
HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmid Construction
2.3. Chromatin Immunoprecipitation (ChIP)
2.4. Coimmunoprecipitation (CoIP)
2.5. Western Blot
2.6. Pulldown of Bacterially-Expressed Recombinant Proteins
2.7. Photocrosslinking in Live Mammalian Cells
2.8. Mass Spectrometry
3. Results
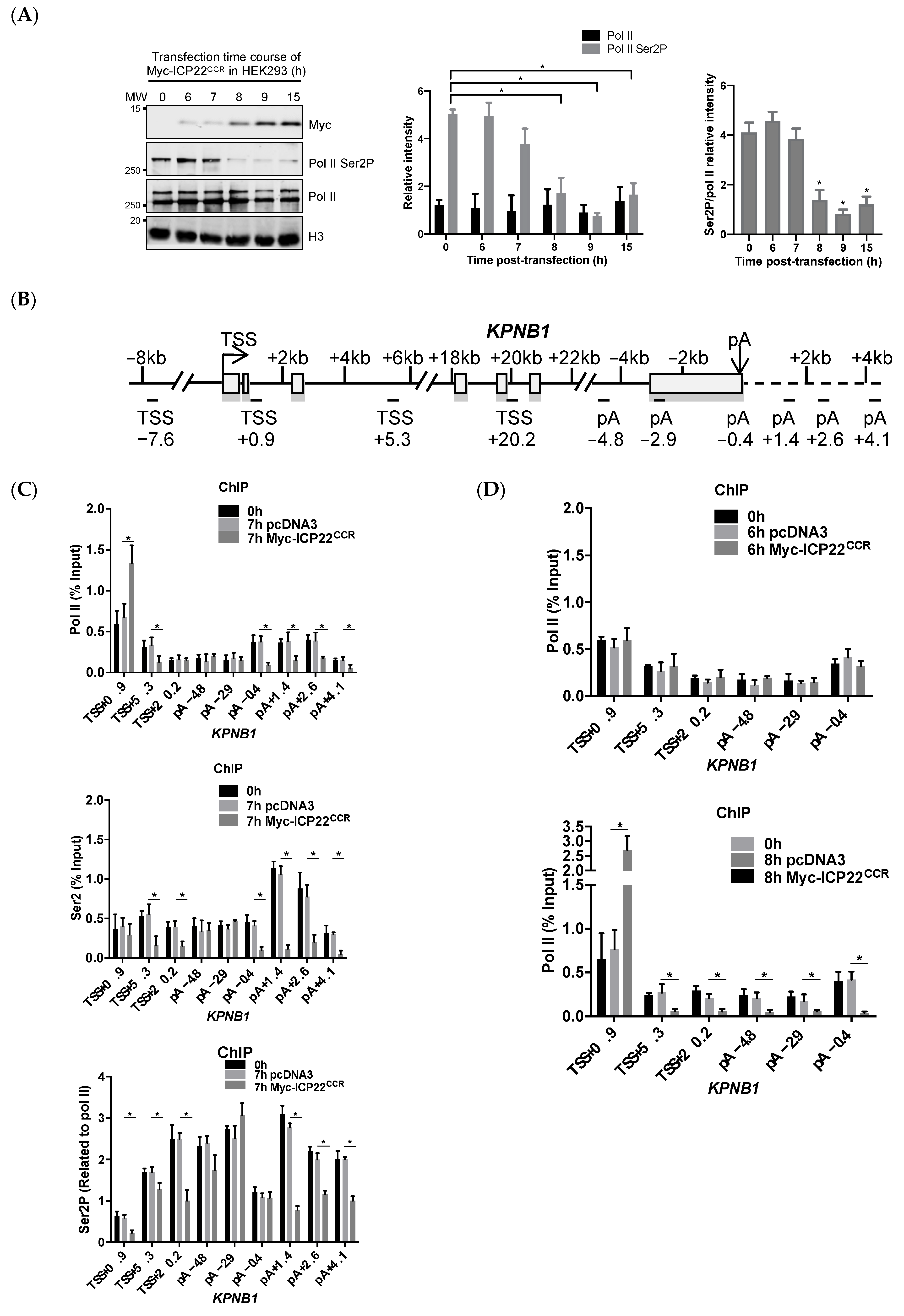
3.1. ICP22 Inhibits Pol II Elongation at the Beginning and End of Host Cell Genes
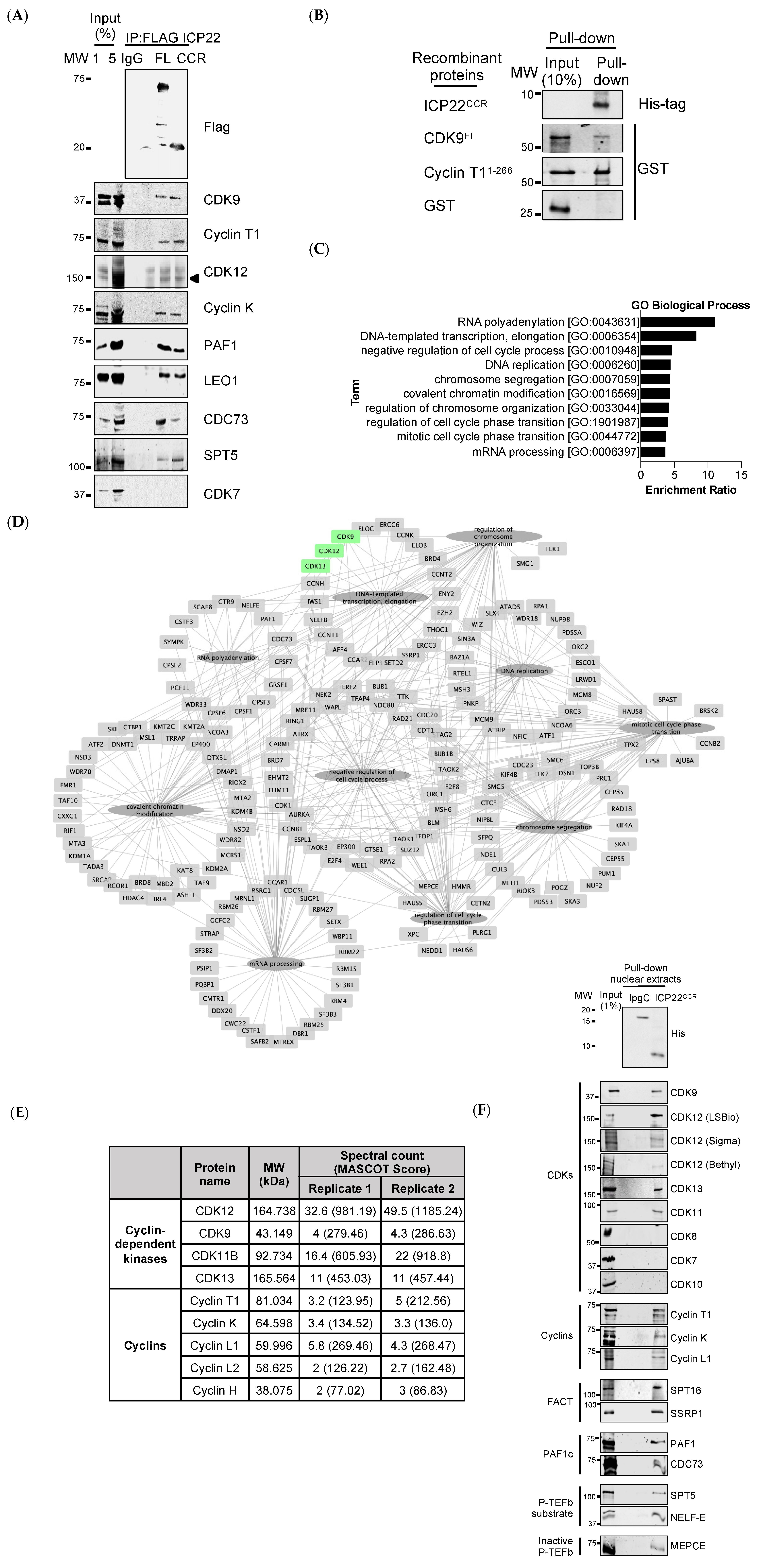
3.2. ICP22 Interacts with Cellular Transcription Elongation Factors
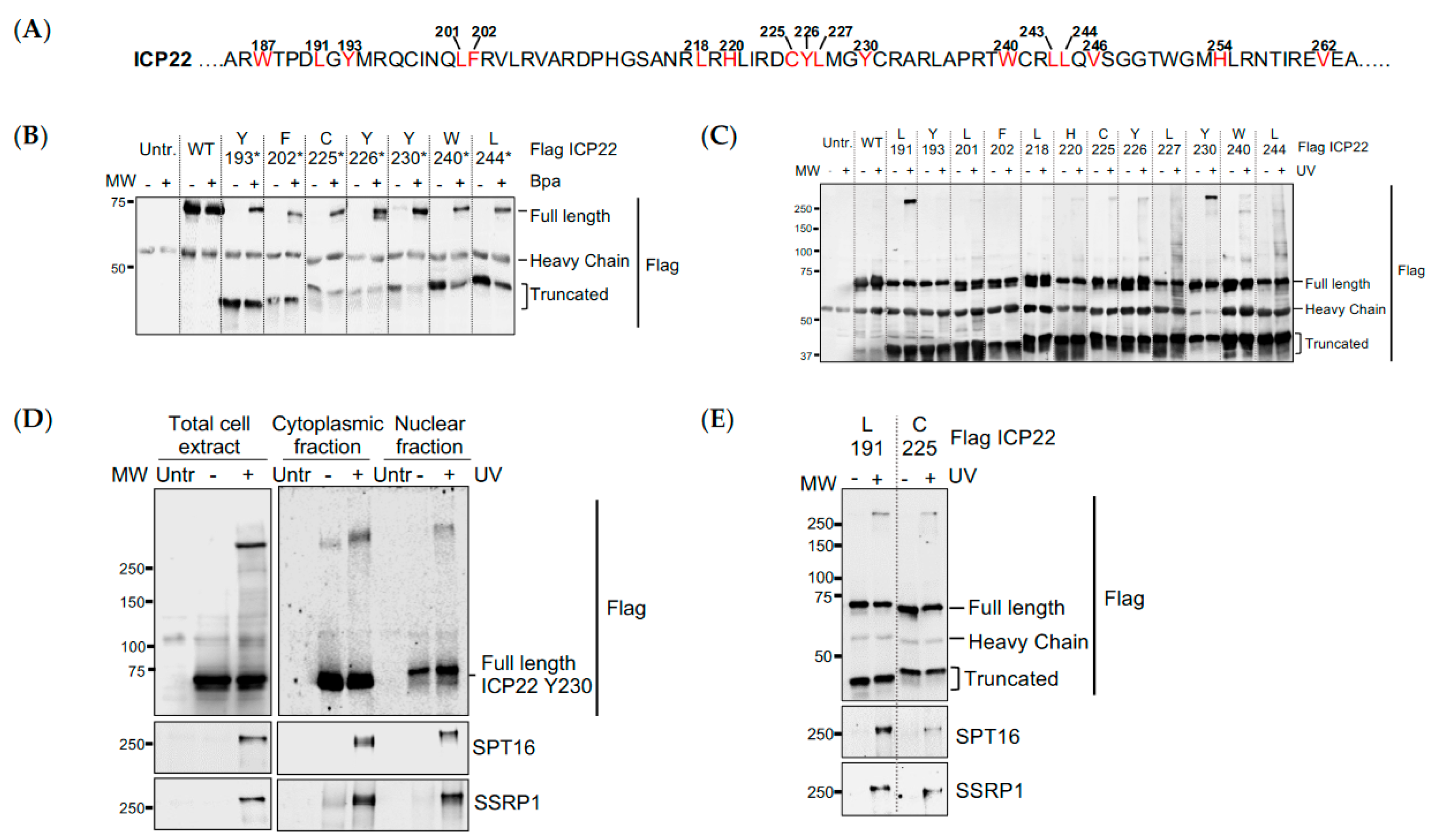
3.3. UV Crosslinking Reveals that ICP22 Interacts Directly with the FACT Complex
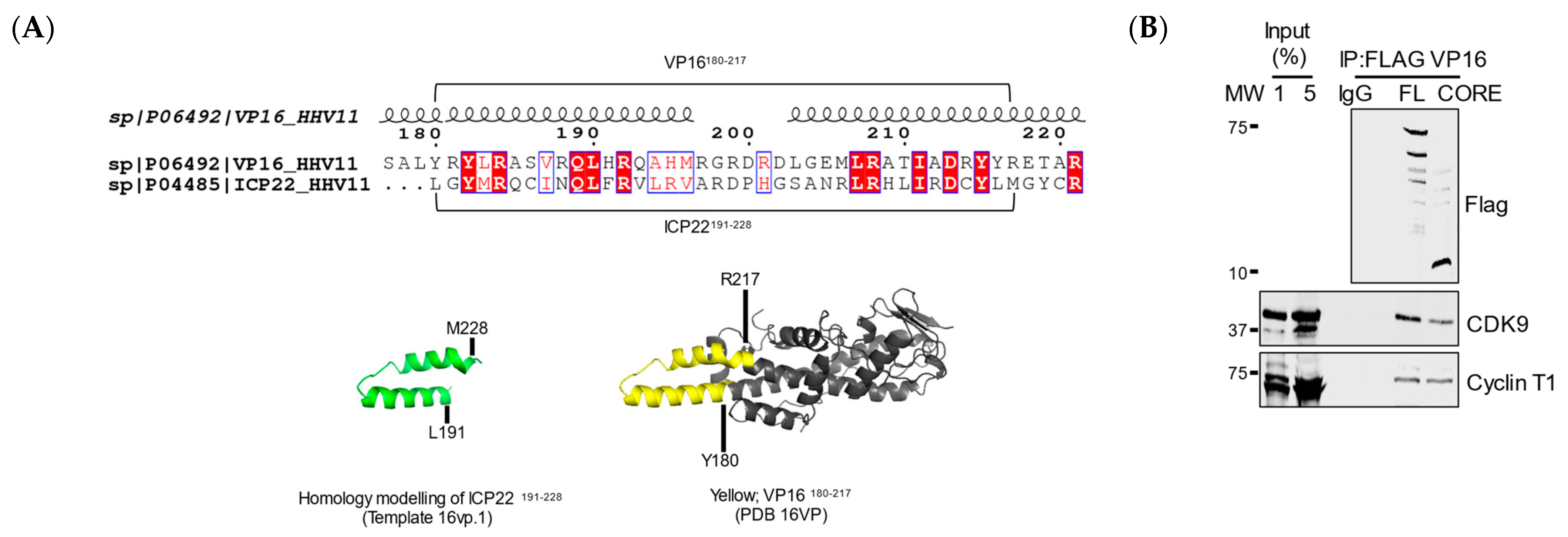
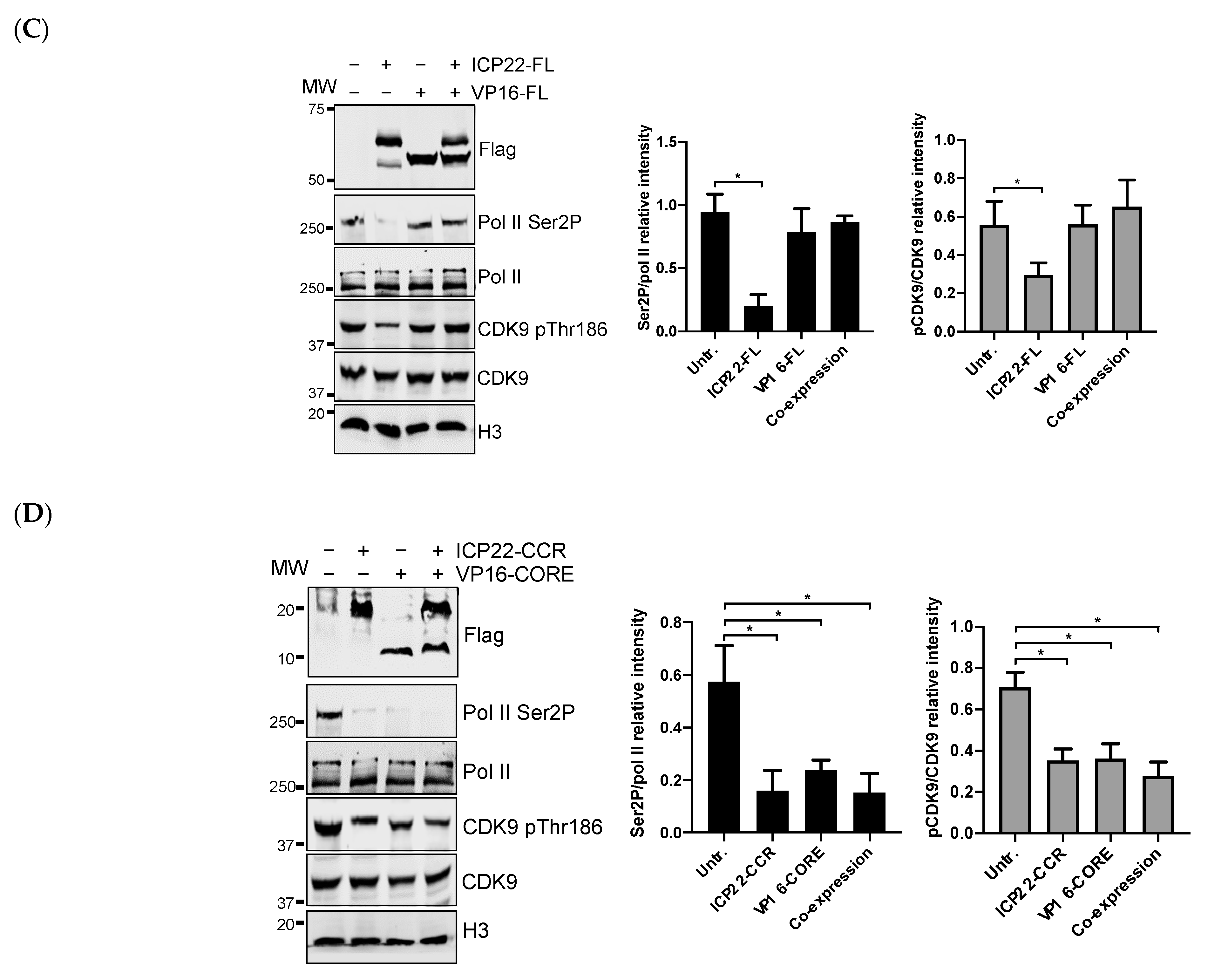
3.4. VP16 Relieves ICP22-Mediated Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- La Boissière, S.; Hughes, T.; O’Hare, P. HCF-dependent nuclear import of VP16. EMBO J. 1999, 18, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Babb, R.; Huang, C.C.; Aufiero, D.J.; Herr, W. DNA Recognition by the Herpes Simplex Virus Transactivator VP16: A Novel DNA-Binding Structure. Mol. Cell. Biol. 2001, 21, 4700–4712. [Google Scholar] [CrossRef][Green Version]
- Poffenberger, K.L.; Idowu, A.D.; Fraser-Smith, E.B.; Raichlen, P.E.; Herman, R.C. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch. Virol. 1994, 139, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Matundan, H.H.; Jaggi, U.; Wang, S.; Ghiasi, H. Loss ofICP22in HSV-1 Elicits Immune Infiltration and Maintains Stromal Keratitis Despite Reduced Primary and Latent Virus Infectivity. Investig. Opthalmol. Vis. Sci. 2019, 60, 3398–3406. [Google Scholar] [CrossRef]
