Abstract
Influenza viruses cause annual epidemics and occasional pandemics. The high diversity of viral envelope proteins permits viruses to escape host immunity. Therefore, the development of a universal vaccine and broadly neutralizing antibodies (bnAbs) is essential for controlling various mutant viruses. Here, we review some potentially valuable bnAbs for influenza; one is a novel passive immunotherapy using a variable domain of heavy chain-only antibody (VHH), and the other is polymeric immunoglobulin A (pIgA) induced by intranasal vaccination. Recently, it was reported that a tetravalent multidomain antibody (MDAb) was developed by genetic fusion of four VHHs, which are bnAbs against the influenza A or B viruses. The transfer of a gene encoding the MDAb–Fc fusion protein provided cross-protection against both influenza A and B viruses in vivo. An intranasal universal influenza vaccine, which can induce neutralizing pIgAs in the upper respiratory tract, is currently undergoing clinical studies. A recent study has revealed that tetrameric IgAs formed in nasal mucosa are more broadly protective against influenza than the monomeric and dimeric forms. These broadly neutralizing antibodies have high potential to control the currently circulating influenza virus.
1. Introduction
Cross protection against infectious diseases is extremely important due to the existence of multiple virus subtypes. Vaccines have successfully eradicated smallpox and can control other viral infections [1]. However, not all current vaccines are universal. For example, seasonal viruses such as influenza spread around the world because antigenic drift and antigenic shift, mainly of membrane proteins such as hemagglutinin (HA), permit the virus to escape host immunity [2,3]. There are fewer therapeutic monoclonal antibodies (mAbs) against viral infections than there are against cancer and autoimmune disease [4,5], mainly because viral antigens are continuously evolving [6]. Therefore, the development of a universal vaccine or broadly neutralizing mAbs (bnAbs) is important to assist with the control of viral infections.
Here, we review how cross-protection can be achieved, via passive immunotherapy or vaccination, against influenza, a representative viral infection. Recently, several bnAbs against the influenza virus that produce cross-protection have been isolated [7]. First, we focus on the engineering of novel antibodies, such as multidomain antibodies (MDAbs), which link camelid single-domain antibodies to protect against influenza A and B infections. It has been reported that an MDAb neutralized both influenza A and B viruses [8]. Current passive immunotherapy using MDAbs may provide broad protection against numerous virus infections. It is well known that influenza viruses invade the respiratory tract, where polymeric IgAs (pIgAs), which play a key protective role against the virus, are produced [9]. Second, we focus on intranasal vaccines that induce pIgA generation in the upper respiratory mucosa and protect against influenza virus infection [9,10]. Recently, it was revealed that tetrameric IgA has better neutralizing activity than monomeric or dimeric IgAs [11].
2. Antibody Engineering for Cross Protection against Influenza Virus Infection
2.1. Structural and Physiological Features of HA as a Therapeutic Target
Influenza viruses infect the epithelial cells of the respiratory mucosa, to which they bind by their surface glycoprotein, hemagglutinin (HA), to the cell receptor, sialic acid [10]. Human influenza is caused by influenza virus types A and B [12]. There are 18 HA subtypes of influenza type A [13]. On the basis of HA antigenic differences and phylogenetic sequence relatedness, influenza A viruses are also classified into group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18) and group 2 (H3, H4, H7, H10, H14, and H15) [14]. Influenza B viruses are not divided into subclasses, but into two major phylogenetic lineages, B/Victoria/2/87 and B/Yamagata/16/88, respectively termed the Victoria and Yamagata lineages in the 1980s [15,16,17]. Currently, human influenza is caused by two circulating influenza A viruses, H1N1 and H3N2, and influenza B virus, which lead to seasonal epidemics and occasional pandemics. HA is the major target of the host humoral immune response and vaccination [18].
HA is composed of a globular head domain (HA1), containing the receptor-binding site (RBS), and a stalk domain (HA2), containing the transmembrane domain (Figure 1A) [19,20]. In the 1980s, five antigenic sites A–E (A–D, Webster and Laver; E. Skehel et al.) have been identified on the HA1 surface of the H3N2 influenza virus [21,22,23]. Wiley and colleagues compiled a directory of amino acids in each of the antigenic sites and characterized the structural features and physical boundaries of each site [24]. Antigenic site B significantly overlaps with the RBS [24,25]. A recent report showed that antigenic site B has been immunodominant over site A in recently circulating H3N2 viruses [21]. Therefore, it was suggested that genetic mutations in antigenic site B drive the antigenic drift that facilitates immune evasion.
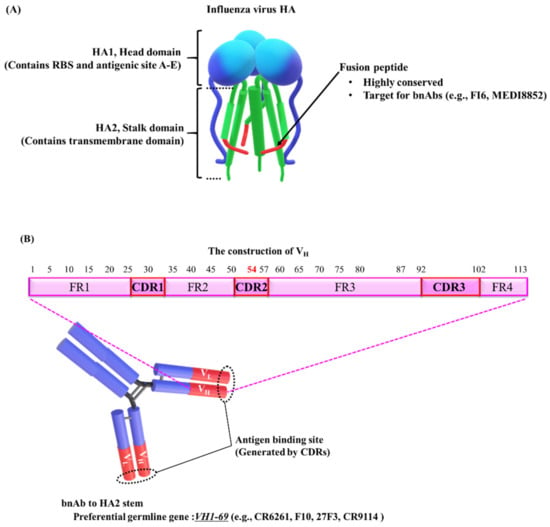
Figure 1.
Schematic diagram of HA, the target for broadly neutralizing antibodies (bnAbs). (A) The model shows hemagglutinin (HA) structure (prefusion). HA is composed of a globular head domain (HA1) and a stalk domain (HA2) with a highly conserved sequence, the fusion peptide. The HA structure model was obtained from [81,82,83,84]. (B) The VH1-69 germline gene generally dominates the human bnAb response to the HA2 stalk domain. The variable domain of heavy chains (VH) consists of four framework regions (FR1-4) and three complementary determining regions (CDR1-3). The phenylalanine at position 54 (F54) is conserved in VH1-69 and is required for initial development of most VH1-69 antibodies. However, in the process of affinity maturation, F54 and accumulated mutations are functionally redundant. The numbers indicate the residue positions [54].
N-glycosylation of HA1 also helps the influenza virus to escape the host immune system [25,26]. Antibodies have limited access to major antigenic sites shielded by N-glycans on the HA1 domain. It has been suggested that the oligosaccharide chains accumulate in antigenic sites and contribute to immune evasion [23,27,28].
Influenza B viruses generally circulate in humans, while influenza A viruses can infect other species [29]. The antigenicity of HA is different in influenza A and B viruses [30]. Therefore, it is difficult to protect against influenza B virus infection, even with a universal vaccine or bnAbs against influenza A viruses [8,31].
