEVC
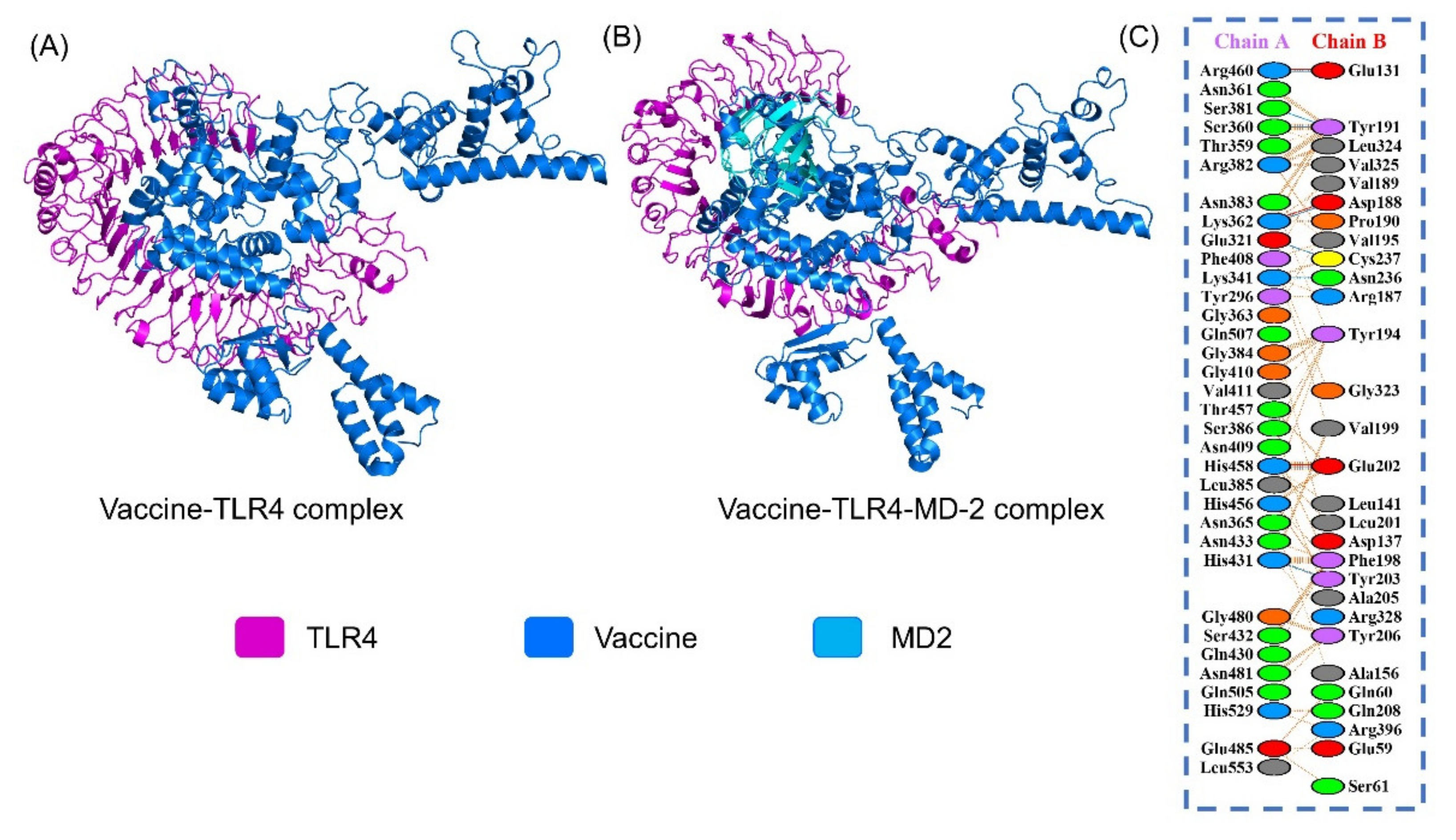
2.3.1. Molecular Docking of the Vaccine
2.3.2. Binding Free Energy Calculation
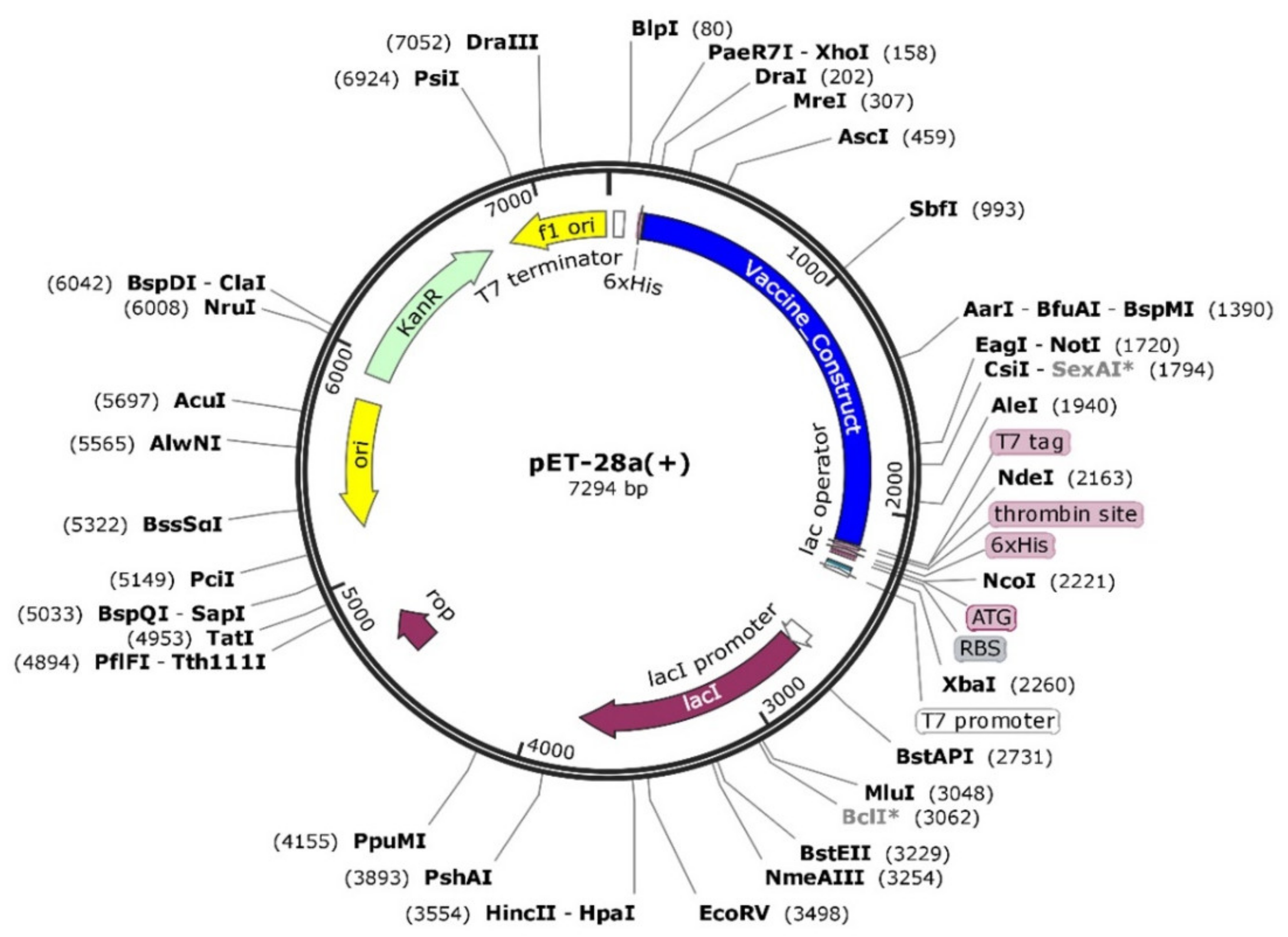
2.3.3. In Silico Cloning and Expression
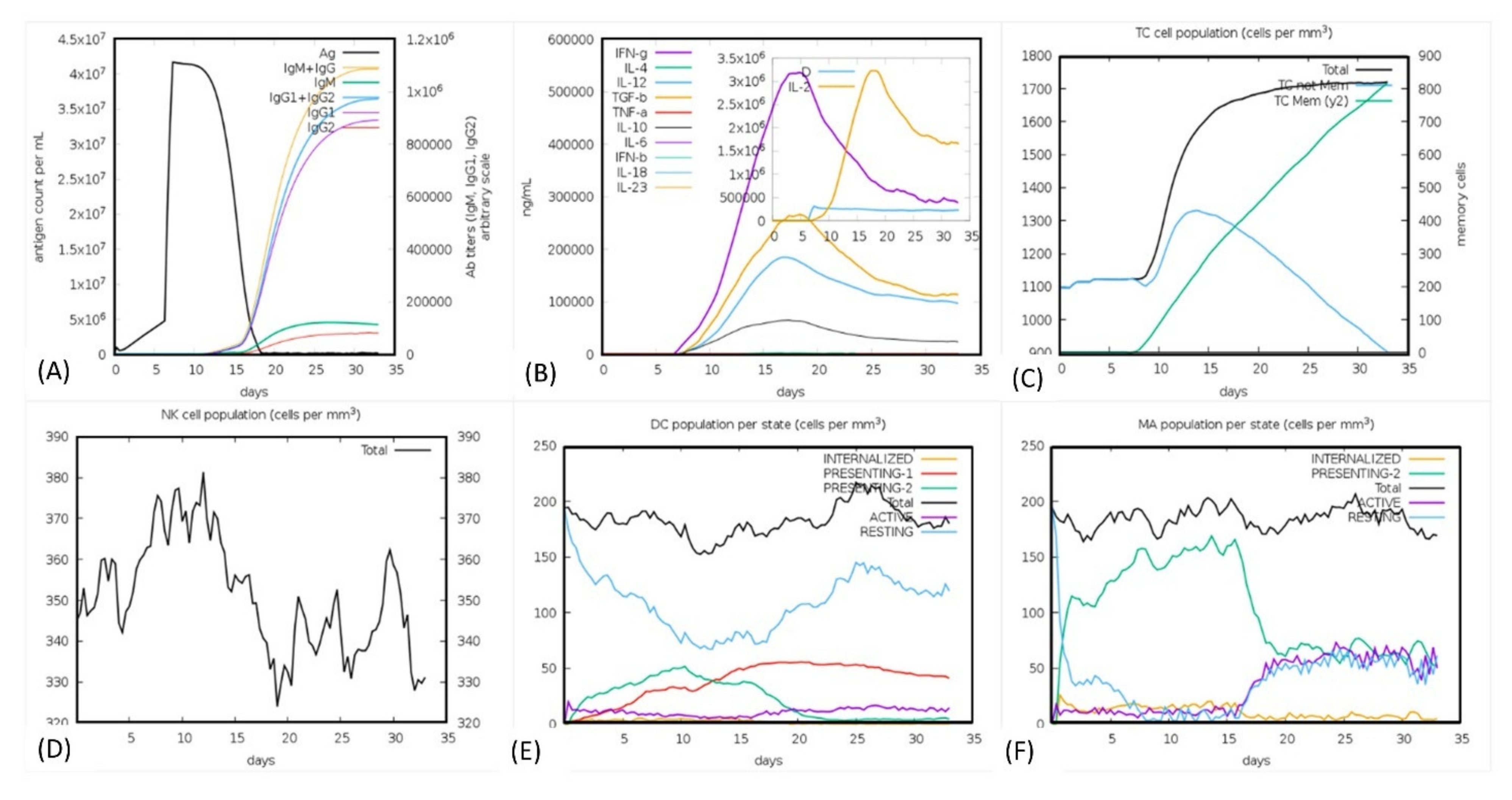
2.3.4. Immune Simulation
3. Results
3.1. Sequence Retrieval and Antigenicity Profiling
3.2. Unveiling Cytotoxic T Lymphocytes (CTL) Epitopes
3.3. Prioritizing Helper T Lymphocytes (HTL) Epitopes
3.4. Prediction of B Cell Epitopes
3.5. Multi-Epitope Peptide Vaccine Design
3.6. Prediction of Vaccine 3D Structure and Validation
3.7. D Structure Validation
3.8. Prediction of Allergenicity
3.9. Antigenicity of the Vaccine Construct
3.10. Physiochemical Properties of the Designed Vaccine
3.11. Prediction of Secondary Structure Elements
3.12. Protein Folding Evaluation through All-Atoms MD Simulation
3.13. Protein-Peptide Docking of the Vaccine to TLR4
3.14. Binding Free Energy Evaluation of MEVC-TLR4
3.15. Codon Optimization and In Silico Cloning
3.16. Immune Simulation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansfield, K.; Johnson, N.; Phipps, L.; Stephenson, J.; Fooks, A.; Solomon, T. Tick-borne encephalitis virus—A review of an emerging zoonosis. J. Gen. Virol. 2009, 90, 1781–1794. [Google Scholar] [CrossRef]
- Gould, E.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Süss, J. Tick-borne encephalitis in europe and beyond—The epidemiological situation as of 2007. Eurosurveillance 2008, 13, 717–727. [Google Scholar] [CrossRef]
- Bogovic, P.; Strle, F. Tick-borne encephalitis: A review of epidemiology, clinical characteristics, and management. World J. Clin. Cases 2015, 3, 430. [Google Scholar] [CrossRef] [PubMed]