- Cun, W.; Guo, L.; Zhang, Y.; Liu, L.; Wang, L.; Li, J.; Dong, C.; Wang, J.; Li, Q. Transcriptional regulation of the Herpes Simplex Virus 1α-gene by the viral immediate-early protein ICP22 in association with VP16. Sci. China Ser. C Life Sci. 2009, 52, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wu, W.-J.; Liu, L.-D.; Wang, L.-C.; Zhang, Y.; Wu, L.-Q.; Guan, Y.; Li, Q.-H. Herpes Simplex Virus 1 ICP22 Inhibits the Transcription of Viral Gene Promoters by Binding to and Blocking the Recruitment of P-TEFb. PLoS ONE 2012, 7, e45749. [Google Scholar] [CrossRef] [PubMed]
- Prod’Hon, C.; Machuca, I.; Berthommé, H.; Epstein, A.; Jacquemont, B. Characterization of Regulatory Functions of the HSV-1 Immediate-Early Protein ICP22. Virology 1996, 226, 393–402. [Google Scholar] [CrossRef][Green Version]
- Bowman, J.J.; Orlando, J.S.; Davido, D.J.; Kushnir, A.S.; Schaffer, P.A. Transient Expression of Herpes Simplex Virus Type 1 ICP22 Represses Viral Promoter Activity and Complements the Replication of an ICP22 Null Virus. J. Virol. 2009, 83, 8733–8743. [Google Scholar] [CrossRef] [PubMed]
- Schwyzer, M.; Wirth, U.V.; Vogt, B.; Fraefel, C. BICP22 of bovine herpesvirus 1 is encoded by a spliced 1.7 kb RNA which exhibits immediate early and late transcription kinetics. J. Gen. Virol. 1994, 75, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Ogle, W.O.; Roizman, B. Functional Anatomy of Herpes Simplex Virus 1 Overlapping Genes Encoding Infected-Cell Protein 22 and U S 1.5 Protein. J. Virol. 1999, 73, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Zaborowska, J.; Baumli, S.; Laitem, C.; O’Reilly, D.; Thomas, P.H.; O’Hare, P.; Murphy, S. Herpes Simplex Virus 1 (HSV-1) ICP22 Protein Directly Interacts with Cyclin-Dependent Kinase (CDK)9 to Inhibit RNA Polymerase II Transcription Elongation. PLoS ONE 2014, 9, e107654. [Google Scholar] [CrossRef]
