Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of FP8-Displaying Recombinant VLPs
2.2. Production and Purification of FP8-Displaying Recombinant VLPs
2.3. Conjugation of FP8 to Qβ Bacteriophage VLPs
2.4. Characterization of FP8-Displaying VLPs
2.5. Mice Immunizations
2.6. Characterization of Antibody Responses
2.7. Immunoglobulin Purification
2.8. HIV-1 Neutralization Assay
2.9. Statistical Analysis
3. Results and Discussion
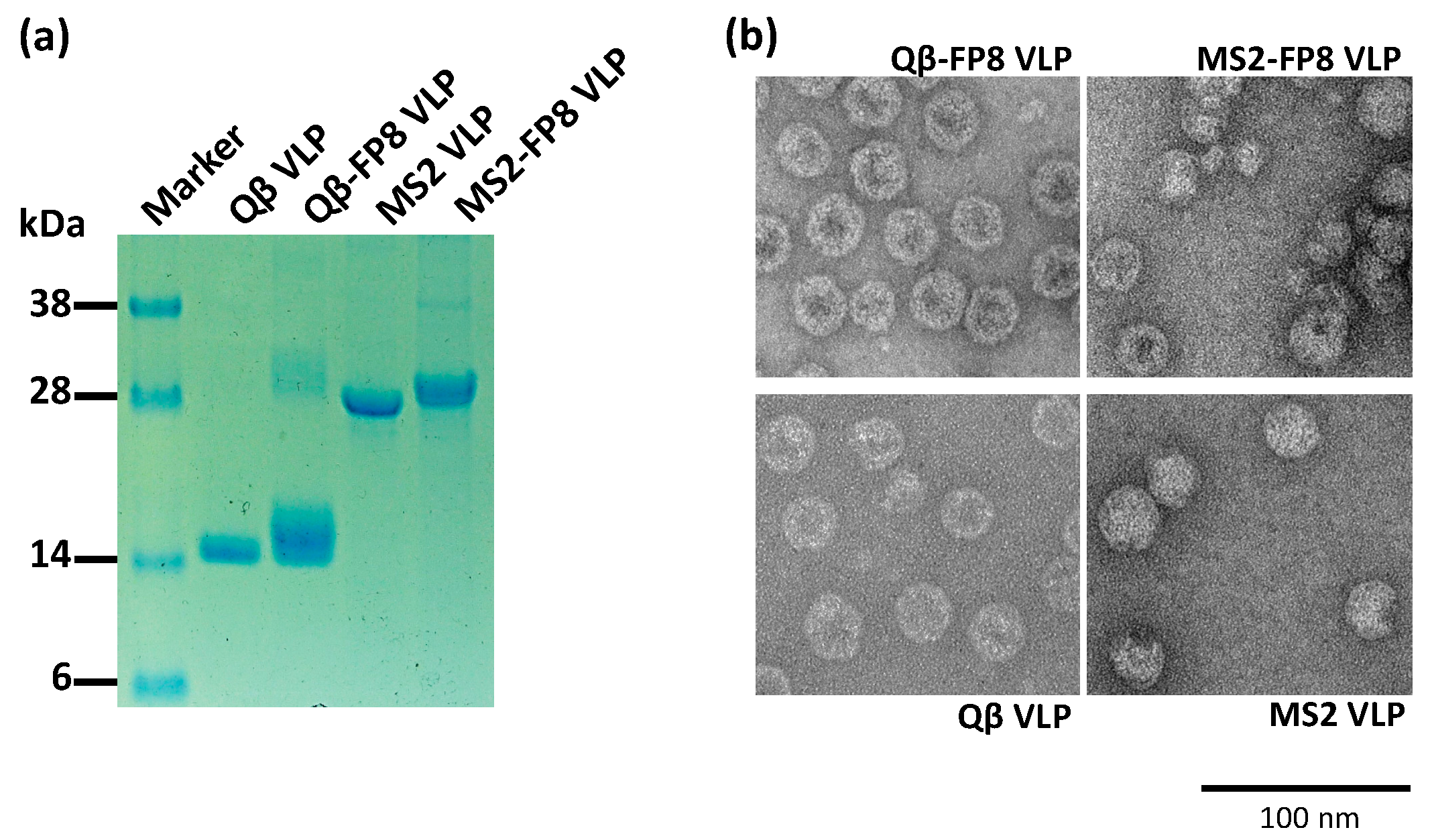
3.1. Engineering and Characterization of FP8-Displaying VLPs
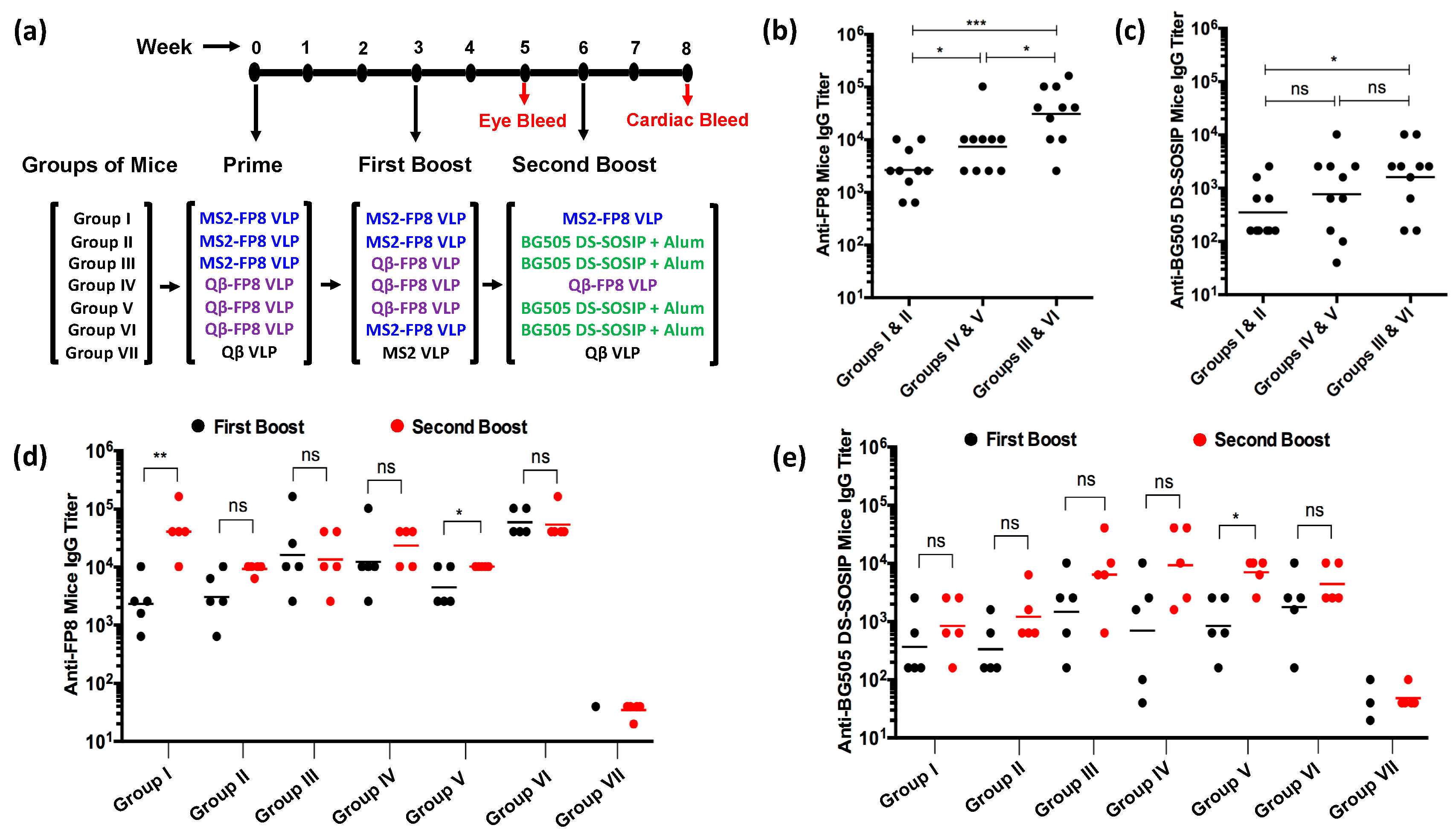
3.2. Immunogenicity of FP8-VLPs
3.3. FP8-VLPs Elicit HIV-1 Neutralizing Antibodies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Escolano, A.; Steichen, J.M.; Dosenovic, P.; Kulp, D.W.; Golijanin, J.; Sok, D.; Freund, N.T.; Gitlin, A.D.; Oliveira, T.; Araki, T.; et al. Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell 2016, 166, 1445–1458.e12. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Verkoczy, L.K.; Jiang, C.; Zhang, J.; Parks, R.; Chen, H.; Housman, M.; Bouton-Verville, H.; Shen, X.; Trama, A.M.; et al. Vaccine Induction of Heterologous Tier 2 HIV-1 Neutralizing Antibodies in Animal Models. Cell Rep. 2017, 21, 3681–3690. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Acharya, P.; Kong, R.; Cheng, C.; Chuang, G.Y.; Liu, K.; Louder, M.K.; O’Dell, S.; Rawi, R.; Sastry, M.; et al. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat. Med. 2018, 24, 857–867. [Google Scholar] [CrossRef]
- Dashti, A.; DeVico, A.L.; Lewis, G.K.; Sajadi, M.M. Broadly Neutralizing Antibodies against HIV: Back to Blood. Trends Mol. Med. 2019, 25, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef]
- Bonsignori, M.; Zhou, T.; Sheng, Z.; Chen, L.; Gao, F.; Joyce, M.G.; Ozorowski, G.; Chuang, G.Y.; Schramm, C.A.; Wiehe, K.; et al. Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 2016, 165, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef]
- Landais, E.; Murrell, B.; Briney, B.; Murrell, S.; Rantalainen, K.; Berndsen, Z.T.; Ramos, A.; Wickramasinghe, L.; Smith, M.L.; Eren, K.; et al. HIV Envelope Glycoform Heterogeneity and Localized Diversity Govern the Initiation and Maturation of a V2 Apex Broadly Neutralizing Antibody Lineage. Immunity 2017, 47, 990–1003.e9. [Google Scholar] [CrossRef]
