ELISA-Based Assay for Studying Major and Minor Group Rhinovirus–Receptor Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Propagation of RV Strains
2.2. Purification of RV Strains
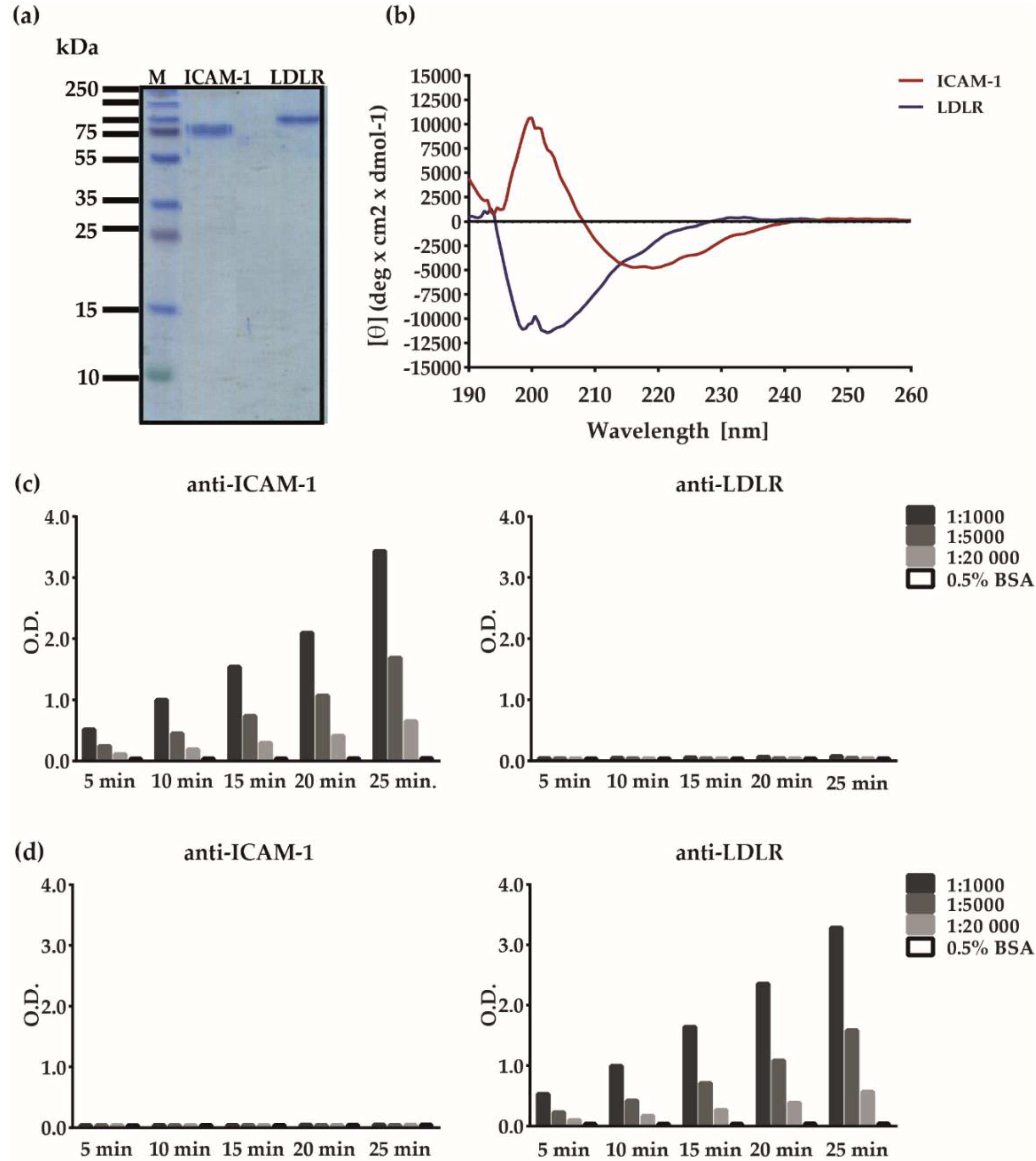
2.3. Analysis of Virus Preparations
2.4. Analysis of Recombinant Human Receptor Proteins
2.5. Receptor- and Virus-Specific Antibodies
2.6. ELISA-Based Virus–Receptor Interaction Assay
2.7. ELISA-Based Inhibition of Virus–Receptor Interaction by Anti-Receptor Antibodies, Recombinant Receptors, or RV-Specific Antibodies
2.8. Cell Culture-Based Virus Neutralization Assay
3. Results
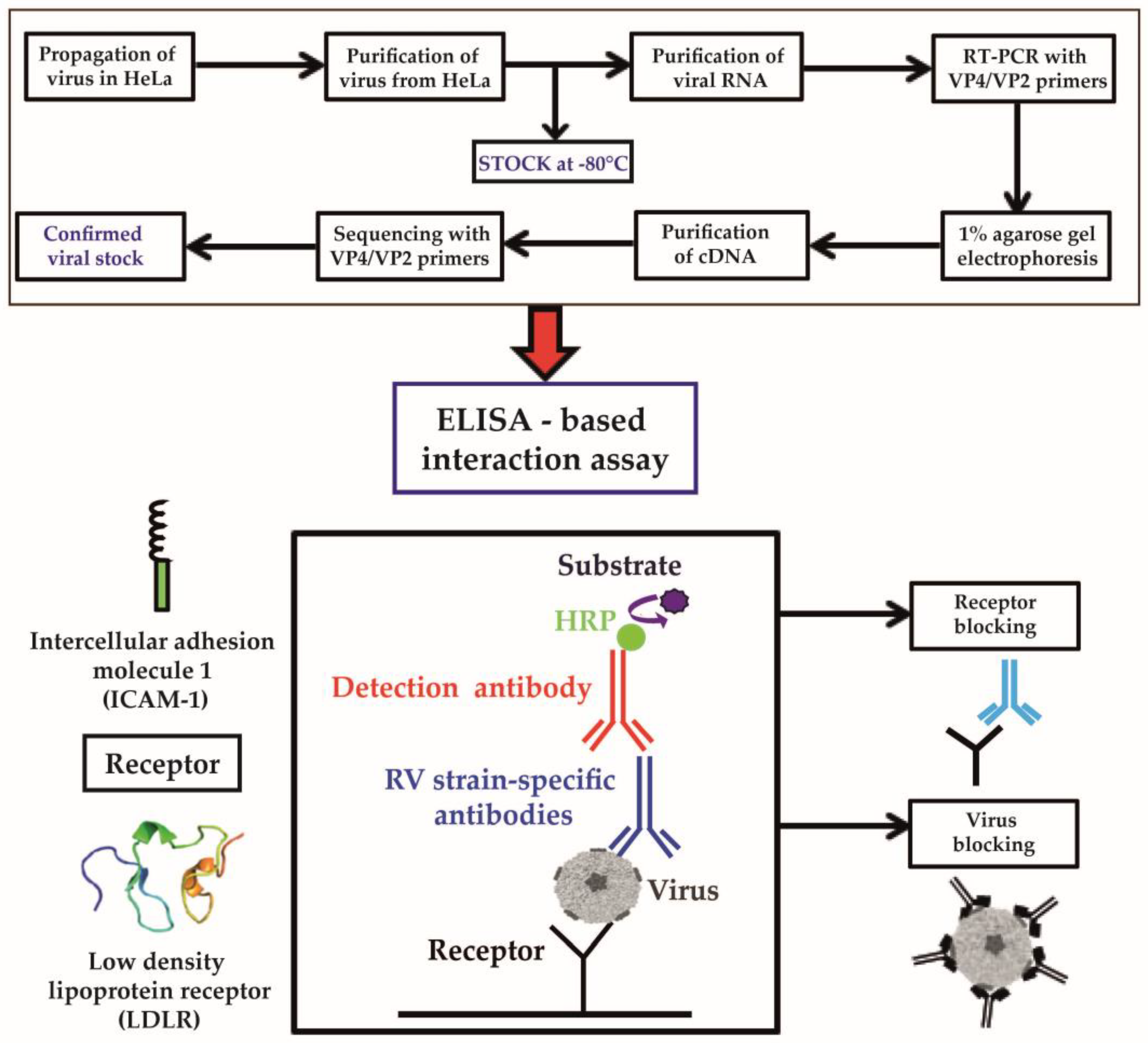
3.1. Development of an ELISA-Based RV–Receptor Interaction Assay
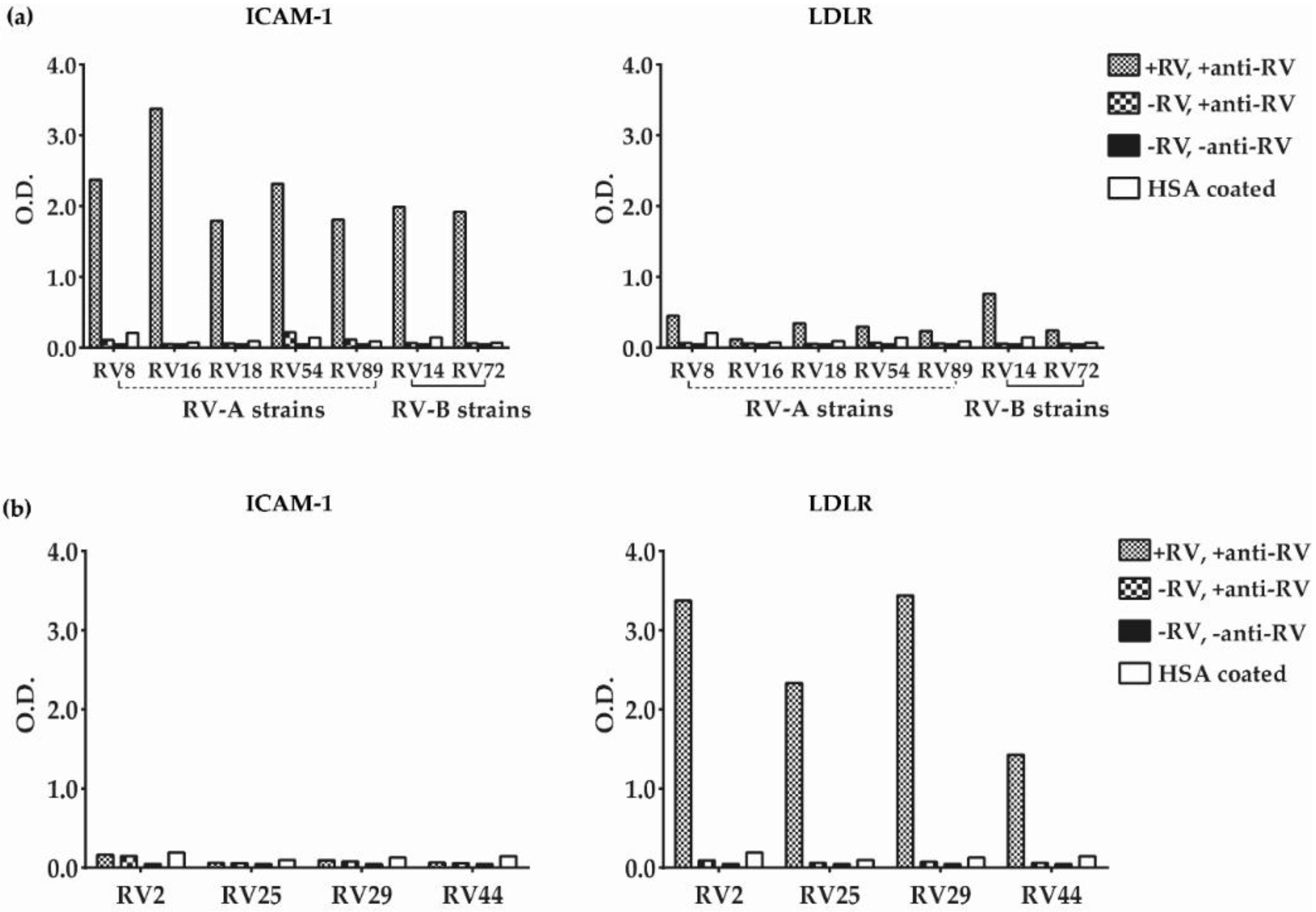
3.2. Major and Minor Group RV Strains Bind Specifically to Their Corresponding Receptor Proteins in the ELISA Assay
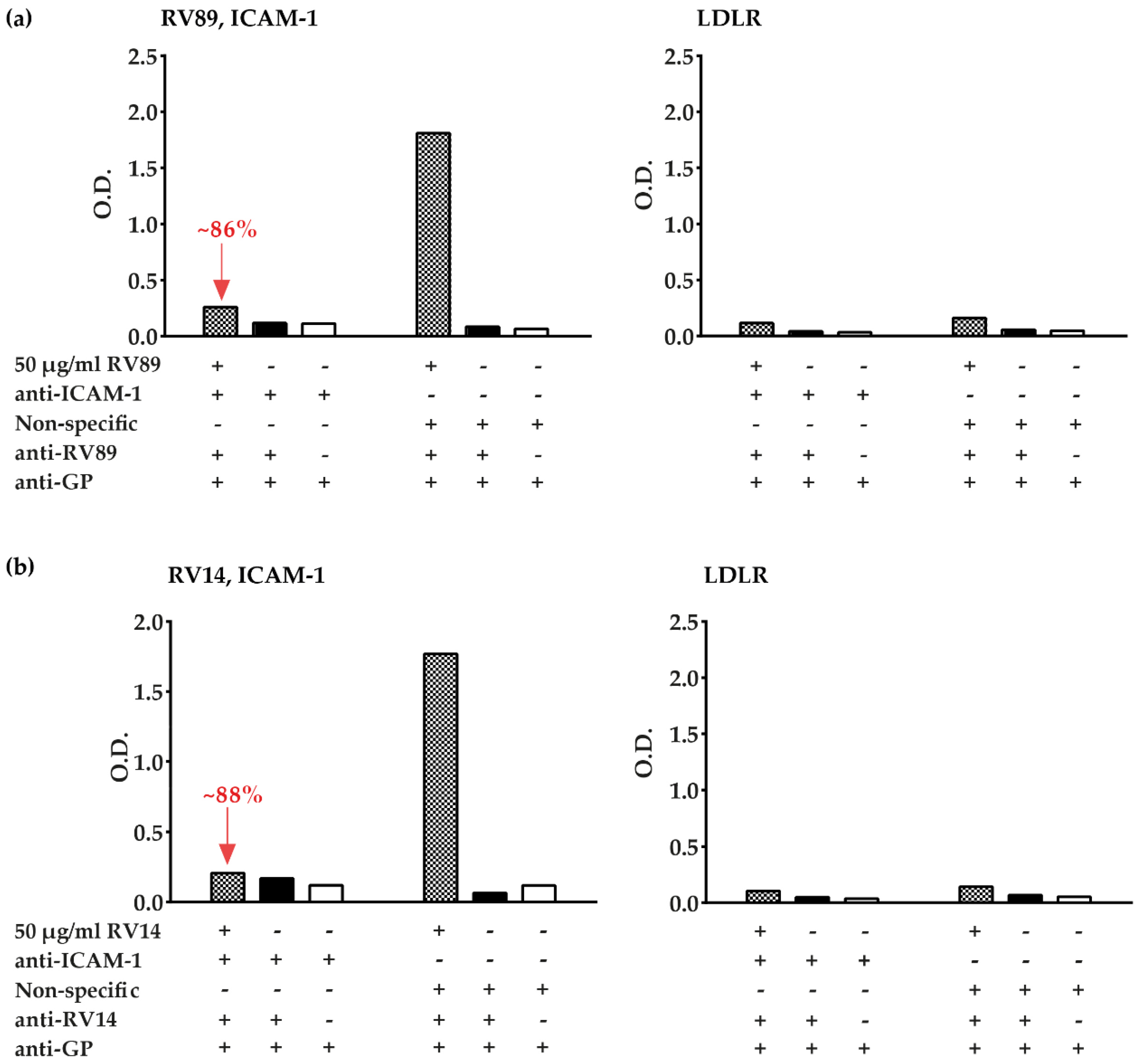
3.3. Polyclonal Antibodies Produced against ICAM-1 Strongly and Specifically Inhibit Major Group RV Binding to ICAM-1
