
The Immunology of Hepatocellular Carcinoma

 ,
, 

Abstract
:1. Introduction
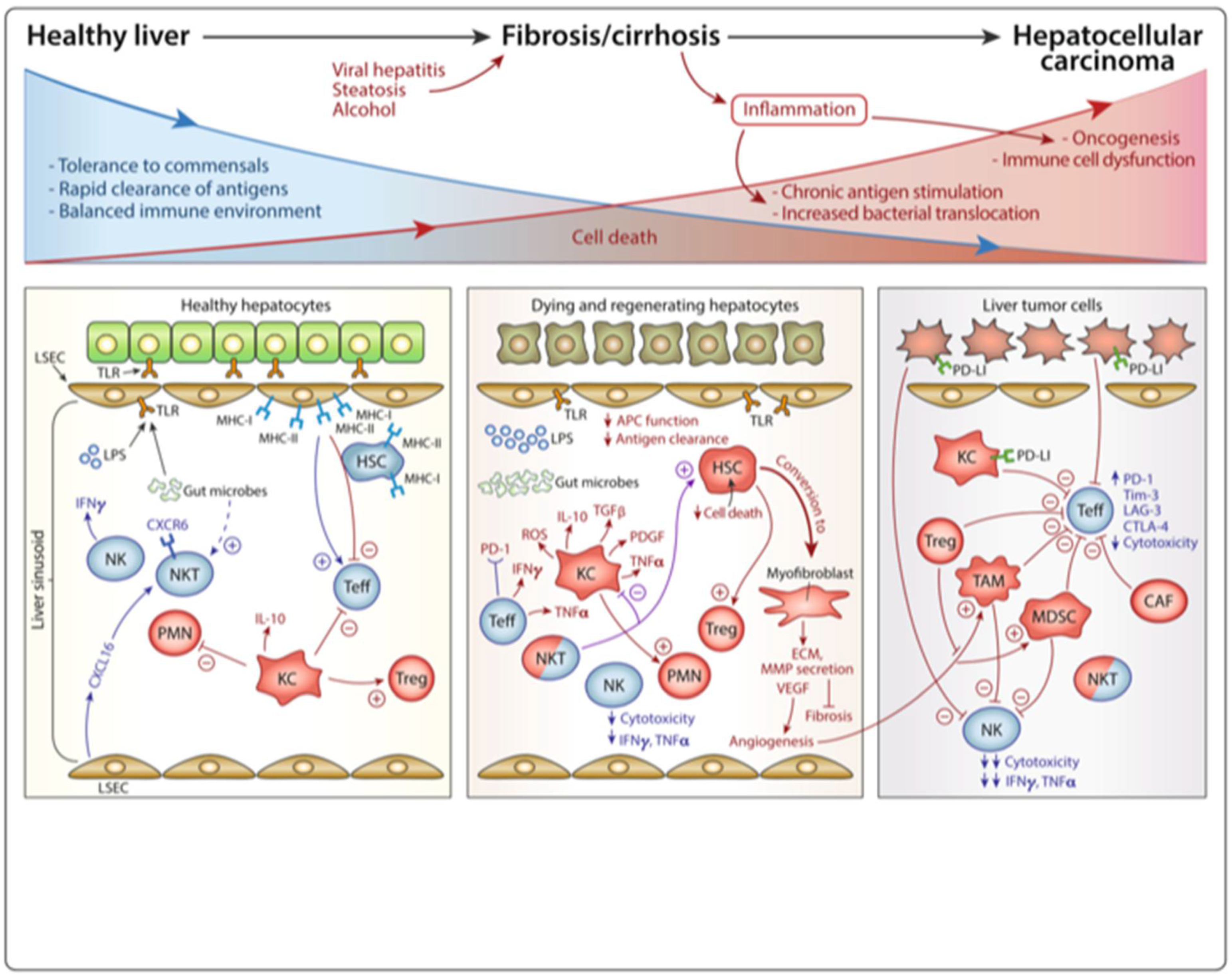
2. Landscape of the Immune Microenvironment in HCC
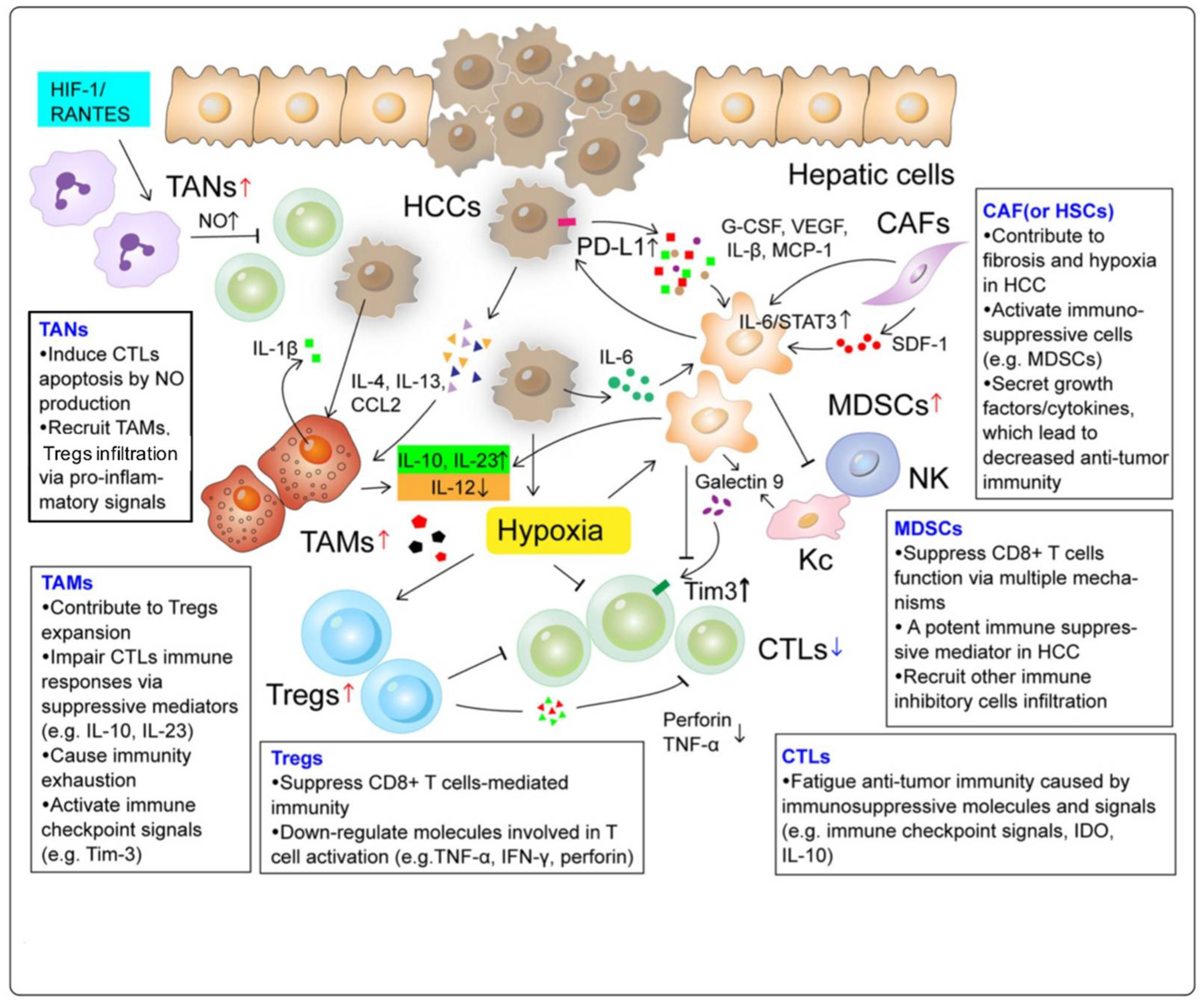
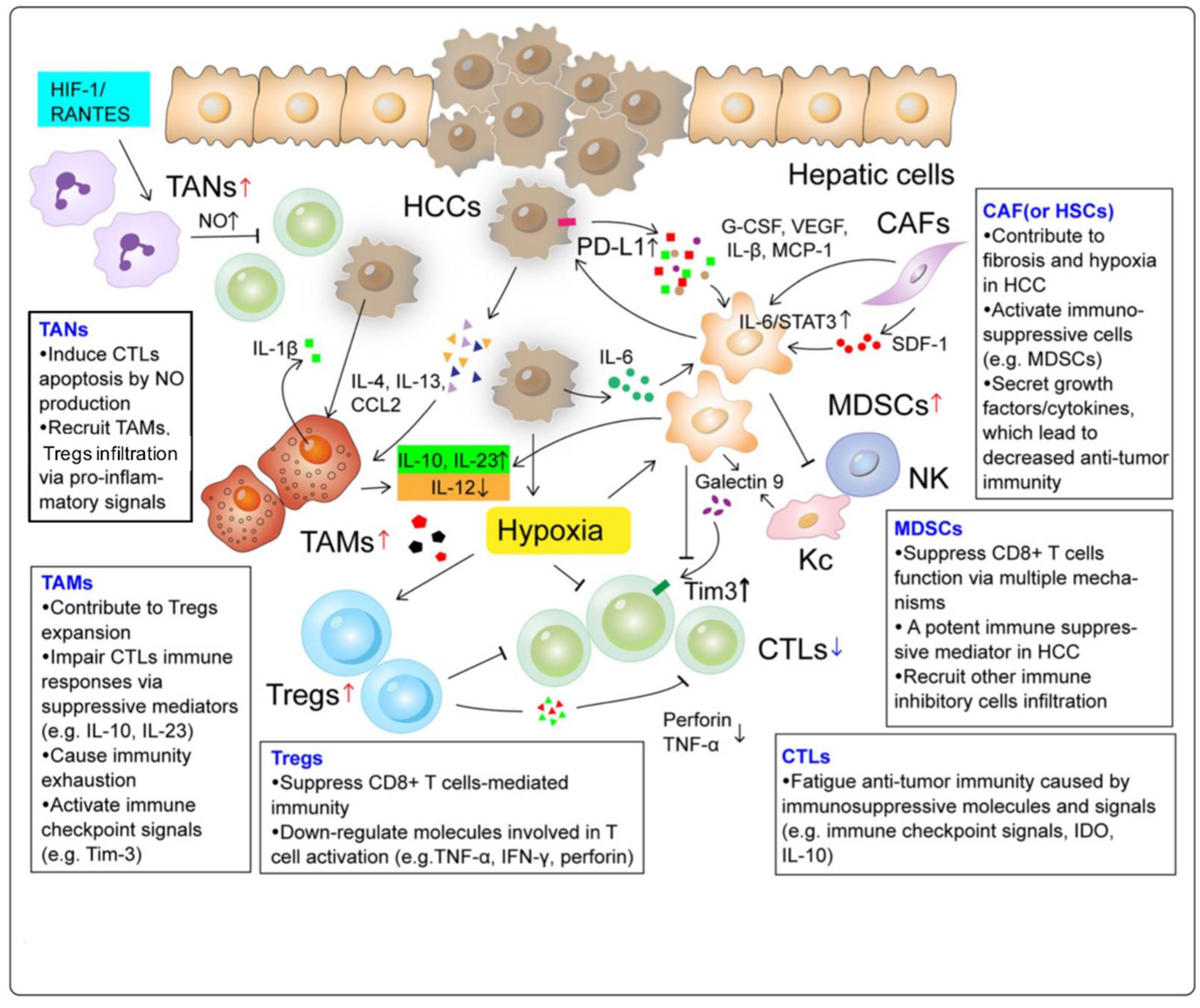
2.1. Tumor-Associated Macrophages (TAMs)
2.2. Tumor-Associated Neutrophils (TANs)
2.3. Myeloid-Derived Suppressor Cells (MDSCs)
2.4. Regulatory T Cells (Tregs)
3. Immunotherapy
3.1. Indirect Therapy
3.1.1. Vaccines
Oncolytic Virus Vaccines
Dendritic Cells Vaccines
Antigen Peptide Vaccines
3.1.2. Immune Checkpoint Inhibitors