- Maruzuru, Y.; Fujii, H.; Oyama, M.; Kozuka-Hata, H.; Kato, A.; Kawaguchi, Y. Roles of p53 in Herpes Simplex Virus 1 Replication. J. Virol. 2013, 87, 9323–9332. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. The Histone Acetyltransferase CLOCK Is an Essential Component of the Herpes Simplex Virus 1 Transcriptome That Includes TFIID, ICP4, ICP27, and ICP22. J. Virol. 2011, 85, 9472–9477. [Google Scholar] [CrossRef] [PubMed]
- Fox, H.L.; Dembowski, J.A.; DeLuca, N.A. A Herpesviral Immediate Early Protein Promotes Transcription Elongation of Viral Transcripts. mBio 2017, 8, e00745-17. [Google Scholar] [CrossRef] [PubMed]
- Durand, L.O.; Advani, S.J.; Poon, A.P.W.; Roizman, B. The Carboxyl-Terminal Domain of RNA Polymerase II Is Phosphorylated by a Complex Containing cdk9 and Infected-Cell Protein 22 of Herpes Simplex Virus. J. Virol. 2005, 79, 6757–6762. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, Q.; Wang, M.; Chen, S.; Jia, R.; Yang, Q.; Zhu, D.; Liu, M.; Zhao, X.; Zhang, S.; et al. Multifaceted Roles of ICP22/ORF63 Proteins in the Life Cycle of Human Herpesviruses. Front. Microbiol. 2021, 12, 1370. [Google Scholar] [CrossRef]
- Adlakha, M.; Livingston, C.M.; Bezsonova, I.; Weller, S.K. The Herpes Simplex Virus 1 Immediate Early Protein ICP22 Is a Functional Mimic of a Cellular J Protein. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Corden, J.L. RNA Polymerase II C-Terminal Domain: Tethering Transcription to Transcript and Template. Chem. Rev. 2013, 113, 8423–8455. [Google Scholar] [CrossRef]