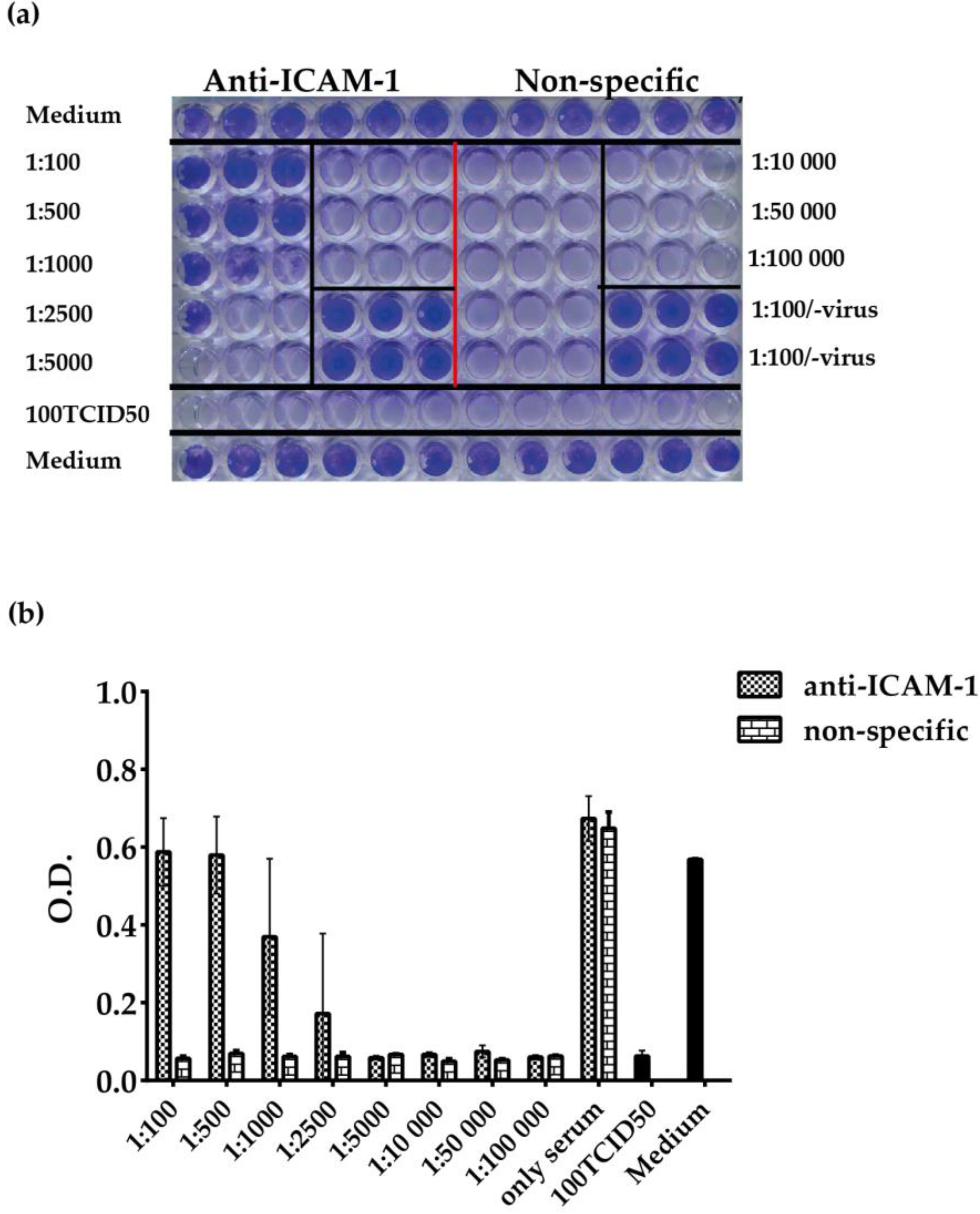
3.4. ELISA-Based RV–Receptor Interaction is More Sensitive in Showing Anti-ICAM-1 Effects than an Established Cell Culture-Based Virus Neutralization Assay
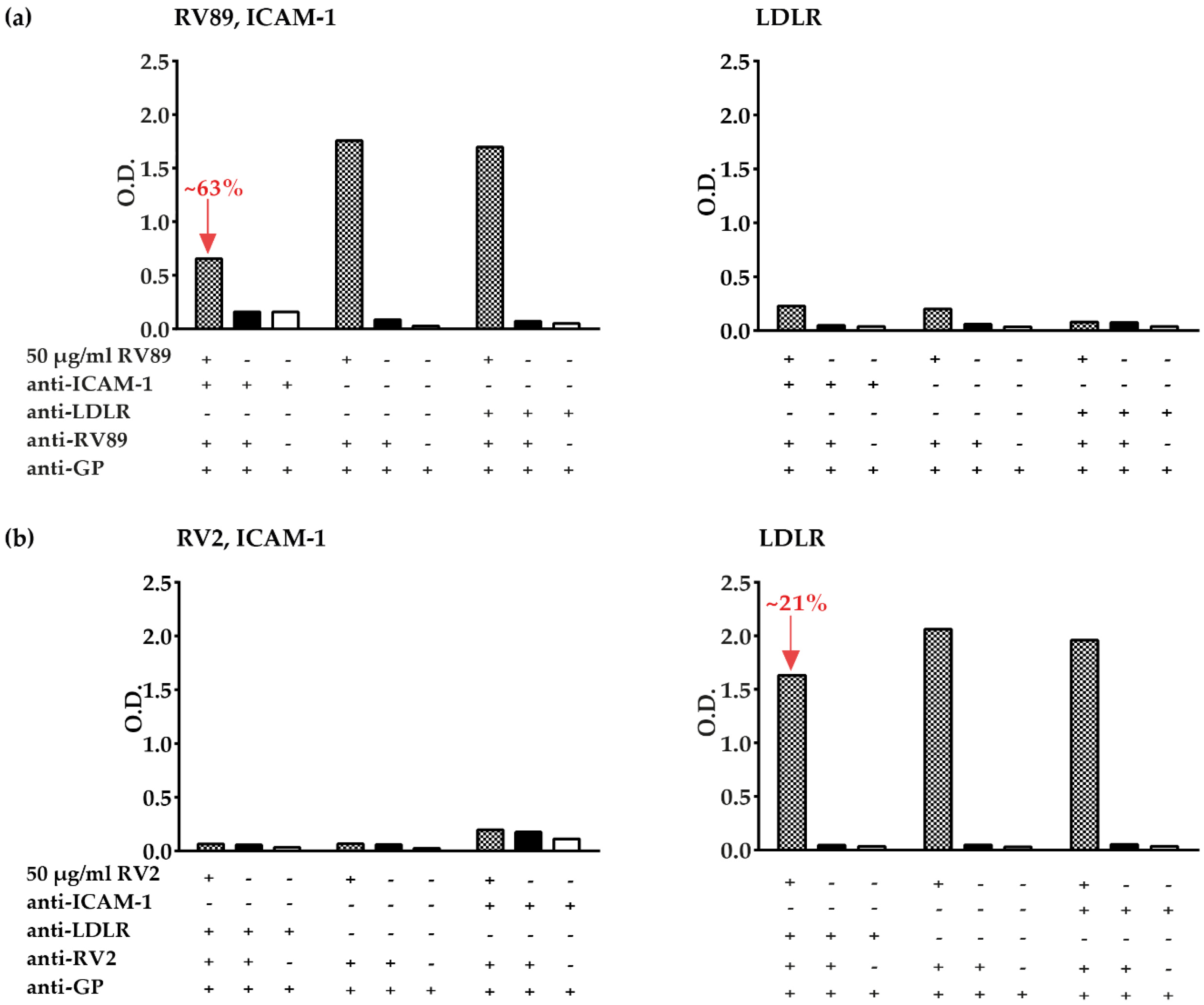
3.5. Inhibition of Major and Minor Group RV Binding to Their Receptors with Anti-ICAM-1 and Anti-LDLR Antibodies
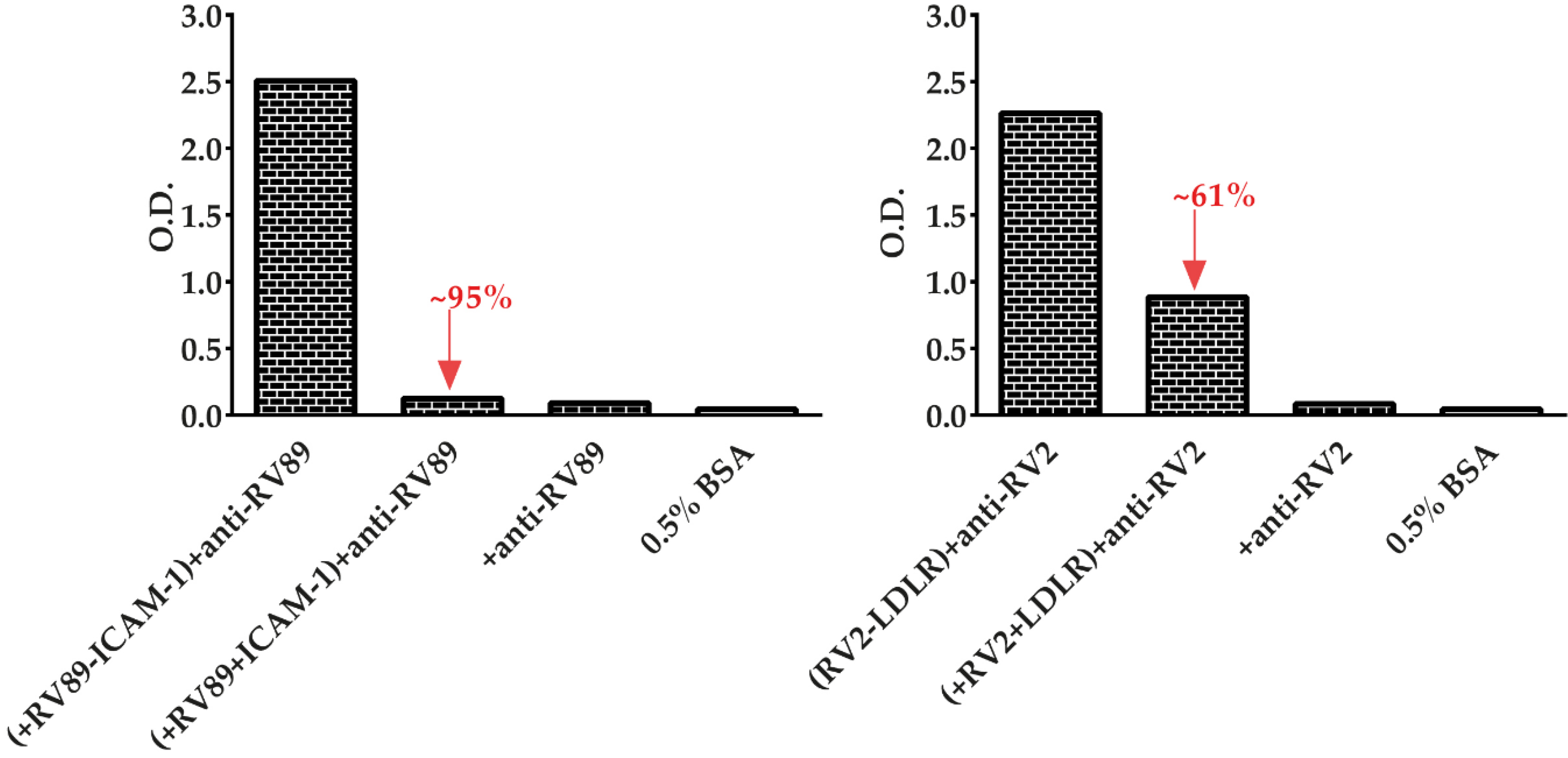
3.6. Soluble Recombinant Receptors Specifically Inhibit Major and Minor RV Binding to Their Receptors
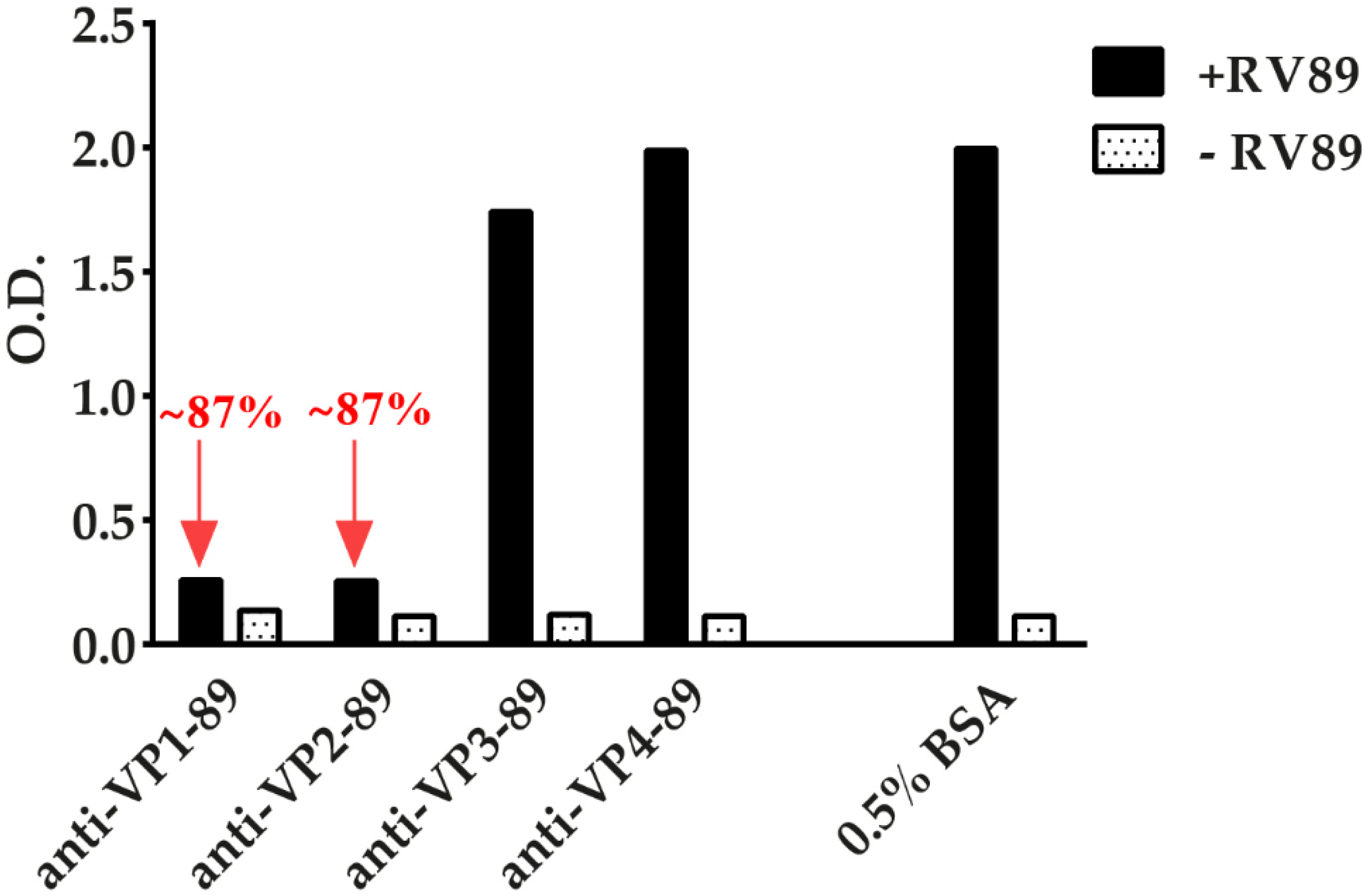
3.7. ELISA-Based RV–Receptor Interaction Assay Allows Identifying Virus-Neutralizing Antibodies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edwards, M.R.; Bartlett, N.W.; Hussell, T.; Openshaw, P.; Johnston, S.L. The microbiology of asthma. Nat. Rev. Microbiol. 2012, 10, 459–471. [Google Scholar] [CrossRef]