Programmed Cell Death-1 (PD-1) Inhibition
Cytotoxic T Lymphocyte–Associated Antigen 4 (CTLA-4) Inhibition
3.2. Direct Therapy
3.2.1. CAR-T Cells
3.2.2. CIK Cells
3.2.3. TCR Engineered T Cell Therapy
3.2.4. TILs
4. Combination Immunotherapy
4.1. Dual Immune Checkpoint Inhibitors—Combined PD-1 and CTLA-4 Inhibition
4.2. NK Cells and Immune Checkpoint Inhibitors
4.3. Targeted Therapies and Immune Checkpoint Inhibitors—Combined VEGF and PD-1 Inhibition
4.4. Locoregional Therapies and Immune Checkpoint Inhibitors
4.5. Bispecific Antibodies
5. Potential Prognostic Biomarkers for Immunotherapy
6. The Future of Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Townsend, C.M.; Beauchamp, R.D.; Mattox, K.L.; Evers, B.M. Sabiston Textbook of Surgery; Elsevier Saunders: Philadelphia, PA, USA, 2017. [Google Scholar]
- Brunicardi, F.C.; Andersen, D.K.; Billiar, T.R.; Dunn, D.L.; Hunter, J.G.; Kao, L.; Matthews, J.B.; Pollock, R.E. Chapter 1: Leadership in Surgery. In Schwartz Principles of Surgery; 2019; Available online: https://accessmedicine.mhmedical.com/content.aspx?bookid=2576§ionid=210404908 (accessed on 13 October 2021).