- Zaborowska, J.; Egloff, S.; Murphy, S. The Pol II CTD: New Twists in the Tail. Nat. Struct. Mol. Biol. 2016, 23, 771–777. [Google Scholar] [CrossRef]
- Chen, F.; Woodfin, A.R.; Gardini, A.; Rickels, R.A.; Marshall, S.A.; Smith, E.R.; Shiekhattar, R.; Shilatifard, A. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 2015, 162, 1003–1015. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Boehning, M.; Wigge, C.; Linden, A.; Urlaub, H.; Cramer, P. Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 2018, 560, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT Facilitates Transcription-Dependent Nucleosome Alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef]
- Wada, T.; Orphanides, G.; Hasegawa, J.; Kim, D.-K.; Shima, D.; Yamaguchi, Y.; Fukuda, A.; Hisatake, K.; Oh, S.; Reinberg, D.; et al. FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Mol. Cell 2000, 5, 1067–1072. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef]
- Bösken, C.A.; Farnung, L.; Hintermair, C.; Schachter, M.M.; Vogel-Bachmayr, K.; Blazek, D.; Anand, K.; Fisher, R.P.; Eick, D.; Geyer, M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014, 5, 3505. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Thérien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D. Analysis of the Large Inactive P-TEFb Complex Indicates That It Contains One 7SK Molecule, a Dimer of HEXIM1 or HEXIM2, and Two P-TEFb Molecules Containing Cdk9 Phosphorylated at Threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef]