2.2. Broadly Neutralizing Monoclonal Antibodies (bnAbs) Against Influenza Virus
The globular head domain HA1 contains major antigenic sites that are highly immunogenic and are able to elicit high serum antibody titers, and undergo antigenic variation [32,33,34,35]. The antibodies against RBS in HA1 can inhibit the attachment of the influenza virus to host cell receptors [18,35]. Mutations in this region would allow the virus to escape the host immune response [35]. However, a conserved RBS epitope, which is a potential target for a universal vaccine or bnAbs, was recently identified [36,37,38,39,40].
HA2, the stalk domain, also contains a highly conserved region, the fusion peptide, which is a possible therapeutic target (Figure 1A) [33,34]. The fusion peptide mediates the attachment of the viral envelope to the endosomal host membrane via a pH-triggered conformational change of HA. Neutralizing antibodies bind to the fusion peptide and prevent the conformation change responsible for membrane fusion, thus inhibiting infection [14]. However, HA2 stalk-specific antibodies are rarely produced during natural infections or conventional vaccinations [14,32]. On the basis of the possibility that HA2 is masked by the membrane-distal portion of HA, Palese and colleagues proposed a unique vaccine, lacking the globular head, HA1 [32]. Other studies using human subjects have suggested that the preexisting memory B cells against HA1 epitopes predominate, inhibiting the induction of protective antibodies against HA2 upon revaccination with similar strains [41]. The development of universal vaccines is required to overcome these obstacles.
Several bnAbs targeting the fusion peptide have been generated (e.g., FI6 [42] and MEDI8852 [43]) and have been shown to successfully neutralize multiple influenza A virus subtypes of group 1 and group 2 HAs (Figure 1A) [14,44]. In general, human antibodies responding to the HA2 stalk domain (e.g., CR6261 [45], F10 [46], 27F3 [47], and CR9114 [37]) are mainly encoded by the VH1-69 germline gene (Figure 1B) [43,46,47,48]. A striking feature of the VH1-69-encoded antibodies is that the conserved hydrophobic residues are exposed, particularly the phenylalanine at position 54 (F54), located at the tip of the heavy-chain complementary determining region 2 (HCDR2) loop (Figure 1B) [45,46,47]. It is common for antibodies to use the conserved hydrophobic regions to target the conserved hydrophobic residues of viral proteins [49]. The VH1-69 germline gene also encodes bnAbs that recognize HA1 [50] or the corresponding targets of other viruses. These bnAbs include gp120, a membrane protein of human immunodeficiency virus type 1 (HIV-1) [51], and the RBS of severe acute respiratory syndrome coronavirus (SARS-CoV) [52] and Middle East respiratory syndrome (MERS)-CoV [53]. Lanzavecchia and colleagues found functional redundancy of the accumulated mutations and F54 in the process of affinity maturation (Figure 1B) [54]. Using genetic analysis of isolated B cell clones as previously described [54], the group also established MEDI8852, a bnAb that recognizes groups 1 and 2 of influenza A viruses [43]. They reconstructed the genealogical trees of the variable region from isolated B cell clones carrying the same germline gene segments. They then selected one clone, FY1, on the basis of its potency, breadth of reactivity to HA, and low number of somatic mutations, for affinity optimization with parsimonious mutagenesis [55], termed ex vivo affinity maturation. They generated MEDI8852 by modifying FY1 using ex vivo affinity maturation and succeeded in improving its affinity to H3 HA by 14-fold and that to H1 HA by 5-fold as compared to the affinity of the original. The clone contains a rare germline gene, VH6-1, but not a common germline gene, VH1-69. Their approach may be used to generate rare but potent bnAbs against the influenza virus.
2.3. Multidomain Antibody (MDAb) Engineering Using Nanobodies from Camelid Antibodies
Human IgGs consist of two identical heavy chains with one variable domain (VH), three constant domains (CH1, CH2, and CH3), and two identical light chains (Figure 2A) [56]. In 1993, Hamers et al. discovered that camelid antibodies have a totally different structure [57]. Camelid has the same structure as human IgGs, but IgG2 and IgG3 lack light chains and CH1 domains; nevertheless, IgG2 and IgG3 can bind a number of antigens (Figure 2B). Similar to a conventional VH, the camelid variable domain of a heavy chain-only antibody (VHH) (Figure 2C) consists of four constant framework regions (FR1–4) separated by three complementary determining region (CDR) loops (CDR1, 2, and 3) [58,59,60]. The CDR3 of VHH is more variable in both length and sequence than the other regions, and is similar to human VH [56,60,61]. VHH forms a single-domain paratope with a molecular weight of approximately 15 kDa [58,62]. VHH is 10 times smaller than a whole IgG antibody (around 150 kDa) and half of a single-chain variable fragment (scFv) consist of a VH and VL-linked domain and a linker oligopeptide of around 30 kDa [58,63,64]. Therefore, VHHs are called “nanobodies” [64].
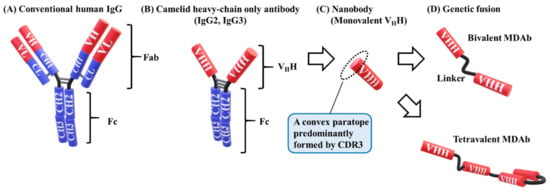
Figure 2.
Model of human and camelid IgG. (A) Conventional human IgG consists of two identical heavy chains with one variable domain (VH), three constant domains (CH1, CH2, and CH3), and two identical light chains with one variable domain (VL) and one constant domain (CL). Fab contains the antigen-binding sites, and Fc is involved in effector functions. (B) The structure of camelid IgG1 (not shown) is similar to that of human IgG. Camelid IgG2 and IgG3 lack light chains and CH1 domains. The hinge region of IgG2 is longer than that of IgG3 (the model is representative) [85]. (C) VHH, known as a nanobody, consists of a variable domain of camelid IgG (IgG2 or IgG3) and forms a paratope with a single domain. The molecular weight is approximately 15 kDa. A convex paratope is predominantly formed by the CDR3 loop to bind to clefts or pockets of the antigen. (D) VHH is easily modified via genetic multimerization to a bivalent or tetravalent multidomain antibody (MDAb).
Compared with conventional IgG antibodies, VHHs have several advantages. They are compact, and small VHHs are remarkably stable under extreme conditions [60,62,65]. VHHs also have a convex paratope, formed predominantly by the CDR3 loop, which allows binding to the clefts and pockets of the antigen to avoid recognition by conventional antibodies that have a dimeric concave or flat paratope (Figure 2C) [65,66]. They are cost-effective due to their high stability and ease of manufacture in microorganisms [67]. These advantages allow for the widespread use of VHHs, including passive immunotherapy for infectious diseases [65].