- Hansel, T.T.; Johnston, S.L.; Openshaw, P.J. Microbes and mucosal immune responses in asthma. Lancet 2013, 381, 861–873. [Google Scholar] [CrossRef]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Niespodziana, K.; Borochova, K.; Pazderova, P.; Schlederer, T.; Astafyeva, N.; Baranovskaya, T.; Barbouche, M.-R.; Beltiukov, E.; Berger, A.; Borzova, E. Towards personalization of asthma treatment according to trigger factors. J. Allergy Clin. Immunol. 2020, 145, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Gorbalenya, A.E.; Harvala, H.; Hovi, T.; Knowles, N.J.; Lindberg, A.M.; Oberste, M.S.; Palmenberg, A.C.; Reuter, G.; Skern, T. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020, 165, 793–797. [Google Scholar] [CrossRef]
- Greve, J.M.; Davis, G.; Meyer, A.M.; Forte, C.P.; Yost, S.C.; Marlor, C.W.; Kamarck, M.E.; McClelland, A. The major human rhinovirus receptor is ICAM-1. Cell 1989, 56, 839–847. [Google Scholar] [CrossRef]
- Hofer, F.; Gruenberger, M.; Kowalski, H.; Machat, H.; Huettinger, M.; Kuechler, E.; Blaas, D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1839–1842. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Watters, K.; Ashraf, S.; Griggs, T.F.; Devries, M.K.; Jackson, D.J.; Palmenberg, A.C.; Gern, J.E. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc. Natl. Acad. Sci. USA 2015, 112, 5485–5490. [Google Scholar] [CrossRef]
- Nakauchi, M.; Nagata, N.; Takayama, I.; Saito, S.; Kubo, H.; Kaida, A.; Oba, K.; Odagiri, T.; Kageyama, T. Propagation of Rhinovirus C in Differentiated Immortalized Human Airway HBEC3-KT Epithelial Cells. Viruses 2019, 11, 216. [Google Scholar] [CrossRef]
- Johnston, S.L.; Sanderson, G.; Pattemore, P.K.; Smith, S.; Bardin, P.G.; Bruce, C.B.; Lambden, P.R.; Tyrrell, D.A.; Holgate, S.T. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J.Clin. Mmicrobiol. 1993, 31, 111–117. [Google Scholar] [CrossRef]