- Laitem, C.; Zaborowska, J.; Isa, N.F.; Kufs, J.; Dienstbier, M.; Murphy, S. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II–transcribed genes. Nat. Struct. Mol. Biol. 2015, 22, 396–403. [Google Scholar] [CrossRef]
- Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef]
- Kobbi, L.; Demey-Thomas, E.; Braye, F.; Proux, F.; Kolesnikova, O.; Vinh, J.; Poterszman, A.; Bensaude, O. An evolutionary conserved Hexim1 peptide binds to the Cdk9 catalytic site to inhibit P-TEFb. Proc. Natl. Acad. Sci. USA 2016, 113, 12721–12726. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Hauschild, J.-P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- A Rice, S.; Long, M.C.; Lam, V.; A Schaffer, P.; A Spencer, C. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 1995, 69, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Tellier, M.; Zaborowska, J.; Caizzi, L.; Mohammad, E.; Velychko, T.; Schwalb, B.; Ferrer-Vicens, I.; Blears, D.; Nojima, T.; Cramer, P.; et al. CDK12 globally stimulates RNA polymerase II transcription elongation and carboxyl-terminal domain phosphorylation. Nucleic Acids Res. 2020, 48, 7712–7727. [Google Scholar] [CrossRef]
- Durand, L.O.; Roizman, B. Role of cdk9 in the Optimization of Expression of the Genes Regulated by ICP22 of Herpes Simplex Virus 1. J. Virol. 2008, 82, 10591–10599. [Google Scholar] [CrossRef]
- Adam, P.R.; Patil, M.K.; Dickenson, N.E.; Choudhari, S.; Barta, M.; Geisbrecht, B.V.; Picking, W.L.; Picking, W.D. Binding Affects the Tertiary and Quaternary Structures of the Shigella Translocator Protein IpaB and Its Chaperone IpgC. Biochemistry 2012, 51, 4062–4071. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, T.; Peterlin, B. VP16 and Ubiquitin: Binding of P-TEFb via Its Activation Domain and Ubiquitin Facilitates Elongation of Transcription of Target Genes. Curr. Biol. 2004, 14, 1112–1116. [Google Scholar] [CrossRef]