One disadvantage of VHHs is their rapid removal from circulation via renal filtration within a couple of hours after injection [58,68,69], with a threshold in the range of 40–60 kDa [70,71,72]. The short half-life limits the therapeutic application of VHHs, and they require repeated administration. The half-life of conventional human IgG is approximately 3 weeks, due to the presence of a neonatal Fc receptor, FcRn [56,73]. Therefore, the fusion of a human Fc segment to a VHH will extend its half-life [58,62,65,74]. Enlarging the size of VHHs from 15 kDa to 80 kDa also prolongs their retention in the body [75]. It has been reported that intranasal inoculation with VHH-Fc fusions against neuraminidase (NA), another membrane protein, succeeded in neutralizing a lethal influenza virus infection in mice [76].
Single-domain VHHs are easily modified via genetic multimerization to form bispecific, bivalent, or tetravalent molecules to enhance function (Figure 2D) [77,78]. Bivalent MDAbs against tumor necrosis factor (TNF) molecules exhibited 100–500-fold greater binding and neutralizing ability than monovalent VHHs [79]. The length of a linker sequence, such as the glycine–serine (GS) linker, affected the potency (16-fold) [80]. The linker of a bivalent MDAb contains up to several dozens of amino acids in order to avoid steric clashes with the antigens. A bispecific MDAb, which contains two VHHs, each recognizing a different epitope, is more potent than a monomeric VHH [80]. An up to 10-fold greater potency can be obtained, depending on the relative position of VHH molecules.
Laursen et al. prepared several broadly neutralizing VHHs against influenza A and B viruses by immunizing llamas with recombinant HA [8]. They isolated two neutralizing VHHs against the influenza A virus (SD36 and SD38), and two against the influenza B virus (SD83 and SD84) (Table 1 and Figure 3). Next, they generated bivalent MDAbs with SD38 linked to SD36 and SD83 linked to SD84 by 18-, 38-, and 60-residue linkers. Although the length of the linker did not significantly affect the neutralizing activity, bivalent MDAbs were notably more potent at neutralizing influenza virus strains than monovalent VHHs. Laursen et al. generated a tetravalent MDAb, MD2407, via a genetic fusion of SD38, SD36, SD83, and SD84, using 10-residue linkers (Figure 3). The linkers were too short to allow the VHHs to bind the same HA molecule. In addition, they combined the tetravalent MDAb and human IgG1 Fc to obtain a tetravalent MDAb–Fc fusion protein (MD3606) (Figure 3). MD2407 and MD3606 neutralized all influenza A and B viruses tested, except for the H12 virus. One reagent provided broad protection against both influenza A and B viruses.

Table 1.
Neutralizing potency of isolated VHHs against the influenza A or B viruses [8]. Neutralizing titer was determined by in vitro microtiter assay.
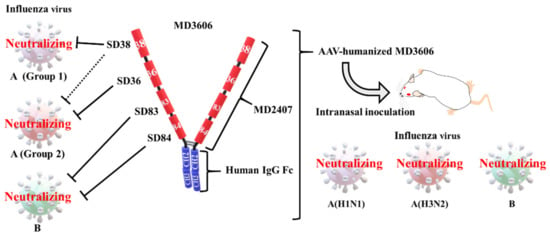
Figure 3.
Cross-protection with a heavy chain only antibody (VHH)–Fc fusion construct. All four VHHs were obtained by immunizing llamas with recombinant hemagglutinin (HA). VHH (SD38) can neutralize group 1 influenza A viruses and weakly neutralize some group 2 viruses, VHH (SD36) neutralizes group 2 influenza A viruses, and VHH (SD83 and SD84) neutralizes influenza B viruses [8]. A tetravalent multidomain antibody (MDAb; MD2407) was generated by connecting VHHs with (GGGGS)2-linkers. An MDAb–Fc fusion (MD3606) was generated by attaching MD2407 to the human IgG Fc region. MD2407 and MD3606 neutralized influenza A and B viruses in vitro. Intranasal injection with an AAV vector encoding a humanized MD3606, administered 7 days before exposure, provided broad cross-protection against a lethal dose of H1N1, H3N2, and influenza B virus.
3. Recent Passive Immunization via Antibody–Gene Transfer
Several factors hinder the widespread use of antibody drugs, such as the necessity of weekly or biweekly infusions due to short half-life (approximately 20 days) [86], high production cost, laborious purification procedures, and quality control [87]. To address the above problems, a novel passive immunization method using plasmids or viral vectors encoding the antibody gene has been introduced. Long-term neutralizing antibodies are produced by the host cells following a single administration. In addition, gene-based antibody drugs are cost-effective in terms of production, purification, and administration [88,89].
Previously, for the first time, we induced over 10 μg/mL of the neutralizing anti-HA antibodies to BALB/c mice using electro-gene transfer with a plasmid encoding the neutralizing antibody gene [90]. The expression was stable for at least 70 days after inoculation. These potent and stable neutralizing antibodies provided long-lasting protection against influenza infection. We also administered passive immune treatment via hydrodynamic injection, which involves a rapid injection of a large volume of plasmid DNA solution into a mouse tail vein [91]. Neutralizing antibodies were detected within 4 hours of injection. We then successfully treated a lethal influenza virus infection 2 days after the challenge. Although we have not carried out the cross-protection experiments yet, another group has demonstrated broad cross-protection with plasmid DNA encoding two bnAbs that target influenza A and B viruses [92]. Several reports have described potent and stable cross-protection against influenza virus infection achieved via antibody–gene transfer with adeno-associated virus (AAV) vectors [89]. One study indicated that passive immunization via intranasal delivery of an AAV vector encoding bnAb FI6 could broadly protect against influenza virus infections [93]. Laursen et al. generated an AAV vector encoding a humanized tetravalent MDAb–Fc fusion protein (MD3606) (Figure 3) [8]. These authors demonstrated broad cross-protection via intranasal injection of mice with the AAV construct 7 days before the challenge with a lethal dose of H1N1, H3N2, or influenza B virus. However, AAV vector immunogenicity and rare gene toxicity due to the integration of DNA into the host genome are concerning [94]. Passive immunotherapy has a long history, dating back to the use of antisera against tetanus and diphtheria, first developed by Kitasato and Behring in the 19th century [95,96]. In the future, passive immunotherapy using antibody gene-encoding MDAbs, such as MD3606, could broadly protect against influenza virus infections.