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Sharma, R.; Allara, E.; Yen, C.; Arizumi, T.; Kubota, K.; Bettinger, D.; Jang, J.W.; Smirne, C.; Kim, Y.W.; et al. The ALBI grade provides objective hepatic reserve estimation across each BCLC stage of hepatocellular carcinoma. J. Hepatol. 2017, 66, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couri, T.; Pillai, A. Goals and targets for personalized therapy for HCC. Hepatol. Int. 2019, 13, 125–137. [Google Scholar] [CrossRef]
- Heinrich, B.; Czauderna, C.; Marquardt, J.U. Immunotherapy of Hepatocellular Carcinoma. Oncol. Res. Treat. 2018, 41, 292–297. [Google Scholar] [CrossRef]
- Abd El Aziz, M.A.; Facciorusso, A.; Nayfeh, T.; Saadi, S.; Elnaggar, M.; Cotsoglou, C.; Sacco, R. Immune Checkpoint Inhibitors for Unresectable Hepatocellular Carcinoma. Vaccines 2020, 8, 616. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Tovoli, F.; De Lorenzo, S.; Trevisani, F. Immunotherapy with Checkpoint Inhibitors for Hepatocellular Carcinoma: Where Are We Now? Vaccines 2020, 8, 578. [Google Scholar] [CrossRef]
- Tahmasebi Birgani, M.; Carloni, V. Tumor Microenvironment, a Paradigm in Hepatocellular Carcinoma Progression and Therapy. Int. J. Mol. Sci. 2017, 18, 405. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, Q.; Liang, N.; Xue, H.; Yang, T.; Chen, X.; Qiu, Z.; Zeng, C.; Sun, T.; Yuan, W.; et al. Oncogenic driver genes and tumor microenvironment determine the type of liver cancer. Cell Death Dis. 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Lamm, R.; Altshuler, P.; Dang, H.; Shah, A.P. Hepatocellular Carcinoma-The Influence of Immunoanatomy and the Role of Immunotherapy. Int. J. Mol. Sci. 2020, 21, 6757. [Google Scholar] [CrossRef]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell. Biochem. 2018, 119, 9419–9432. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.R.; Li, J.H.; Zhang, R.; Chen, R.X.; Wang, Y.H. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol. 2018, 16, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.S.; Yi, H.S. Hepatic Immune Microenvironment in Alcoholic and Nonalcoholic Liver Disease. BioMed Res. Int. 2017, 2017, 6862439. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. CR 2015, 34, 141. [Google Scholar] [CrossRef] [Green Version]
- Dardalhon, V.; Anderson, A.C.; Karman, J.; Apetoh, L.; Chandwaskar, R.; Lee, D.H.; Cornejo, M.; Nishi, N.; Yamauchi, A.; Quintana, F.J.; et al. Tim-3/galectin-9 pathway: Regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J. Immunol. (Baltimore Md. 1950) 2010, 185, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tang, J.; Chen, Y.; Deng, L.; Ji, J.; Xie, Y.; Wang, K.; Jia, W.; Chu, W.M.; Sun, B. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat. Commun. 2017, 8, 15129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.; Eshrat, F.; Al-Jumayli, M.; Saeed, A.; Saeed, A. Immuno-Oncotherapeutic Approaches in Advanced Hepatocellular Carcinoma. Vaccines 2020, 8, 447. [Google Scholar] [CrossRef]
- Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Jiang, X.; Li, Q.; Liu, H.; Huang, L.; Wu, J.; Celis, E.; et al. Identification of α-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology (Baltim Md.) 2018, 68, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, F.; Abe, M.; Hirooka, M.; Ikeda, Y.; Hiasa, Y.; Lee, Y.; Jung, N.C.; Lee, W.B.; Lee, H.S.; Bae, Y.S.; et al. Phase I/II study of immunotherapy using tumor antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Int. J. Oncol. 2012, 41, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J. Hepatol. 2017, 67, 739–748. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Yoshikawa, T.; Fujinami, N.; Saito, K.; Mizuno, S.; Sawada, Y.; Endo, I.; Nakatsura, T. Immunological efficacy of glypican-3 peptide vaccine in patients with advanced hepatocellular carcinoma. Oncoimmunology 2017, 6, e1346764. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. CR 2018, 37, 110. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, W.; Huang, Y.; Cui, R.; Li, X.; Li, B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Agdashian, D.; ElGindi, M.; Xie, C.; Sandhu, M.; Pratt, D.; Kleiner, D.E.; Figg, W.D.; Rytlewski, J.A.; Sanders, C.; Yusko, E.C.; et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. CII 2019, 68, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Principe, N.; Kidman, J.; Goh, S.; Tilsed, C.M.; Fisher, S.A.; Fear, V.S.; Forbes, C.A.; Zemek, R.M.; Chopra, A.; Watson, M.; et al. Tumor Infiltrating Effector Memory Antigen-Specific CD8(+) T Cells Predict Response to Immune Checkpoint Therapy. Front. Immunol. 2020, 11, 584423. [Google Scholar] [CrossRef]
- Xie, Y.; Xiang, Y.; Sheng, J.; Zhang, D.; Yao, X.; Yang, Y.; Zhang, X. Immunotherapy for Hepatocellular Carcinoma: Current Advances and Future Expectations. J. Immunol. Res. 2018, 2018, 8740976. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science (N. Y.) 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Li, D.; Li, N.; Zhang, Y.F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology 2020, 158, 2250–2265.e2220. [Google Scholar] [CrossRef]
- Sun, B.; Yang, D.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef]
- Li, K.; Qian, S.; Huang, M.; Chen, M.; Peng, L.; Liu, J.; Xu, W.; Xu, J. Development of GPC3 and EGFR-dual-targeting chimeric antigen receptor-T cells for adoptive T cell therapy. Am. J. Transl. Res. 2021, 13, 156–167. [Google Scholar]
- Chen, C.; Li, K.; Jiang, H.; Song, F.; Gao, H.; Pan, X.; Shi, B.; Bi, Y.; Wang, H.; Wang, H.; et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol. Immunother. CII 2017, 66, 475–489. [Google Scholar] [CrossRef]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Zou, F.; Tan, J.; Liu, T.; Liu, B.; Tang, Y.; Zhang, H.; Li, J. The CD39(+) HBV surface protein-targeted CAR-T and personalized tumor-reactive CD8(+) T cells exhibit potent anti-HCC activity. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Wei, D.; Liu, Z.K.; Yong, Y.L.; Wei, W.; Zhang, Z.Y.; Lv, J.J.; Zhang, Z.; Chen, Z.N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.C.; Xiong, W.; Badeti, S.; Yang, Y.; Ma, M.; Liu, T.; Ramos, C.A.; Dotti, G.; Fritzky, L.; Jiang, J.G.; et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020, 11, 4810. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Meng, Y.; Yu, Z.; Wu, Y.; Du, T.; Chen, S.; Meng, F.; Su, N.; Ma, Y.; Li, X.; Sun, S.; et al. Cell-based immunotherapy with cytokine-induced killer (CIK) cells: From preparation and testing to clinical application. Hum. Vaccines Immunother. 2017, 13, 1–9. [Google Scholar] [CrossRef]
- Wang, F.S.; Liu, M.X.; Zhang, B.; Shi, M.; Lei, Z.Y.; Sun, W.B.; Du, Q.Y.; Chen, J.M. Antitumor activities of human autologous cytokine-induced killer (CIK) cells against hepatocellular carcinoma cells in vitro and in vivo. World J. Gastroenterol. 2002, 8, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Yuan, F.J.; Jia, G.F.; Zhang, J.F.; Hu, L.Y.; Huang, L.; Wang, J.; Dai, Z.Q. CIK cells from patients with HCC possess strong cytotoxicity to multidrug-resistant cell line Bel-7402/R. World J. Gastroenterol. 2005, 11, 3339–3345. [Google Scholar] [CrossRef]
- Rong, X.X.; Wei, F.; Lin, X.L.; Qin, Y.J.; Chen, L.; Wang, H.Y.; Shen, H.F.; Jia, L.T.; Xie, R.Y.; Lin, T.Y.; et al. Recognition and killing of cancer stem-like cell population in hepatocellular carcinoma cells by cytokine-induced killer cells via NKG2d-ligands recognition. Oncoimmunology 2016, 5, e1086060. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zhang, W.; Wang, L.; Xiao, C.; Wang, L.; Gong, Y.; Huang, D.; Guo, B.; Li, Q.; Xiang, Y.; et al. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC Cancer 2018, 18, 984. [Google Scholar] [CrossRef] [Green Version]