- Taba, P.; Schmutzhard, E.; Forsberg, P.; Lutsar, I.; Ljøstad, U.; Mygland, Å.; Levchenko, I.; Strle, F.; Steiner, I. Ean consensus review on prevention, diagnosis and management of tick-borne encephalitis. Eur. J. Neurol. 2017, 24, 1214–1261. [Google Scholar] [CrossRef]
- Heinze, D.; Gould, E.; Forrester, N. Revisiting the clinal concept of evolution and dispersal for the tick-borne flaviviruses by using phylogenetic and biogeographic analyses. J. Virol. 2012, 86, 8663–8671. [Google Scholar] [CrossRef]
- Danielová, V.; Daniel, M.; Schwarzová, L.; Materna, J.; Rudenko, N.; Golovchenko, M.; Holubová, J.; Grubhoffer, L.; Kilián, P. Integration of a tick-borne encephalitis virus and borrelia burgdorferi sensu lato into mountain ecosystems, following a shift in the altitudinal limit of distribution of their vector, ixodes ricinus (krkonoše mountains, Czech republic). Vector-Borne Zoonotic Dis. 2010, 10, 223–230. [Google Scholar] [CrossRef]
- Radda, A.; Hofmann, H.; Pretzmann, G. Threshold of viraemia in apodemus flavicollis for infection of ixodes ricinus with tick-borne encephalitis virus. Acta Virol. 1969, 13, 74. [Google Scholar]
- Kubinski, M.; Beicht, J.; Gerlach, T.; Volz, A.; Sutter, G.; Rimmelzwaan, G.F. Tick-borne encephalitis virus: A quest for better vaccines against a virus on the rise. Vaccines 2020, 8, 451. [Google Scholar] [CrossRef] [PubMed]
- Proutski, V.; Gould, E.A.; Holmes, E.C. Secondary structure of the 3′ untranslated region of flaviviruses: Similarities and differences. Nucleic Acids Res. 1997, 25, 1194–1202. [Google Scholar] [CrossRef]
- Kovalev, S.; Mukhacheva, T. Reconsidering the classification of tick-borne encephalitis virus within the siberian subtype gives new insights into its evolutionary history. Infect. Genet. Evol. 2017, 55, 159–165. [Google Scholar] [CrossRef]