- Lemanske, R.F., Jr.; Dick, E.C.; Swenson, C.A.; Vrtis, R.F.; Busse, W.W. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J. Clin. Investig. 1989, 83, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Heymann, P.W.; Kennedy, J.L. Rhinovirus-induced asthma exacerbations during childhood: The importance of understanding the atopic status of the host. J. Allergy Clin. Immunol. 2012, 130, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- Soto-Quiros, M.; Avila, L.; Platts-Mills, T.A.E.; Hunt, J.F.; Erdman, D.D.; Carper, H.; Murphy, D.D.; Odio, S.; James, H.R.; Patrie, J.T. High titers of IgE antibody to dust mite allergen and risk for wheezing among asthmatic children infected with rhinovirus. J. Allergy Clin. Immunol. 2012, 129, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.; Khaitov, M.; Jackson, D.J.; Traub, S.; Trujillo-Torralbo, M.B.; Kudlay, D.A.; Dvornikov, A.S.; Del-Rosario, A.; Valenta, R.; Stanciu, L.A.; et al. M1-like macrophages are potent producers of anti-viral interferons and M1-associated marker-positive lung macrophages are decreased during rhinovirus-induced asthma exacerbations. EBioMedicine 2020, 54, 102734. [Google Scholar] [CrossRef] [PubMed]