- Oakes, J.E.; Hyman, R.W.; Rapp, F. Genome location of polyadenylated transcripts of herpes simplex virus type 1 and type 2 DNA. Virology 1976, 75, 145–154. [Google Scholar] [CrossRef]
- A Rice, S.; Long, M.C.; Lam, V.; A Spencer, C. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 1994, 68, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.A.; Rice, S.A. Herpes Simplex Virus Type 1 Infection Leads to Loss of Serine-2 Phosphorylation on the Carboxyl-Terminal Domain of RNA Polymerase II. J. Virol. 2005, 79, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Orlando, J.S.; Astor, T.L.; Rundle, S.A.; Schaffer, P.A. The Products of the Herpes Simplex Virus Type 1 Immediate-Early U S 1/U S 1.5 Genes Downregulate Levels of S-Phase-Specific Cyclins and Facilitate Virus Replication in S-Phase Vero Cells. J. Virol. 2006, 80, 4005–4016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mostafa, H.H.; Davido, D.J. Herpes Simplex Virus 1 ICP22 but Not U S 1.5 Is Required for Efficient Acute Replication in Mice and VICE Domain Formation. J. Virol. 2013, 87, 13510–13519. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell. Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef]
- Matundan, H.; Ghiasi, H. Herpes Simplex Virus 1 ICP22 Suppresses CD80 Expression by Murine Dendritic Cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ou, M.; Sandri-Goldin, R.M. Inhibition of cdk9 during Herpes Simplex Virus 1 Infection Impedes Viral Transcription. PLoS ONE 2013, 8, e79007. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, A.P.C.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Hsin, J.-P.; Manley, J. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef]