- González-Carmona, M.A.; Märten, A.; Hoffmann, P.; Schneider, C.; Sievers, E.; Schmidt-Wolf, I.G.; Sauerbruch, T.; Caselmann, W.H. Patient-derived dendritic cells transduced with an a-fetoprotein-encoding adenovirus and co-cultured with autologous cytokine-induced lymphocytes induce a specific and strong immune response against hepatocellular carcinoma cells. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 369–379. [Google Scholar] [CrossRef]
- Xu, K.; Meng, Z.; Mu, X.; Sun, B.; Chai, Y. One Single Site Clinical Study: To Evaluate the Safety and Efficacy of Immunotherapy With Autologous Dendritic Cells, Cytokine-Induced Killer Cells in Primary Hepatocellular Carcinoma Patients. Front. Oncol. 2020, 10, 581270. [Google Scholar] [CrossRef]
- Cao, J.; Kong, F.H.; Liu, X.; Wang, X.B. Immunotherapy with dendritic cells and cytokine-induced killer cells for hepatocellular carcinoma: A meta-analysis. World J. Gastroenterol. 2019, 25, 3649–3663. [Google Scholar] [CrossRef]
- Chang, B.; Shen, L.; Wang, K.; Jin, J.; Huang, T.; Chen, Q.; Li, W.; Wu, P. High number of PD-1 positive intratumoural lymphocytes predicts survival benefit of cytokine-induced killer cells for hepatocellular carcinoma patients. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.Z.; Liu, Q.; Zhou, Y.Q.; Zhao, J.J.; Wang, Q.J.; Li, Y.Q.; Tang, Y.; Gu, J.M.; He, J.; Chen, S.P.; et al. CIK cell cytotoxicity is a predictive biomarker for CIK cell immunotherapy in postoperative patients with hepatocellular carcinoma. Cancer Immunol. Immunother. CII 2020, 69, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.C.; Huang, Z.L.; Li, W.; Zhao, M.; Zhou, Q.M.; Xia, J.C.; Wu, P.H. Serum alpha-fetoprotein measurement in predicting clinical outcome related to autologous cytokine-induced killer cells in patients with hepatocellular carcinoma undergone minimally invasive therapy. Chin. J. Cancer 2010, 29, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tian, Y.; Li, Y.; Zhang, W.; Cai, W.; Liu, Y.; Ren, Y.; Liang, Z.; Zhou, P.; Zhang, Y.; et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e001748. [Google Scholar] [CrossRef] [PubMed]
- Docta, R.Y.; Ferronha, T.; Sanderson, J.P.; Weissensteiner, T.; Pope, G.R.; Bennett, A.D.; Pumphrey, N.J.; Ferjentsik, Z.; Quinn, L.L.; Wiedermann, G.E.; et al. Tuning T-Cell Receptor Affinity to Optimize Clinical Risk-Benefit When Targeting Alpha-Fetoprotein-Positive Liver Cancer. Hepatology (Baltim. Md.) 2019, 69, 2061–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, L.; Frigault, M.; Meyer, T.; Feun, L.G.; Bruix, J.; El-Khoueiry, A.; Hausner, P.; Sangro, B.; Pierce, T.T.; Norry, E. Initial safety of AFP SPEAR T-cells in patients with advanced hepatocellular carcinoma. AACR 2019, 79. [Google Scholar] [CrossRef]
- Luo, X.; Cui, H.; Cai, L.; Zhu, W.; Yang, W.C.; Patrick, M.; Zhu, S.; Huang, J.; Yao, X.; Yao, Y.; et al. Selection of a Clinical Lead TCR Targeting Alpha-Fetoprotein-Positive Liver Cancer Based on a Balance of Risk and Benefit. Front. Immunol. 2020, 11, 623. [Google Scholar] [CrossRef]
- Chew, V.; Tow, C.; Teo, M.; Wong, H.L.; Chan, J.; Gehring, A.; Loh, M.; Bolze, A.; Quek, R.; Lee, V.K.; et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J. Hepatol. 2010, 52, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e1110. [Google Scholar] [CrossRef]