- Dai, X.; Shang, G.; Lu, S.; Yang, J.; Xu, J. A new subtype of eastern tick-borne encephalitis virus discovered in qinghai-tibet plateau, china. Emerg. Microbes Infect. 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Bröker, M.; Liang, G. Tick-borne encephalitis in mainland china. Vector-Borne Zoonotic Dis. 2008, 8, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kollaritsch, H.; Paulke-Korinek, M.; Holzmann, H.; Hombach, J.; Bjorvatn, B.; Barrett, A. Vaccines and vaccination against tick-borne encephalitis. Expert Rev. Vaccines 2012, 11, 1103–1119. [Google Scholar] [CrossRef]
- Barrett, P.N.; Schober-Bendixen, S.; Ehrlich, H.J. History of tbe vaccines. Vaccine 2003, 21, S41–S49. [Google Scholar] [CrossRef]
- Khan, S.; Ali, S.S.; Zaheer, I.; Saleem, S.; Ziaullah; Zaman, N.; Iqbal, A.; Suleman, M.; Wadood, A.; Rehman, A.U.; et al. Proteome-wide mapping and reverse vaccinology-based b and t cell multi-epitope subunit vaccine designing for immune response reinforcement against porphyromonas gingivalis. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Ahmad, I.; Ali, S.S.; Shah, I.; Khan, S.; Khan, M.; Ullah, S.; Ali, S.; Khan, J.; Ali, M.; Khan, A.; et al. Computational vaccinology based development of multi-epitope subunit vaccine for protection against the norovirus infections. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gul, H.; Ali, S.S.; Saleem, S.; Khan, S.; Khan, J.; Wadood, A.; Rehman, A.U.; Ullah, Z.; Ali, S.; Khan, H.; et al. Subtractive proteomics and immunoinformatics approaches to explore bartonella bacilliformis proteome (virulence factors) to design b and t cell multi-epitope subunit vaccine. Infect. Genet. Evol. 2020, 85, 104551. [Google Scholar] [CrossRef]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef]