4. A Universal Intranasal Vaccination against Influenza
4.1. Features of Intranasal Vaccination for Influenza Virus Infection
Effective antibody production depends upon the route of vaccine administration. Several types of vaccines are available, such as live attenuated influenza vaccines (LAIVs), inactivated viral vaccines, and subunit vaccines [97]. In general, LAIVs are administered intranasally, whereas current inactivated vaccines are administered subcutaneously or intramuscularly. Current intranasal LAIVs can minimize viral infection and induce the production of both secretory pIgA and IgG in the respiratory tract [98]. However, LAIVs are not approved for high-risk individuals, due to the transmission of the attenuated but live virus [98]. Conventional subcutaneous vaccines can mainly induce serum IgG generation but are not effective against the variant viruses [99]. Adjuvant-combined, nasal-inactivated vaccines, however, induce potent secretory pIgA and serum IgG production. Induced pIgA production provides broadly neutralizing activity against the influenza virus in the upper respiratory tract; that is, pIgAs can effectively protect against both homologous and heterologous influenza virus infections [99]. We first discuss previous research into intranasal vaccination.
A study was conducted by Tamura et al. [98] to compare the efficacy of the intra-nasal and subcutaneous routes of administration of influenza vaccines. Vaccines were prepared against influenza virus strains A/Guizhou-X (H3N2), A/Fukuoka (H3N2), A/Sichuan (H3N2), A/PR8 (H1N1), and B/Ibaraki using the cholera toxin B subunit (CTB) as an adjuvant [100]. Mice were challenged with a non-lethal infection of A/Guizhou-X. All intranasal vaccinations except B/Ibaraki provided secretory IgA cross-reacting to A/Guizhou-X-HA (Table 2). Only homologous vaccination could induce the specific IgG to A/Guizhou-X-HA. The protective effect of the challenged virus reflected the titer of specific antibodies (Table 2). Vaccination with A/PR8, which is the other subtype from the challenged virus, provided low cross-protection, and vaccination with influenza B virus failed to induce cross-protection and induction of secretory IgA. No subcutaneous vaccinations induced the specific IgA, although they could induce a potent specific IgG reacting only to the homologous strain. Overall, intranasal vaccinations led to the production of more potent secretory IgA and cross-protection than conventional vaccinations.
Table 2.
Comparison table of the efficacy between intranasal (i.n.) and subcutaneous (s.c.) vaccines for influenza [98]. N.S., not significant. HA (A/Guizhou-X)-Reactive IgA in nasal wash; +, −5 ng/mouse; ++, 5–10 ng/mouse; +++, 10–15 ng/mouse. HA (A/Guizhou-X)-Reactive IgG in nasal wash; ++++, 20–25 ng/mouse; +++++, 40–45 ng/mouse. EID50, 50% egg infectious dose. Protection against A/Guizhou-X in nasal wash; N.S., approximately 1 × 104 EID50; ++, approximately 1 × 102 EID50; +++, approximately 1 × 10 EID50.
It is also important to have a suitable adjuvant that is safe for human use in the clinical application of intranasal influenza vaccines. Several studies have been conducted into reducing side effects by the introduction of mutations in CTB [101] or complement component C3d [102]. Ichinohe et al. demonstrated another effective adjuvant, a synthetic double-stranded RNA (dsRNA) polyriboinosinic polyribocytidylic acid (poly (I:C)) for intranasal vaccination [103]. Clinical studies have also suggested that secretory pIgAs, generated in response to inactivated nasally administered vaccine, play a significant role in providing protection from heterologous influenza viruses [104,105]. An intranasally delivered inactivated vaccine could be valuable in promoting some of the favorable immune responses elicited by LAIVs, without the risk of live virus to immunocompromised individuals. In the next section, we focus on the mechanism of secretory pIgA protection against influenza virus infections.
4.2. Secretory pIgAs against Influenza Virus Infections
Among the isotypes, IgA is the principal immunoglobulin found on mucosal surfaces [106]. Characteristic features differentiate IgA from others, notably its quaternary structure [107]. While IgAs almost always exist in the monomeric form in human serum, those in the lamina propria of mucosal tissue covalently link with the heavy chains to the J chain to form polymers [108]. pIgA binds to the polymeric immunoglobulin receptor (pIgR) on the basolateral surfaces of mucosal epithelial cells via the J chain. The complex is transported across epithelial cells to the apical surface, where pIgR is cleaved, releasing a secretory component (SC), which remains associated with pIgA in the lumen. Secretory pIgA exists mainly in the dimeric form, with low levels of the tetrameric form [11,107,109]. Studies have been conducted to generate recombinant secretory pIgA, but the main focus was on the dimeric form [110,111,112].
Suzuki et al. showed that tetrameric IgA (tet-IgA) offers more protection against influenza A viruses in nasal mucosa than monomeric or dimeric forms [11]. To evaluate the polymerization of secretory pIgA, and the molecular mechanism of protection against influenza infection, several antibodies with identical variable regions are needed in monomer, dimer, and tetramer form. Saito et al. recently generated recombinant secretory pIgAs [113]. They produced recombinant monoclonal human tet-IgAs by co-expressing the heavy, light, and J chains along with SC in mammalian cells. They compared the reactivity and functionality of the generated bnAbs, including monomeric IgA, dimeric IgA, and tet-IgA. Compared to the other forms, recombinant tet-IgAs exhibited enhanced neutralizing activity against low- but not high-affinity virus strains. It has been suggested that tetramerization significantly enhances the neutralization breadth of IgA. Overall, to prevent an influenza outbreak, a safe vaccine should provide broad, long-term cross-protection. Compared with conventional vaccines, a nasally administered inactivated vaccine is expected to provide universal protection against influenza infections.
5. Conclusions
We present an overview of novel passive immunotherapy using tetravalent MDAbs involving four VHHs and tet-IgA induced by intranasal vaccination for influenza. Our conclusions include the following three points. First, antigen design based on physical features is important. Next, we discussed the conformation of the antibody for cross-protection. Finally, we focused on the technology of MDAb engineering.
Several conserved HA targets have been identified (e.g., fusion peptides in the stalk domain), and many bnAbs are proceeding to clinical trials [14]. There are also ongoing clinical trials of intranasal vaccinations with inducible neutralizing pIgAs against HA to achieve cross-protection [105]. However, it is difficult to obtain HA2 stalk-specific antibodies with natural infections or conventional vaccinations. Considering the highly biased usage of the hydrophobic VH1-69 gene for antibodies against the HA2 stalk [41], the hydrophobic epitopes (e.g., HA2) might have the drawback; although the antibodies targeting the hydrophobic epitope have a tendency to broad reactivity, they are rarely induced by conventional vaccination. Antigen design based on physical features such as hydrophobicity, clefts, and pockets, is also essential to the development of bnAbs.
The larger secretory pIgAs induced by intranasal vaccination had high neutralizing efficacy against an influenza virus infection [11]. Recombinant technology producing tetrameric IgA (tet-IgA) could be also powerful tool, inducing passive immunization. Several studies have already succeeded in inducing potent neutralizing IgG antibodies using antibody–gene transfer with plasmid or adeno-associated virus (AAV) vectors [86]. Isolated monoclonal pentameric IgMs also broadly protect against the influenza B virus [114], although it is difficult to induce potent neutralizing polymeric IgMs via vaccination [11]. Monoclonal IgGs were demonstrated to spontaneously assemble into hexamers [115]. However, IgG hexamers have not been shown to effectively neutralize influenza. Further research is needed to understand the relationships between the quaternary structure and the neutralizing effects of antibodies.