- Bonsignori, M.; Kreider, E.F.; Fera, D.; Meyerhoff, R.R.; Bradley, T.; Wiehe, K.; Alam, S.M.; Aussedat, B.; Walkowicz, W.E.; Hwang, K.K.; et al. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- MacLeod, D.T.; Choi, N.M.; Briney, B.; Garces, F.; Ver, L.S.; Landais, E.; Murrell, B.; Wrin, T.; Kilembe, W.; Liang, C.H.; et al. Early Antibody Lineage Diversification and Independent Limb Maturation Lead to Broad HIV-1 Neutralization Targeting the Env High-Mannose Patch. Immunity 2016, 44, 1215–1226. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Xu, K.; Zhou, T.; Acharya, P.; Lemmin, T.; Liu, K.; Ozorowski, G.; Soto, C.; Taft, J.D.; Bailer, R.T.; et al. Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 2016, 352, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Kelsoe, G.; Haynes, B.F. What Are the Primary Limitations in B-Cell Affinity Maturation, and How Much Affinity Maturation Can We Drive with Vaccination? Breaking through Immunity’s Glass Ceiling. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Dingens, A.S.; Acharya, P.; Haddox, H.K.; Rawi, R.; Xu, K.; Chuang, G.Y.; Wei, H.; Zhang, B.; Mascola, J.R.; Carragher, B.; et al. Complete functional mapping of infection- and vaccine-elicited antibodies against the fusion peptide of HIV. PLoS Pathog. 2018, 14, e1007159. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Duan, H.; Sheng, Z.; Xu, K.; Acharya, P.; Chen, X.; Cheng, C.; Dingens, A.S.; Gorman, J.; Sastry, M.; et al. Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 2019, 178, 567–584.e19. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Cottrell, C.A.; Ozorowski, G.; van Gils, M.J.; Kumar, S.; Wu, N.C.; Sarkar, A.; Torres, J.L.; de Val, N.; Copps, J.; et al. Conformational Plasticity in the HIV-1 Fusion Peptide Facilitates Recognition by Broadly Neutralizing Antibodies. Cell Host Microbe 2019, 25, 873–883.e5. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sarkar, A.; Pugach, P.; Sanders, R.W.; Moore, J.P.; Ward, A.B.; Wilson, I.A. Capturing the inherent structural dynamics of the HIV-1 envelope glycoprotein fusion peptide. Nat. Commun. 2019, 10, 763. [Google Scholar] [CrossRef]
- Keating, G.M.; Noble, S. Recombinant hepatitis B vaccine (Engerix-B): A review of its immunogenicity and protective efficacy against hepatitis B. Drugs 2003, 63, 1021–1051. [Google Scholar] [CrossRef]
- Schiller, J.; Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 2018, 36, 4768–4773. [Google Scholar] [CrossRef]
- Wu, T.; Li, S.W.; Zhang, J.; Ng, M.H.; Xia, N.S.; Zhao, Q. Hepatitis E vaccine development: A 14 year odyssey. Hum. Vaccines Immunother. 2012, 8, 823–827. [Google Scholar] [CrossRef]