- Ali, A.; Khan, A.; Kaushik, A.C.; Wang, Y.; Ali, S.S.; Junaid, M.; Saleem, S.; Cho, W.C.; Mao, X.; Wei, D.-Q. Immunoinformatic and systems biology approaches to predict and validate peptide vaccines against epstein–barr virus (ebv). Sci. Rep. 2019, 9, 720. [Google Scholar] [CrossRef]
- Uddin, R.; Siddiqui, Q.N.; Azam, S.S.; Saima, B.; Wadood, A. Identification and characterization of potential druggable targets among hypothetical proteins of extensively drug resistant mycobacterium tuberculosis (xdr kzn 605) through subtractive genomics approach. Eur. J. Pharm. Sci. 2018, 114, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Weinreich Olsen, A.; Hansen, P.R.; Holm, A.; Andersen, P. Efficient protection against mycobacterium tuberculosis by vaccination with a single subdominant epitope from the esat-6 antigen. Eur. J. Immunol. 2000, 30, 1724–1732. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Kathwate, G.H. In silico design and characterization of multiepitopes vaccine for SARS-CoV2 from its spike proteins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Singh, S.; Malik, B.; Sharma, D. Metabolic pathway analysis of s. Pneumoniae: An in silico approach towards drug-design. J. Bioinform. Comput. Biol. 2007, 5, 135–153. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. Ncbi blast: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic t-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. Ellipro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P. Prediction methods for b-cell epitopes. In Immunoinformatics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 387–394. [Google Scholar]
- Sabourin, M.; Tuzon, C.T.; Fisher, T.S.; Zakian, V.A. A flexible protein linker improves the function of epitope-tagged proteins in saccharomyces cerevisiae. Yeast 2007, 24, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.-G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A potential protein adjuvant derived from mycobacterium tuberculosis rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G. Algpred: Prediction of allergenic proteins and mapping of ige epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef]
- Zaharieva, N.; Dimitrov, I.; Flower, D.; Doytchinova, I. Vaxijen dataset of bacterial immunogens: An update. Curr. Comput. Aided Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the expasy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The psipred protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Peng, J.; Xu, J. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. Prosa-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Lengths, M.; Angles, M. Limitations of structure evaluation tools errat. Quick Guidel. Comput. Drug Des. 2018, 16, 75. [Google Scholar]
- Bhardwaj, V.K.; Purohit, R. Targeting the protein-protein interface pocket of aurora-a-tpx2 complex: Rational drug design and validation. J. Biomol. Struct. Dyn. 2021, 39, 3882–3891. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with amber on gpus. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. Ptraj and cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Patchdock and symmdock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. Firedock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the mm/pbsa and mm/gbsa methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Modeling 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. Jcat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- G Biotech. Snapgene viewer. Glick B, editor 3.