One of the advantages of VHHs is that they are compact, allowing easy genetic modification. Another is their stability under extreme conditions. They are a unique form of paratope consisting of a single domain. It would be valuable to identify the unique germline gene of VHH which is homologous to VH1-69 in human IgG and obtain the sequence for ex vivo affinity maturation. Passive immunization with multivalent MDAbs based on VHH could control circulating influenza virus. If we succeeded in generating multivalent MDAb with several VHHs against each influenza A virus (group 1 or group 2), or B virus, even one reagent would induce cross-protection for whole influenza subtypes. These strategies could help to prevent future influenza pandemics.
Author Contributions
Writing—original draft preparation, M.B., T.Y.; writing—review and editing, M.B., T.Y., J.C., S.A.-T.; visualization, T.Y.; supervision, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This review was funded by JSPS KAKENHI grant number 19K07491 (to T.Y), Takeda Science Foundation (to T.Y), and a grant from Unit Support, Aichi Medical University (to T.Y and S.A.-T.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAV | Adeno-associated virus |
| bnAb | Broadly neutralizing mAb |
| CDR | Complementary determining region |
| CTB | Cholera toxin B subunit |
| FR | Framework regions |
| F54 | Phenylalanine at position 54 |
| HA | Hemagglutinin |
| HIV-1 | Human immunodeficiency virus type 1 |
| MDAb | Multidomain antibody |
| i.n. | Intranasal |
| LAIV | Live attenuated influenza vaccine |
| mAb | Monoclonal antibody |
| MERS-CoV | Middle East respiratory syndrome CoV |
| NA | Neuraminidase |
| pIgA | Polymeric IgA |
| pIgR | Polymeric immunoglobulin receptor |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SC | Secretory component |
| s.c. | Subcutaneous |
| tet-IgA | Tetrameric IgA |
| VHH | The variable domain of heavy chain-only antibody |
References
- Mast, E.E.; Cochi, S.L.; Kew, O.M.; Cairns, K.L.; Bloland, P.B.; Martin, R. Fifty Years of Global Immunization at CDC, 1966-2015. Public Health Rep. 2017, 132, 1–26. [Google Scholar] [CrossRef]
- Si, L.; Xu, H.; Zhou, X.; Zhang, Z.; Tian, Z.; Wang, Y.; Wu, Y.; Zhang, B.; Niu, Z.; Zhang, C.; et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 2016, 354, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Sandbulte, M.R.; Westgeest, K.B.; Gao, J.; Xu, X.; Klimov, A.I.; Russell, C.A.; Burke, D.F.; Smith, D.J.; Fouchier, R.A.; Eichelberger, M.C. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 20748–20753. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.F.; Wang, R.; Ling, S.; Wang, S. Antibody Engineering for Pursuing a Healthier Future. Front. Microbiol. 2017, 8, 495. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Saylor, C.; Dadachova, E.; Casadevall, A. Monoclonal antibody-based therapies for microbial diseases. Vaccine 2009, 27, G38–G46. [Google Scholar] [CrossRef]
- Sano, K.; Ainai, A.; Suzuki, T.; Hasegawa, H. The road to a more effective influenza vaccine: Up to date studies and future prospects. Vaccine 2017, 35, 5388–5395. [Google Scholar] [CrossRef]
- Laursen, N.S.; Friesen, R.H.E.; Zhu, X.; Jongeneelen, M.; Blokland, S.; Vermond, J.; van Eijgen, A.; Tang, C.; van Diepen, H.; Obmolova, G.; et al. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 2018, 362, 598–602. [Google Scholar] [CrossRef]
- Tamura, S.; Tanimoto, T.; Kurata, T. Mechanisms of broad cross-protection provided by influenza virus infection and their application to vaccines. Jpn J. Infect. Dis. 2005, 58, 195–207. [Google Scholar]
- Tamura, S.; Kurata, T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J. Infect. Dis. 2004, 57, 236–247. [Google Scholar]
- Suzuki, T.; Kawaguchi, A.; Ainai, A.; Tamura, S.; Ito, R.; Multihartina, P.; Setiawaty, V.; Pangesti, K.N.; Odagiri, T.; Tashiro, M.; et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc. Natl. Acad. Sci. USA 2015, 112, 7809–7814. [Google Scholar] [CrossRef]
- Nachbagauer, R.; Palese, P. Is a Universal Influenza Virus Vaccine Possible? Annu Rev. Med. 2020, 71, 315–327. [Google Scholar] [CrossRef] [PubMed]
- CDC. Types of Influenza Viruses. 2019. Available online: https://www.cdc.gov/flu/about/viruses/types.htm (accessed on 28 May 2020).