- Jiang, S.S.; Tang, Y.; Zhang, Y.J.; Weng, D.S.; Zhou, Z.G.; Pan, K.; Pan, Q.Z.; Wang, Q.J.; Liu, Q.; He, J.; et al. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget 2015, 6, 41339–41349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, G.; Eshhar, Z. Therapeutic Potential of T Cell Chimeric Antigen Receptors (CARs) in Cancer Treatment: Counteracting Off-Tumor Toxicities for Safe CAR T Cell Therapy. Annu. Rev. Pharm. Toxicol. 2016, 56, 59–83. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.J.; Tang, Y.M. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014, 343, 172–178. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L.; et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef]
- Ho, W.J.; Sharma, G.; Zhu, Q.; Stein-O’Brien, G.; Durham, J.; Anders, R.; Popovic, A.; Mo, G.; Kamel, I.; Weiss, M.; et al. Integrated immunological analysis of a successful conversion of locally advanced hepatocellular carcinoma to resectability with neoadjuvant therapy. J. Immunother. Cancer 2020, 8, e000932. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Yau, T.; Hsu, C.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Subgroup analyses from CheckMate 040. J. Clin. Oncol. 2020, 38, 512. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Am. Soc. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef] [Green Version]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Wang, C.; Jin, G.; Ruan, H.; Gu, D.; Wei, L.; Wang, H.; Wang, N.; Arunachalam, E.; Zhang, Y.; et al. Regulator of Calcineurin 1 Gene Isoform 4, Down-regulated in Hepatocellular Carcinoma, Prevents Proliferation, Migration, and Invasive Activity of Cancer Cells and Metastasis of Orthotopic Tumors by Inhibiting Nuclear Translocation of NFAT1. Gastroenterology 2017, 153, 799–811.e733. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.; Finn, R.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.; Merle, P.J.A. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. Am. Soc. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y. First-line avelumab+ axitinib in patients with advanced hepatocellular carcinoma: Results from a phase 1b trial (VEGF Liver 100). Am. Soc. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Greten, T.F.; Mauda-Havakuk, M.; Heinrich, B.; Korangy, F.; Wood, B.J. Combined locoregional-immunotherapy for liver cancer. J. Hepatol. 2019, 70, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Pinato, D.J.; Cole, T.; Bengsch, B.; Tait, P.; Sayed, A.A.; Abomeli, F.; Gramenitskaya, D.; Allara, E.; Thomas, R.; Ward, C.J.A. A phase Ib study of pembrolizumab following trans-arterial chemoembolization (TACE) in hepatocellular carcinoma (HCC): PETAL. Ann. Oncol. 2019, 30, v288. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Cheung, N.V. Immunotherapy of hepatocellular carcinoma using chimeric antigen receptors and bispecific antibodies. Cancer Lett. 2017, 399, 44–52. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sano, Y.; Komatsu, S.I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 2017, 9, eaal4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (Lond. Engl.) 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet. Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Chang, B.; Huang, T.; Wei, H.; Shen, L.; Zhu, D.; He, W.; Chen, Q.; Zhang, H.; Li, Y.; Huang, R.; et al. The correlation and prognostic value of serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death-ligand 1 (sPD-L1) in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. CII 2019, 68, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.J.; Danilova, L.; Lim, S.J.; Verma, R.; Xavier, S.; Leatherman, J.M.; Sztein, M.B.; Fertig, E.J.; Wang, H.; Jaffee, E.; et al. Viral status, immune microenvironment and immunological response to checkpoint inhibitors in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000394. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaoka, T.; Ando, Y.; Yamauchi, M.; Suehiro, Y.; Yamaoka, K.; Kosaka, Y.; Fuji, Y.; Uchikawa, S.; Morio, K.; Fujino, H.; et al. Incidence of microsatellite instability-high hepatocellular carcinoma among Japanese patients and response to pembrolizumab. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2020, 50, 885–888. [Google Scholar] [CrossRef]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (N. Y.) 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.F.; Gao, C.; Huang, X.Y.; Lu, J.C.; Guo, X.J.; Shi, G.M.; Cai, J.B.; Ke, A.W. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science (N. Y.) 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feun, L.G.; Li, Y.Y.; Wu, C.; Wangpaichitr, M.; Jones, P.D.; Richman, S.P.; Madrazo, B.; Kwon, D.; Garcia-Buitrago, M.; Martin, P.; et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 2019, 125, 3603–3614. [Google Scholar] [CrossRef]
- Qayyum, A.; Hwang, K.P.; Stafford, J.; Verma, A.; Maru, D.M.; Sandesh, S.; Sun, J.; Pestana, R.C.; Avritscher, R.; Hassan, M.M.; et al. Immunotherapy response evaluation with magnetic resonance elastography (MRE) in advanced HCC. J. Immunother. Cancer 2019, 7, 329. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| NCT Number | Treatment [70] | Setting | Design |
|---|---|---|---|
| NCT01658878 | Nivolumab | Second line in sorafenib pretreated patients | Phase I-II dose escalation and expansion |
| NCT01658878 | Nivolumab + Ipilimumab | Second line in sorafenib pretreated patients | Phase I-II |
| NCT02576509 | Nivolumab vs Sorafenib | First line treatment | Phase III |
| NCT02702414 | Pembrolizumab | Second line in sorafenib pretreated patients | Phase II |
| NCT02702401 | Pembrolizumab vs placebo | Second line treatment | Phase III |
| NCT03006926 | Pembrolizumab + Lenvatinib | First line treatment | Phase Ib |
| NCT03289533 | Avelumab + Axitinib | First line treatment | Phase Ib |
| NCT03510871 | Nivolumab or Nivolumab + Ipilimumab | Perioperative treatment, resectable HCC | Phase II |
| NCT02715531 | Atezolizumab + Bevacizumab vs Atezolizumab alone | First line treatment | Phase Ib |
| NCT03434379 | Atezolizumab + Bevacizumab vs Sorafenib | First line treatment | Phase III |
| NCT01693562 | Durvalumab | Mainly second line in sorafenib pretreated patients | Phase I-II |
| NCT02519348 | Durvalumab + Tremelimumab | Mainly second line in sorafenib pretreated patients | Phase I |
| NCT02572687 | Durvalumab + Ramucirumab | Second line treatment | Phase I |
| NCT01008358 | Tremelimumab | Pretreated advanced HCC from HCV | Phase II |
| NCT01853618 | Tremelimumab + ablation | Locally advanced HCC | Phase I-II |
| NCT02989922 | Camrelizumab | Second line treatment | Phase II |
| NCT03463876 | Camrelizumab + Apatinib | Advanced HCC | Phase II |
| NCT03092895 | Camrelizumab + FOLFOX4 or GEMOX regimen | First line treatment | Phase II |
| NCT02407990 | Tislelizumab | Sorafenib-refractory HCC | Phase I dose escalation and expansion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawal, G.; Xiao, Y.; Rahnemai-Azar, A.A.; Tsilimigras, D.I.; Kuang, M.; Bakopoulos, A.; Pawlik, T.M. The Immunology of Hepatocellular Carcinoma. Vaccines 2021, 9, 1184. https://doi.org/10.3390/vaccines9101184
Lawal G, Xiao Y, Rahnemai-Azar AA, Tsilimigras DI, Kuang M, Bakopoulos A, Pawlik TM. The Immunology of Hepatocellular Carcinoma. Vaccines. 2021; 9(10):1184. https://doi.org/10.3390/vaccines9101184
Chicago/Turabian StyleLawal, Gbemisola, Yao Xiao, Amir A. Rahnemai-Azar, Diamantis I. Tsilimigras, Ming Kuang, Anargyros Bakopoulos, and Timothy M. Pawlik. 2021. "The Immunology of Hepatocellular Carcinoma" Vaccines 9, no. 10: 1184. https://doi.org/10.3390/vaccines9101184
APA StyleLawal, G., Xiao, Y., Rahnemai-Azar, A. A., Tsilimigras, D. I., Kuang, M., Bakopoulos, A., & Pawlik, T. M. (2021). The Immunology of Hepatocellular Carcinoma. Vaccines, 9(10), 1184. https://doi.org/10.3390/vaccines9101184