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; ul Qamar, M.T.; Saleem, S.; Ahmad, S.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; et al. Mutational landscape of pirin and elucidation of the impact of most detrimental missense variants that accelerate the breast cancer pathways: A computational modelling study. Front. Mol. Biosci. 2021, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 13276–13281. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3clpro). J. Biomol. Struct. Dyn. 2020, 39, 4659–4670. [Google Scholar] [CrossRef]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the SARS-CoV-2 new variants is associated with k417n/t, e484k, and n501y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Khan, A.; Heng, W.; Wang, Y.; Qiu, J.; Wei, X.; Peng, S.; Saleem, S.; Khan, M.; Ali, S.S.; Wei, D.-Q. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3clpro). Phytother. Res. 2021, 1–3. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Saleem, S.; Nizam-Uddin, N.; Mohammad, A.; Khan, T.; Ahmad, S.; Arshad, M.; Ali, S.S.; Suleman, M.; et al. Immunogenomics guided design of immunomodulatory multi-epitope subunit vaccine against the SARS-CoV-2 new variants, and its validation through in silico cloning and immune simulation. Comput. Biol. Med. 2021, 133, 104420. [Google Scholar] [CrossRef]
- Lin, W.J.; Yeh, W.C. Implication of toll-like receptor and tumor necrosis factor alpha signaling in septic shock. Shock 2005, 24, 206–209. [Google Scholar] [CrossRef]
- Beard, C.B.; Nelson, C.A.; Mead, P.S.; Petersen, L.R. Bartonella spp. Bacteremia and rheumatic symptoms in patients from lyme disease–endemic region. Emerg. Infect. Dis. 2012, 18, 1918. [Google Scholar] [CrossRef]
- McGowin, C.L.; Totten, P.A. The unique microbiology and molecular pathogenesis of mycoplasma genitalium. J. Infect. Dis. 2017, 216, S382–S388. [Google Scholar] [CrossRef]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.-Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrhpvs e6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef]
- Black, M.; Trent, A.; Tirrell, M.; Olive, C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev. Vaccines 2010, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Apellániz, B.; Nieva, J.L. The use of liposomes to shape epitope structure and modulate immunogenic responses of peptide vaccines against hiv mper. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 99, pp. 15–54. [Google Scholar]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore helicobacter pylori proteome (virulence factors) to design b and t cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring leishmania secretory proteins to design b and t cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Langshaw, E.; Hartas, J.; Lam, A.; Batzloff, M.R.; Good, M.F. A synthetic m protein peptide synergizes with a cxc chemokine protease to induce vaccine-mediated protection against virulent streptococcal pyoderma and bacteremia. J. Immunol. 2015, 194, 5915–5925. [Google Scholar] [CrossRef]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a peptide-based vaccine against shigella sonnei: A subtractive reverse vaccinology based approach. Biologicals 2017, 50, 87–99. [Google Scholar] [CrossRef]
- Rahlenbeck, S.; Fingerle, V.; Doggett, S. Prevention of tick-borne diseases: An overview. Br. J. Gen. Pract. 2016, 66, 492–494. [Google Scholar] [CrossRef]