- Sedeyn, K.; Saelens, X. New antibody-based prevention and treatment options for influenza. Antiviral Res. 2019, 170, 104562. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Biere, B.; Bauer, B.; Schweiger, B. Differentiation of influenza B virus lineages Yamagata and Victoria by real-time PCR. J. Clin. Microbiol. 2010, 48, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Caini, S.; Kusznierz, G.; Garate, V.V.; Wangchuk, S.; Thapa, B.; de Paula Junior, F.J.; Ferreira de Almeida, W.A.; Njouom, R.; Fasce, R.A.; Bustos, P.; et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE 2019, 14, e0222381. [Google Scholar] [CrossRef]
- Subbarao, K.; Joseph, T. Scientific barriers to developing vaccines against avian influenza viruses. Nat. Rev. Immunol. 2007, 7, 267–278. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Popova, L.; Smith, K.; West, A.H.; Wilson, P.C.; James, J.A.; Thompson, L.F.; Air, G.M. Immunodominance of antigenic site B over site A of hemagglutinin of recent H3N2 influenza viruses. PLoS ONE 2012, 7, e41895. [Google Scholar] [CrossRef]
- Webster, R.G.; Laver, W.G. Determination of the number of nonoverlapping antigenic areas on Hong Kong (H3N2) influenza virus hemagglutinin with monoclonal antibodies and the selection of variants with potential epidemiological significance. Virology 1980, 104, 139–148. [Google Scholar] [CrossRef]
- Skehel, J.J.; Stevens, D.J.; Daniels, R.S.; Douglas, A.R.; Knossow, M.; Wilson, I.A.; Wiley, D.C. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl. Acad. Sci. USA 1984, 81, 1779–1783. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981, 289, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Otwinowski, J.; Thompson, A.J.; Nycholat, C.M.; Nourmohammad, A.; Wilson, I.A. Major antigenic site B of human influenza H3N2 viruses has an evolving local fitness landscape. Nat. Commun. 2020, 11, 1233. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; David, A.; Schmidt, L.; Gibbs, J.S.; Puigbo, P.; Ince, W.L.; Bennink, J.R.; Yewdell, J.W. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc. Natl. Acad. Sci. USA 2011, 108, E1417–E1422. [Google Scholar] [CrossRef]
- Wanzeck, K.; Boyd, K.L.; McCullers, J.A. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am. J. Respir. Crit. Care Med. 2011, 183, 767–773. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Ulett, K.B.; Boyd, K.L.; Madsen, J.; Hawgood, S.; McCullers, J.A. N-linked glycosylation attenuates H3N2 influenza viruses. J. Virol. 2007, 81, 8593–8600. [Google Scholar] [CrossRef]
- Baker, S.F.; Nogales, A.; Finch, C.; Tuffy, K.M.; Domm, W.; Perez, D.R.; Topham, D.J.; Martinez-Sobrido, L. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J. Virol. 2014, 88, 10778–10791. [Google Scholar] [CrossRef]
- Wei, C.J.; Crank, M.C.; Shiver, J.; Graham, B.S.; Mascola, J.R.; Nabel, G.J. Next-generation influenza vaccines: opportunities and challenges. Nat. Rev. Drug Discov. 2020, 19, 239–252. [Google Scholar] [CrossRef]
- Asahi, Y.; Yoshikawa, T.; Watanabe, I.; Iwasaki, T.; Hasegawa, H.; Sato, Y.; Shimada, S.; Nanno, M.; Matsuoka, Y.; Ohwaki, M.; et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J. Immunol. 2002, 168, 2930–2938. [Google Scholar] [CrossRef]
- Steel, J.; Lowen, A.C.; Wang, T.T.; Yondola, M.; Gao, Q.; Haye, K.; Garcia-Sastre, A.; Palese, P. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Tewawong, N.; Suwannakarn, K.; Prachayangprecha, S.; Korkong, S.; Vichiwattana, P.; Vongpunsawad, S.; Poovorawan, Y. Molecular epidemiology and phylogenetic analyses of influenza B virus in Thailand during 2010 to 2014. PLoS ONE 2015, 10, e0116302. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Kondrashkina, E.; Wang, Q. Structural basis for the divergent evolution of influenza B virus hemagglutinin. Virology 2013, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Skehel, J.J. Variation and infectivity neutralization in influenza. Immunology 2006, 119, 1–7. [Google Scholar] [CrossRef]
- Raymond, D.D.; Bajic, G.; Ferdman, J.; Suphaphiphat, P.; Settembre, E.C.; Moody, M.A.; Schmidt, A.G.; Harrison, S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc. Natl. Acad. Sci. USA 2018, 115, 168–173. [Google Scholar] [CrossRef]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef]
- Iba, Y.; Fujii, Y.; Ohshima, N.; Sumida, T.; Kubota-Koketsu, R.; Ikeda, M.; Wakiyama, M.; Shirouzu, M.; Okada, J.; Okuno, Y.; et al. Conserved neutralizing epitope at globular head of hemagglutinin in H3N2 influenza viruses. J. Virol. 2014, 88, 7130–7144. [Google Scholar] [CrossRef]
- Whittle, J.R.; Zhang, R.; Khurana, S.; King, L.R.; Manischewitz, J.; Golding, H.; Dormitzer, P.R.; Haynes, B.F.; Walter, E.B.; Moody, M.A.; et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 2011, 108, 14216–14221. [Google Scholar] [CrossRef]
- Watanabe, A.; McCarthy, K.R.; Kuraoka, M.; Schmidt, A.G.; Adachi, Y.; Onodera, T.; Tonouchi, K.; Caradonna, T.M.; Bajic, G.; Song, S.; et al. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell 2019, 177, 1124–1135.e1116. [Google Scholar] [CrossRef]
- Andrews, S.F.; Huang, Y.; Kaur, K.; Popova, L.I.; Ho, I.Y.; Pauli, N.T.; Henry Dunand, C.J.; Taylor, W.M.; Lim, S.; Huang, M.; et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 2015, 7, 316ra192. [Google Scholar] [CrossRef]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Kallewaard, N.L.; Corti, D.; Collins, P.J.; Neu, U.; McAuliffe, J.M.; Benjamin, E.; Wachter-Rosati, L.; Palmer-Hill, F.J.; Yuan, A.Q.; Walker, P.A.; et al. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell 2016, 166, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, D.A.; Lee, F.E. Antibodies against conserved antigens provide opportunities for reform in influenza vaccine design. Front. Immunol. 2011, 2, 76. [Google Scholar] [CrossRef] [PubMed]
- Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L.L.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 2008, 3, e3942. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef]
- Lang, S.; Xie, J.; Zhu, X.; Wu, N.C.; Lerner, R.A.; Wilson, I.A. Antibody 27F3 Broadly Targets Influenza A Group 1 and 2 Hemagglutinins through a Further Variation in VH1-69 Antibody Orientation on the HA Stem. Cell Rep. 2017, 20, 2935–2943. [Google Scholar] [CrossRef]
- Wrammert, J.; Koutsonanos, D.; Li, G.M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011, 208, 181–193. [Google Scholar] [CrossRef]
- Chen, F.; Tzarum, N.; Wilson, I.A.; Law, M. VH1-69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr. Opin. Virol. 2019, 34, 149–159. [Google Scholar] [CrossRef]