- Chackerian, B. Virus-like particles: Flexible platforms for vaccine development. Expert Rev. Vaccines 2007, 6, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Frietze, K.M.; Peabody, D.S.; Chackerian, B. Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol. 2016, 18, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Lua, L.H.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P. Bioengineering virus-like particles as vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Pumpens, P.; Renhofa, R.; Dishlers, A.; Kozlovska, T.; Ose, V.; Pushko, P.; Tars, K.; Grens, E.; Bachmann, M.F. The True Story and Advantages of RNA Phage Capsids as Nanotools. Intervirology 2016, 59, 74–110. [Google Scholar] [CrossRef]
- Peabody, D.S.; Manifold-Wheeler, B.; Medford, A.; Jordan, S.K.; do Carmo Caldeira, J.; Chackerian, B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J. Mol. Biol. 2008, 380, 252–263. [Google Scholar] [CrossRef]
- Tumban, E.; Peabody, J.; Tyler, M.; Peabody, D.S.; Chackerian, B. VLPs displaying a single L2 epitope induce broadly cross-neutralizing antibodies against human papillomavirus. PLoS ONE 2012, 7, e49751. [Google Scholar] [CrossRef]
- O’Rourke, J.P.; Peabody, D.S.; Chackerian, B. Affinity selection of epitope-based vaccines using a bacteriophage virus-like particle platform. Curr. Opin. Virol. 2015, 11, 76–82. [Google Scholar] [CrossRef]
- Jegerlehner, A.; Storni, T.; Lipowsky, G.; Schmid, M.; Pumpens, P.; Bachmann, M.F. Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. Eur. J. Immunol. 2002, 32, 3305–3314. [Google Scholar] [CrossRef]
- Crossey, E.; Amar, M.J.A.; Sampson, M.; Peabody, J.; Schiller, J.T.; Chackerian, B.; Remaley, A.T. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine 2015, 33, 5747–5755. [Google Scholar] [CrossRef]
- Maphis, N.M.; Peabody, J.; Crossey, E.; Jiang, S.; Jamaleddin Ahmad, F.A.; Alvarez, M.; Mansoor, S.K.; Yaney, A.; Yang, Y.; Sillerud, L.O.; et al. Qss Virus-like particle-based vaccine induces robust immunity and protects against tauopathy. NPJ Vaccines 2019, 4, 26. [Google Scholar] [CrossRef]
- Kundig, T.M.; Senti, G.; Schnetzler, G.; Wolf, C.; Prinz Vavricka, B.M.; Fulurija, A.; Hennecke, F.; Sladko, K.; Jennings, G.T.; Bachmann, M.F. Der p 1 peptide on virus-like particles is safe and highly immunogenic in healthy adults. J. Allergy Clin. Immunol. 2006, 117, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Maurer, P.; Bachmann, M.F. Immunization against angiotensins for the treatment of hypertension. Clin. Immunol. 