- Khan, A.; Khan, M.; Saleem, S.; Babar, Z.; Ali, A.; Khan, A.A.; Sardar, Z.; Hamayun, F.; Ali, S.S.; Wei, D.-Q. Phylogenetic analysis and structural perspectives of rna-dependent rna-polymerase inhibition from SARS-CoV-2 with natural products. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 335–348. [Google Scholar] [CrossRef]
- Khan, A.; Khan, M.T.; Saleem, S.; Junaid, M.; Ali, A.; Ali, S.S.; Khan, M.; Wei, D.-Q. Structural insights into the mechanism of rna recognition by the n-terminal rna-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput. Struct. Biotechnol. J. 2020, 18, 2174–2184. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue No. | Peptide Sequence | MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | Transport Affinity | Prediction Score | MHC-I Binding | Antigenecity Score |
|---|---|---|---|---|---|---|---|---|
| 2237 | SSDDNKLAY | 0.7693 | 3.2662 | 0.9150 | 2.9740 | 3.5521 | YES | Non-antigenic |
| 184 | NVDGVYLEY | 0.6968 | 2.9587 | 0.9788 | 3.0560 | 3.2583 | YES | 0.876 |
| 3035 | STLNGGLFY | 0.6183 | 2.6251 | 0.9456 | 3.0220 | 2.9180 | YES | Non-antigenic |
| 98 | ATDWMSWLL | 0.5851 | 2.4842 | 0.9229 | 0.9550 | 2.6704 | YES | Non-antigenic |
| 1788 | SIAARGHLY | 0.4418 | 1.8759 | 0.9337 | 3.1000 | 2.1710 | YES | 0.524 |
| 3262 | ETACLSKAY | 0.4369 | 1.8550 | 0.2200 | 2.8480 | 2.0304 | YES | 0.691 |
| 2130 | CVSALDVFY | 0.4011 | 1.7029 | 0.7949 | 3.1500 | 1.9797 | YES | Non-antigenic |
| 3018 | GVEGISLNY | 0.3920 | 1.6642 | 0.9715 | 2.8940 | 1.9546 | YES | 1.4 |
| 451 | KTILTMGEY | 0.3915 | 1.6621 | 0.7760 | 2.9120 | 1.9241 | YES | 0.494 |
| 422 | KVEPHTGDY | 0.3583 | 1.5213 | 0.9717 | 3.0730 | 1.8207 | YES | 0.707 |
| 971 | HTDQSLWMR | 0.3781 | 1.6055 | 0.7247 | 1.2050 | 1.7744 | YES | Non-antigenic |
| 3077 | ATTIMQKAY | 0.3578 | 1.5192 | 0.4362 | 3.0590 | 1.7375 | YES | 0.454 |
| 1955 | RQDGRTDEY | 0.3297 | 1.3998 | 0.9504 | 3.0380 | 1.6942 | YES | Non-antigenic |
| 800 | EVSEWYDNY | 0.3269 | 1.3879 | 0.9476 | 2.9870 | 1.6794 | YES | Non-antigenic |
| 2530 | CTREEFFVY | 0.3072 | 1.3045 | 0.9681 | 3.0520 | 1.6023 | YES | 0.617 |
| 1783 | WTDPHSIAA | 0.3502 | 1.4869 | 0.4722 | 0.7830 | 1.5185 | YES | Non-antigenic |
| 2214 | MAGVALIFY | 0.2864 | 1.2161 | 0.5939 | 3.0020 | 1.4553 | YES | 0.532 |
| 2855 | MTDTTAFGQ | 0.3395 | 1.4416 | 0.0703 | 0.1020 | 1.4470 | YES | Non-antigenic |
| 2813 | RTWQYWGSY | 0.2555 | 1.0848 | 0.9775 | 3.2670 | 1.3948 | YES | Non-antigenic |
| 3342 | VMEWRDVPY | 0.2301 | 0.9770 | 0.9072 | 3.0960 | 1.2679 | YES | 1.79 |
| 980 | SMKNDTGTY | 0.2236 | 0.9492 | 0.9495 | 3.2290 | 1.2530 | YES | Non-antigenic |
| 1143 | VALFVVLEY | 0.2002 | 0.8501 | 0.9747 | 3.0660 | 1.1496 | YES | 0.652 |
| 1235 | VTTYFLLLV | 0.2186 | 0.9279 | 0.7355 | 0.3550 | 1.0560 | YES | Non-antigenic |
| 598 | PTDSGHDTV | 0.2370 | 1.0062 | 0.3920 | 0.2090 | 1.0545 | YES | 0.425 |
| 553 | VAHIEGTKY | 0.1785 | 0.7579 | 0.9739 | 2.9240 | 1.0502 | YES | Non-antigenic |
| 2133 | ALDVFYTLM | 0.2087 | 0.8861 | 0.9760 | 0.3460 | 1.0498 | YES | Non-antigenic |
| 1634 | AQGVVVGLY | 0.1806 | 0.7667 | 0.8269 | 3.1220 | 1.0469 | YES | 0.718 |
| 1677 | WTSKGQITV | 0.2126 | 0.9025 | 0.8831 | 0.2320 | 1.0466 | YES | 1.19 |
| 448 | SSEKTILTM | 0.2101 | 0.8922 | 0.9417 | 0.2630 | 1.0466 | YES | Non-antigenic |
| 411 | VYDANKIVY | 0.1765 | 0.7494 | 0.8988 | 3.0520 | 1.0369 | YES | Non-antigenic |
| 994 | VTDLRNCSW | 0.1987 | 0.8437 | 0.9180 | 0.7970 | 1.0212 | YES | 1.377 |