- Yu, F.; Song, H.; Wu, Y.L.; Chang, S.Y.; Wang, L.L.; Li, W.; Hong, B.B.; Xia, S.; Wang, C.Y.; Khurana, S.; et al. A Potent Germline-like Human Monoclonal Antibody Targets a pH-Sensitive Epitope on H7N9 Influenza Hemagglutinin. Cell Host Microbe 2017, 22, 471. [Google Scholar] [CrossRef]
- Smith, S.A.; Burton, S.L.; Kilembe, W.; Lakhi, S.; Karita, E.; Price, M.; Allen, S.; Derdeyn, C.A. VH1-69 Utilizing Antibodies Are Capable of Mediating Non-neutralizing Fc-Mediated Effector Functions Against the Transmitted/Founder gp120. Front. Immunol. 2018, 9, 3163. [Google Scholar] [CrossRef]
- Prabakaran, P.; Zhu, Z.; Chen, W.; Gong, R.; Feng, Y.; Streaker, E.; Dimitrov, D.S. Origin, diversity, and maturation of human antiviral antibodies analyzed by high-throughput sequencing. Front. Microbiol. 2012, 3, 277. [Google Scholar] [CrossRef]
- Ying, T.; Prabakaran, P.; Du, L.; Shi, W.; Feng, Y.; Wang, Y.; Wang, L.; Li, W.; Jiang, S.; Dimitrov, D.S.; et al. Junctional and allele-specific residues are critical for MERS-CoV neutralization by an exceptionally potent germline-like antibody. Nat. Commun. 2015, 6, 8223. [Google Scholar] [CrossRef] [PubMed]
- Pappas, L.; Foglierini, M.; Piccoli, L.; Kallewaard, N.L.; Turrini, F.; Silacci, C.; Fernandez-Rodriguez, B.; Agatic, G.; Giacchetto-Sasselli, I.; Pellicciotta, G.; et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 2014, 516, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Balint, R.F.; Larrick, J.W. Antibody engineering by parsimonious mutagenesis. Gene 1993, 137, 109–118. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Chapter 5: Antibodies and Antigens. In Cellular and Molecular Immunology, 9th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- De Vlieger, D.; Ballegeer, M.; Rossey, I.; Schepens, B.; Saelens, X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies (Basel) 2018, 8, 1. [Google Scholar] [CrossRef]
- Desmyter, A.; Decanniere, K.; Muyldermans, S.; Wyns, L. Antigen specificity and high affinity binding provided by one single loop of a camel single-domain antibody. J. Biol. Chem. 2001, 276, 26285–26290. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Ruuls, R.C.; Nijman, I.J.; Niewold, T.A.; Frenken, L.G.; de Geus, B. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 2000, 37, 579–590. [Google Scholar] [CrossRef]
- De Greve, H.; Virdi, V.; Bakshi, S.; Depicker, A. Simplified monomeric VHH-Fc antibodies provide new opportunities for passive immunization. Curr. Opin. Biotechnol. 2020, 61, 96–101. [Google Scholar] [CrossRef]
- Hudson, P.J.; Kortt, A.A. High avidity scFv multimers; diabodies and triabodies. J. Immunol. Methods 1999, 231, 177–189. [Google Scholar] [CrossRef]
- Chaisri, U.; Chaicumpa, W. Evolution of Therapeutic Antibodies, Influenza Virus Biology, Influenza, and Influenza Immunotherapy. Biomed. Res. Int. 2018, 2018, 9747549. [Google Scholar] [CrossRef] [PubMed]
- Vanlandschoot, P.; Stortelers, C.; Beirnaert, E.; Ibanez, L.I.; Schepens, B.; Depla, E.; Saelens, X. Nanobodies(R): new ammunition to battle viruses. Antiviral. Res. 2011, 92, 389–407. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, J.A.; Law, D.A. Nanobodies - from llamas to therapeutic proteins. Drug Discov. Today Technol. 2010, 7, e95–e146. [Google Scholar] [CrossRef] [PubMed]
- Gainkam, L.O.; Huang, L.; Caveliers, V.; Keyaerts, M.; Hernot, S.; Vaneycken, I.; Vanhove, C.; Revets, H.; De Baetselier, P.; Lahoutte, T. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J. Nucl. Med. 2008, 49, 788–795. [Google Scholar] [CrossRef]
- Mejias, M.P.; Hiriart, Y.; Lauche, C.; Fernandez-Brando, R.J.; Pardo, R.; Bruballa, A.; Ramos, M.V.; Goldbaum, F.A.; Palermo, M.S.; Zylberman, V. Development of camelid single chain antibodies against Shiga toxin type 2 (Stx2) with therapeutic potential against Hemolytic Uremic Syndrome (HUS). Sci. Rep. 2016, 6, 24913. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef]
- Tassano, M.R.; Audicio, P.F.; Gambini, J.P.; Fernandez, M.; Damian, J.P.; Moreno, M.; Chabalgoity, J.A.; Alonso, O.; Benech, J.C.; Cabral, P. Development of 99mTc(CO)(3)-dendrimer-FITC for cancer imaging. Bioorg Med. Chem. Lett. 2011, 21, 5598–5601. [Google Scholar] [CrossRef]
- Tabrizi, M.A.; Tseng, C.M.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Huang, C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr. Opin. Biotechnol. 2009, 20, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Rotman, M.; Welling, M.M.; van den Boogaard, M.L.; Moursel, L.G.; van der Graaf, L.M.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. Fusion of hIgG1-Fc to 111In-anti-amyloid single domain antibody fragment VHH-pa2H prolongs blood residential time in APP/PS1 mice but does not increase brain uptake. Nucl. Med. Biol. 2015, 42, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.M.; Ibanez, L.I.; Van den Hoecke, S.; De Baets, S.; Smet, A.; Roose, K.; Schepens, B.; Descamps, F.J.; Fiers, W.; Muyldermans, S.; et al. Single-domain antibodies targeting neuraminidase protect against an H5N1 influenza virus challenge. J. Virol. 2014, 88, 8278–8296. [Google Scholar] [CrossRef] [PubMed]
- Saerens, D.; Ghassabeh, G.H.; Muyldermans, S. Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 2008, 8, 600–608. [Google Scholar] [CrossRef]
- Shriver-Lake, L.C.; Zabetakis, D.; Goldman, E.R.; Anderson, G.P. Evaluation of anti-botulinum neurotoxin single domain antibodies with additional optimization for improved production and stability. Toxicon 2017, 135, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent Llama Single-Domain Antibody Fragments against Tumor Necrosis Factor Have Picomolar Potencies due to Intramolecular Interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef]
- Benhaim, M.A.; Mangala Prasad, V.; Garcia, N.K.; Guttman, M.; Lee, K.K. Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Science Advances 2020, 6, eaaz8822. [Google Scholar] [CrossRef]
- Floyd, D.L.; Ragains, J.R.; Skehel, J.J.; Harrison, S.C.; van Oijen, A.M. Single-particle kinetics of influenza virus membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 15382–15387. [Google Scholar] [CrossRef]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed]
- Shaker, G.H. Evaluation of antidiphtheria toxin nanobodies. Nanotechnol. Sci. Appl. 2010, 3, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Chiba, J.; Akashi-Takamura, S. Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection. Vaccines 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Bah, M.A.; Weiner, D.B. In Vivo Delivery of Nucleic Acid-Encoded Monoclonal Antibodies. BioDrugs 2020, 10, 1–21. [Google Scholar] [CrossRef]