- Kemble, D.J.; McCullough, L.L.; Whitby, F.G.; Formosa, T.; Hill, C.P. FACT Disrupts Nucleosome Structure by Binding H2A-H2B with Conserved Peptide Motifs. Mol. Cell 2015, 60, 294–306. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Li, D.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 14, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Ransom, M.; Dennehey, B.; Tyler, J.K. Chaperoning Histones during DNA Replication and Repair. Cell 2010, 140, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Genet. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.-Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During Lytic Infection Herpes Simplex Virus Type 1 Is Associated with Histones Bearing Modifications That Correlate with Active Transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef] [PubMed]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. Herpes simplex virus 1 activates cdc2 to recruit topoisomerase II for post-DNA synthesis expression of late genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4825–4830. [Google Scholar] [CrossRef]
- E Sears, A.; Halliburton, I.W.; Meignier, B.; Silver, S.; Roizman, B. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: Growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 1985, 55, 338–346. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.; Zhang, N.; Wei, H.; Tan, Y.Z.; Zhang, Z.; Carragher, B.; Potter, C.S.; D’Arcy, S.; Luger, K. FACT caught in the act of manipulating the nucleosome. Nature 2019, 577, 426–431. [Google Scholar] [CrossRef]
- Winkler, D.D.; Luger, K. The Histone Chaperone FACT: Structural Insights and Mechanisms for Nucleosome Reorganization. J. Biol. Chem. 2011, 286, 18369–18374. [Google Scholar] [CrossRef]
- Pham, N.; Parker, R.B.; Kohler, J.J. Photocrosslinking approaches to interactome mapping. Curr. Opin. Chem. Biol. 2012, 17, 90–101. [Google Scholar] [CrossRef]
- Blau, J.; Xiao, H.; McCracken, S.; O’Hare, P.; Greenblatt, J.; Bentley, D. Three functional classes of transcriptional activation domain. Mol. Cell. Biol. 1996, 16, 2044–2055. [Google Scholar] [CrossRef]
- Jiang, H.; Xu, S.; Chen, Y.; Li, H.; Tian, L.; Zhou, H.; Zhao, Z.; Yang, C.; Zhong, Z.; Cai, G.; et al. The structural basis of human Spt16 N-terminal domain interaction with histone (H3-H4)2 tetramer. Biochem. Biophys. Res. Commun. 2018, 508, 864–870. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isa, N.F.; Bensaude, O.; Aziz, N.C.; Murphy, S. HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation. Vaccines 2021, 9, 1054. https://doi.org/10.3390/vaccines9101054
Isa NF, Bensaude O, Aziz NC, Murphy S. HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation. Vaccines. 2021; 9(10):1054. https://doi.org/10.3390/vaccines9101054
Chicago/Turabian StyleIsa, Nur Firdaus, Olivier Bensaude, Nadiah C. Aziz, and Shona Murphy. 2021. "HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation" Vaccines 9, no. 10: 1054. https://doi.org/10.3390/vaccines9101054
APA StyleIsa, N. F., Bensaude, O., Aziz, N. C., & Murphy, S. (2021). HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation. Vaccines, 9(10), 1054. https://doi.org/10.3390/vaccines9101054