2010, 134, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Maurer, P.; Jennings, G.T.; Willers, J.; Rohner, F.; Lindman, Y.; Roubicek, K.; Renner, W.A.; Muller, P.; Bachmann, M.F. A therapeutic vaccine for nicotine dependence: Preclinical efficacy, and Phase I safety and immunogenicity. Eur. J. Immunol. 2005, 35, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Caldeira Jdo, C.; Peabody, J.; Peabody, D.S. Peptide epitope identification by affinity selection on bacteriophage MS2 virus-like particles. J. Mol. Biol. 2011, 409, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Rangel, M.; Hunter, Z.; Peabody, D.S. Virus and virus-like particle-based immunogens for Alzheimer’s disease induce antibody responses against amyloid-beta without concomitant T cell responses. Vaccine 2006, 24, 6321–6331. [Google Scholar] [CrossRef]
- Tumban, E.; Peabody, J.; Peabody, D.S.; Chackerian, B. A pan-HPV vaccine based on bacteriophage PP7 VLPs displaying broadly cross-neutralizing epitopes from the HPV minor capsid protein, L2. PLoS ONE 2011, 6, e23310. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Pancera, M.; Acharya, P.; Georgiev, I.S.; Crooks, E.T.; Gorman, J.; Joyce, M.G.; Guttman, M.; Ma, X.; Narpala, S.; et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat. Struct. Mol. Biol. 2015, 22, 522–531. [Google Scholar] [CrossRef]
- Georgiev, I.S.; Joyce, M.G.; Yang, Y.; Sastry, M.; Zhang, B.; Baxa, U.; Chen, R.E.; Druz, A.; Lees, C.R.; Narpala, S.; et al. Single-Chain Soluble BG505.SOSIP gp140 Trimers as Structural and Antigenic Mimics of Mature Closed HIV-1 Env. J. Virol. 2015, 89, 5318–5329. [Google Scholar] [CrossRef]
- Zhai, L.; Peabody, J.; Pang, Y.S.; Schiller, J.; Chackerian, B.; Tumban, E. A novel candidate HPV vaccine: MS2 phage VLP displaying a tandem HPV L2 peptide offers similar protection in mice to Gardasil-9. Antivir. Res. 2017, 147, 116–123. [Google Scholar] [CrossRef]
- Jennings, G.T.; Bachmann, M.F. The coming of age of virus-like particle vaccines. Biol. Chem. 2008, 389, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Ratto-Kim, S.; Currier, J.R.; Cox, J.H.; Excler, J.L.; Valencia-Micolta, A.; Thelian, D.; Lo, V.; Sayeed, E.; Polonis, V.R.; Earl, P.L.; et al. Heterologous prime-boost regimens using rAd35 and rMVA vectors elicit stronger cellular immune responses to HIV proteins than homologous regimens. PLoS ONE 2012, 7, e45840. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Bolhassani, A.; Shahbazi, S. Prime-boost vaccine strategy against viral infections: Mechanisms and benefits. Vaccine 2016, 34, 413–423. [Google Scholar] [CrossRef]