- Sanders, J.W.; Ponzio, T.A. Vectored immunoprophylaxis: an emerging adjunct to traditional vaccination. Trop. Dis. Travel. Med. Vaccines 2017, 3, 3. [Google Scholar] [CrossRef]
- Hollevoet, K.; Declerck, P.J. State of play and clinical prospects of antibody gene transfer. J. Transl Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Yamazaki, T.; Nagashima, M.; Ninomiya, D.; Arai, Y.; Teshima, Y.; Fujimoto, A.; Ainai, A.; Hasegawa, H.; Chiba, J. Passive immune-prophylaxis against influenza virus infection by the expression of neutralizing anti-hemagglutinin monoclonal antibodies from plasmids. Jpn. J. Infect. Dis. 2011, 64, 40–49. [Google Scholar]
- Yamazaki, T.; Nagashima, M.; Ninomiya, D.; Ainai, A.; Fujimoto, A.; Ichimonji, I.; Takagi, H.; Morita, N.; Murotani, K.; Hasegawa, H.; et al. Neutralizing Antibodies Induced by Gene-Based Hydrodynamic Injection Have a Therapeutic Effect in Lethal Influenza Infection. Front. Immunol. 2018, 9, 47. [Google Scholar] [CrossRef]
- Elliott, S.T.C.; Kallewaard, N.L.; Benjamin, E.; Wachter-Rosati, L.; McAuliffe, J.M.; Patel, A.; Smith, T.R.F.; Schultheis, K.; Park, D.H.; Flingai, S.; et al. DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. NPJ Vaccines 2017, 2, 18. [Google Scholar] [CrossRef]
- Limberis, M.P.; Adam, V.S.; Wong, G.; Gren, J.; Kobasa, D.; Ross, T.M.; Kobinger, G.P.; Tretiakova, A.; Wilson, J.M. Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci. Transl. Med. 2013, 5, 187ra172. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Alves, C. “Blood is a very unusual fluid”. Nat. Immunol. 2016, 17, S5-S5. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E. Emil von Behring: translational medicine at the dawn of immunology. Nat. Rev. Immunol. 2017, 17, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 2019, 19, 383–397. [Google Scholar] [CrossRef]
- Tamura, S.; Ainai, A.; Suzuki, T.; Kurata, T.; Hasegawa, H. Intranasal Inactivated Influenza Vaccines: A Reasonable Approach to Improve the Efficacy of Influenza Vaccine? Jpn J. Infect. Dis. 2016, 69, 165–179. [Google Scholar] [CrossRef]
- Suzuki, T.; Ainai, A.; Hasegawa, H. Functional and structural characteristics of secretory IgA antibodies elicited by mucosal vaccines against influenza virus. Vaccine 2017, 35, 5297–5302. [Google Scholar] [CrossRef]
- Tamura, S.; Funato, H.; Nagamine, T.; Aizawa, C.; Kurata, T. Effectiveness of cholera toxin B subunit as an adjuvant for nasal influenza vaccination despite pre-existing immunity to CTB. Vaccine 1989, 7, 503–505. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Iwasaki, T.; Asanuma, H.; Sato, Y.; Sata, T.; Aizawa, C.; Kurata, T.; Tamura, S. Effects of intranasal administration of cholera toxin (or Escherichia coli heat-labile enterotoxin) B subunits supplemented with a trace amount of the holotoxin on the brain. Vaccine 2001, 19, 1652–1660. [Google Scholar] [CrossRef]
- Watanabe, I.; Ross, T.M.; Tamura, S.; Ichinohe, T.; Ito, S.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T.; Sata, T.; et al. Protection against influenza virus infection by intranasal administration of C3d-fused hemagglutinin. Vaccine 2003, 21, 4532–4538. [Google Scholar] [CrossRef]
- Ichinohe, T.; Watanabe, I.; Ito, S.; Fujii, H.; Moriyama, M.; Tamura, S.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T.; et al. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J. Virol. 2005, 79, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- Ainai, A.; Tamura, S.; Suzuki, T.; van Riet, E.; Ito, R.; Odagiri, T.; Tashiro, M.; Kurata, T.; Hasegawa, H. Intranasal vaccination with an inactivated whole influenza virus vaccine induces strong antibody responses in serum and nasal mucus of healthy adults. Hum. Vaccin. Immunother. 2013, 9, 1962–1970. [Google Scholar] [CrossRef] [PubMed]
- Ainai, A.; van Riet, E.; Ito, R.; Ikeda, K.; Senchi, K.; Suzuki, T.; Tamura, S.I.; Asanuma, H.; Odagiri, T.; Tashiro, M.; et al. Human immune responses elicited by an intranasal inactivated H5 influenza vaccine. Microbiol. Immunol. 2020, 64, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef]
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Chapter 14: Specialized Immunity at Epithelial Barriers and in Immune Privileged Tissues. In Cellular and Molecular Immunology, 9th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Terauchi, Y.; Sano, K.; Ainai, A.; Saito, S.; Taga, Y.; Ogawa-Goto, K.; Tamura, S.I.; Odagiri, T.; Tashiro, M.; Fujieda, M.; et al. IgA polymerization contributes to efficient virus neutralization on human upper respiratory mucosa after intranasal inactivated influenza vaccine administration. Hum. Vaccin Immunother. 2018, 14, 1351–1361. [Google Scholar] [CrossRef]
- Chintalacharuvu, K.R.; Morrison, S.L. Production of secretory immunoglobulin A by a single mammalian cell. Proc. Natl. Acad. Sci. USA 1997, 94, 6364–6368. [Google Scholar] [CrossRef]
- Berdoz, J.; Blanc, C.T.; Reinhardt, M.; Kraehenbuhl, J.P.; Corthesy, B. In vitro comparison of the antigen-binding and stability properties of the various molecular forms of IgA antibodies assembled and produced in CHO cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3029–3034. [Google Scholar] [CrossRef]
- Seibert, C.W.; Rahmat, S.; Krause, J.C.; Eggink, D.; Albrecht, R.A.; Goff, P.H.; Krammer, F.; Duty, J.A.; Bouvier, N.M.; Garcia-Sastre, A.; et al. Recombinant IgA is sufficient to prevent influenza virus transmission in guinea pigs. J. Virol. 2013, 87, 7793–7804. [Google Scholar] [CrossRef]
- Saito, S.; Sano, K.; Suzuki, T.; Ainai, A.; Taga, Y.; Ueno, T.; Tabata, K.; Saito, K.; Wada, Y.; Ohara, Y.; et al. IgA tetramerization improves target breadth but not peak potency of functionality of anti-influenza virus broadly neutralizing antibody. PLoS Pathog. 2019, 15, e1007427. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, M.; Chen, Y.; Zhang, L.; Wang, G.; Chen, J.; Chen, S.; Li, Z.; Wei, F.; Chen, J.; et al. An IgM antibody targeting the receptor binding site of influenza B blocks viral infection with great breadth and potency. Theranostics 2019, 9, 210–231. [Google Scholar] [CrossRef] [PubMed]
- Ido, S.; Kimiya, H.; Kobayashi, K.; Kominami, H.; Matsushige, K.; Yamada, H. Immunoactive two-dimensional self-assembly of monoclonal antibodies in aqueous solution revealed by atomic force microscopy. Nat. Mater. 2014, 13, 264–270. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).