- Niespodziana, K.; Napora, K.; Cabauatan, C.; Focke-Tejkl, M.; Keller, W.; Niederberger, V.; Tsolia, M.; Christodoulou, I.; Papadopoulos, N.G.; Valenta, R. Misdirected antibody responses against an N-terminal epitope on human rhinovirus VP1 as explanation for recurrent RV infections. FASEB J. 2012, 26, 1001–1008. [Google Scholar] [CrossRef]
- Niespodziana, K.; Cabauatan, C.R.; Jackson, D.J.; Gallerano, D.; Trujillo-Torralbo, B.; Del Rosario, A.; Mallia, P.; Valenta, R.; Johnston, S.L. Rhinovirus-induced VP1-specific Antibodies are Group-specific and Associated with Severity of Respiratory Symptoms. EBioMedicine 2015, 2, 64–70. [Google Scholar] [CrossRef]
- Niespodziana, K.; Stenberg-Hammar, K.; Megremis, S.; Cabauatan, C.R.; Napora-Wijata, K.; Vacal, P.C.; Gallerano, D.; Lupinek, C.; Ebner, D.; Schlederer, T.; et al. PreDicta chip-based high resolution diagnosis of rhinovirus-induced wheeze. Nat. Commun. 2018, 9, 2382. [Google Scholar] [CrossRef]