- Cornuz, J.; Zwahlen, S.; Jungi, W.F.; Osterwalder, J.; Klingler, K.; van Melle, G.; Bangala, Y.; Guessous, I.; Muller, P.; Willers, J.; et al. A vaccine against nicotine for smoking cessation: A randomized controlled trial. PLoS ONE 2008, 3, e2547. [Google Scholar] [CrossRef]
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol. 2017, 45, 21–30. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Joyce, M.G. Strategies to guide the antibody affinity maturation process. Curr. Opin. Virol. 2015, 11, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Havenar-Daughton, C.; Abbott, R.K.; Schief, W.R.; Crotty, S. When designing vaccines, consider the starting material: The human B cell repertoire. Curr. Opin. Immunol. 2018, 53, 209–216. [Google Scholar] [CrossRef]
- Andrabi, R.; Bhiman, J.N.; Burton, D.R. Strategies for a multi-stage neutralizing antibody-based HIV vaccine. Curr. Opin. Immunol. 2018, 53, 143–151. [Google Scholar] [CrossRef]
- Briney, B.; Sok, D.; Jardine, J.G.; Kulp, D.W.; Skog, P.; Menis, S.; Jacak, R.; Kalyuzhniy, O.; de Val, N.; Sesterhenn, F.; et al. Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 2016, 166, 1459–1470.e11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Xu, K.; Kong, R.; Chuang, G.Y.; Corrigan, A.R.; Geng, H.; Hill, K.R.; Jafari, A.J.; O’Dell, S.; Ou, L.; et al. Consistent elicitation of cross-clade HIV-neutralizing responses achieved in guinea pigs after fusion peptide priming by repetitive envelope trimer boosting. PLoS ONE 2019, 14, e0215163. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mogus, A.T.; Liu, L.; Jia, M.; Ajayi, D.T.; Xu, K.; Kong, R.; Huang, J.; Yu, J.; Kwong, P.D.; Mascola, J.R.; et al. Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines 2020, 8, 765. https://doi.org/10.3390/vaccines8040765
Mogus AT, Liu L, Jia M, Ajayi DT, Xu K, Kong R, Huang J, Yu J, Kwong PD, Mascola JR, et al. Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines. 2020; 8(4):765. https://doi.org/10.3390/vaccines8040765
Chicago/Turabian StyleMogus, Alemu Tekewe, Lihong Liu, Manxue Jia, Diane T. Ajayi, Kai Xu, Rui Kong, Jing Huang, Jian Yu, Peter D. Kwong, John R. Mascola, and et al. 2020. "Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide" Vaccines 8, no. 4: 765. https://doi.org/10.3390/vaccines8040765
APA StyleMogus, A. T., Liu, L., Jia, M., Ajayi, D. T., Xu, K., Kong, R., Huang, J., Yu, J., Kwong, P. D., Mascola, J. R., Ho, D. D., Tsuji, M., & Chackerian, B. (2020). Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines, 8(4), 765. https://doi.org/10.3390/vaccines8040765