| S. No. | Allele | Start | End | Peptide Sequence | Method | Percentile Rank |
|---|---|---|---|---|---|---|
| 1 | HLA-DRB1*07:01 | 73 | 87 | AALRKIKRTVSALMV | Consensus (comb.lib./smm/nn) | 0.03 |
| 2 | HLA-DRB1*07:01 | 72 | 86 | TAALRKIKRTVSALM | Consensus (comb.lib./smm/nn) | 0.04 |
| 3 | HLA-DRB1*07:01 | 74 | 88 | ALRKIKRTVSALMVG | Consensus (comb.lib./smm/nn) | 0.06 |
| 4 | HLA-DRB1*07:01 | 75 | 89 | LRKIKRTVSALMVGL | Consensus (comb.lib./smm/nn) | 0.06 |
| 5 | HLA-DRB1*07:01 | 76 | 90 | RKIKRTVSALMVGLQ | Consensus (comb.lib./smm/nn) | 0.06 |
| 6 | HLA-DRB5*01:01 | 65 | 79 | SVPLKQATAALRKIK | Consensus (smm/nn/sturniolo) | 0.08 |
| 7 | HLA-DRB5*01:01 | 66 | 80 | VPLKQATAALRKIKR | Consensus (smm/nn/sturniolo) | 0.08 |
| 8 | HLA-DRB1*07:01 | 77 | 91 | KIKRTVSALMVGLQK | Consensus (comb.lib./smm/nn) | 0.13 |
| 9 | HLA-DRB5*01:01 | 64 | 78 | NSVPLKQATAALRKI | Consensus (smm/nn/sturniolo) | 0.14 |
| 10 | HLA-DRB5*01:01 | 63 | 77 | WNSVPLKQATAALRK | Consensus (smm/nn/sturniolo) | 0.43 |
| 11 | HLA-DRB1*15:01 | 35 | 49 | NGLVLMRMMGILWHA | Consensus (smm/nn/sturniolo) | 0.72 |
| 12 | HLA-DRB1*15:01 | 33 | 47 | MPNGLVLMRMMGILW | Consensus (smm/nn/sturniolo) | 0.73 |
| 13 | HLA-DRB1*15:01 | 34 | 48 | PNGLVLMRMMGILWH | Consensus (smm/nn/sturniolo) | 0.73 |
| 14 | HLA-DRB5*01:01 | 68 | 82 | LKQATAALRKIKRTV | Consensus (smm/nn/sturniolo) | 0.75 |
| 15 | HLA-DRB5*01:01 | 67 | 81 | PLKQATAALRKIKRT | Consensus (smm/nn/sturniolo) | 0.81 |
| 16 | HLA-DRB1*15:01 | 36 | 50 | GLVLMRMMGILWHAV | Consensus (smm/nn/sturniolo) | 0.88 |
| 17 | HLA-DRB1*15:01 | 37 | 51 | LVLMRMMGILWHAVA | Consensus (smm/nn/sturniolo) | 0.93 |
| S. No | Position | Epitope | Score |
|---|---|---|---|
| 1 | 3 | KKAILKGKGGGPPRRVSKET | 1 |
| 2 | 1578 | EEKWKGETVQVHAFPPGRAH | 0.999 |
| 3 | 870 | DPTDYRGGVPGLLKKGKDIK | 0.999 |
| 4 | 401 | CEAKKKATGHVYDANKIVYT | 0.999 |
| 5 | 1809 | MTATPPGKSEPFPESNGAIT | 0.999 |
| 6 | 2654 | VMCDIGESSPDAAVEGERTR | 0.998 |
| 7 | 1493 | VFSGQGGRERGDRPFEVKDG | 0.995 |
| 8 | 1518 | SPGLFWGQNQVGVGYGSKGV | 0.995 |
| 9 | 2812 | YRTWQYWGSYRTAPTGSAAS | 0.995 |
| 10 | 2870 | KVDTKAQEPQPGTRVIMRAV | 0.995 |
| 11 | 2049 | TDRSWTWEGPEANAVDEASG | 0.993 |
| 12 | 1987 | DNITTLRGPVATFYGPEQDK | 0.993 |
| 13 | 3362 | LVGRRERAEWAKNIWGAVEK | 0.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleman, M.; ul Qamar, M.T.; Kiran; Rasool, S.; Rasool, A.; Albutti, A.; Alsowayeh, N.; Alwashmi, A.S.S.; Aljasir, M.A.; Ahmad, S.; et al. Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation. Vaccines 2021, 9, 1210. https://doi.org/10.3390/vaccines9111210
Suleman M, ul Qamar MT, Kiran, Rasool S, Rasool A, Albutti A, Alsowayeh N, Alwashmi ASS, Aljasir MA, Ahmad S, et al. Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation. Vaccines. 2021; 9(11):1210. https://doi.org/10.3390/vaccines9111210
Chicago/Turabian StyleSuleman, Muhammad, Muhammad Tahir ul Qamar, Kiran, Samreen Rasool, Aneela Rasool, Aqel Albutti, Noorah Alsowayeh, Ameen S. S. Alwashmi, Mohammad Abdullah Aljasir, Sajjad Ahmad, and et al. 2021. "Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation" Vaccines 9, no. 11: 1210. https://doi.org/10.3390/vaccines9111210
APA StyleSuleman, M., ul Qamar, M. T., Kiran, Rasool, S., Rasool, A., Albutti, A., Alsowayeh, N., Alwashmi, A. S. S., Aljasir, M. A., Ahmad, S., Hussain, Z., Rizwan, M., Ali, S. S., Khan, A., & Wei, D.-Q. (2021). Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation. Vaccines, 9(11), 1210. https://doi.org/10.3390/vaccines9111210