- Stenberg-Hammar, K.; Niespodziana, K.; Soderhall, C.; James, A.; Cabauatan, C.R.; Konradsen, J.R.; Melen, E.; van Hage, M.; Valenta, R.; Hedlin, G. Rhinovirus-specific antibody responses in preschool children with acute wheeze reflect severity of respiratory symptoms. Allergy 2016, 71, 1728–1735. [Google Scholar] [CrossRef]
- Megremis, S.; Niespodziana, K.; Cabauatan, C.; Xepapadaki, P.; Kowalski, M.L.; Jartti, T.; Bachert, C.; Finotto, S.; West, P.; Stamataki, S.; et al. Rhinovirus Species-Specific Antibodies Differentially Reflect Clinical Outcomes in Health and Asthma. Am. J. Respir. Crit. Care Med. 2018, 198, 1490–1499. [Google Scholar] [CrossRef]
- Conant, R.M.; Hamparian, V.V. Rhinoviruses: Basis for a Numbering System: 1. HeLa Cells for Propagation and Serologic Procedures. J. Immun. 1968, 100, 107–113. [Google Scholar]
- Bartlett, N.W.; Singanayagam, A.; Johnston, S.L. Mouse models of rhinovirus infection and airways disease. In Rhinoviruses; Springer: Heidelberg, Germany, 2015; pp. 181–188. [Google Scholar]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Distance Methods for Inferring Phylogenies: A Justification. Evolution 1984, 38, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Gallerano, D.; Devanaboyina, S.C.; Swoboda, I.; Linhart, B.; Mittermann, I.; Keller, W.; Valenta, R. Biophysical characterization of recombinant HIV-1 subtype C virus infectivity factor. Amino Acids 2011, 40, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Edlmayr, J.; Niespodziana, K.; Linhart, B.; Focke-Tejkl, M.; Westritschnig, K.; Scheiblhofer, S.; Stoecklinger, A.; Kneidinger, M.; Valent, P.; Campana, R.; et al. A combination vaccine for allergy and rhinovirus infections based on rhinovirus-derived surface protein VP1 and a nonallergenic peptide of the major timothy grass pollen allergen Phl p 1. J. Immunol. 2009, 182, 6298–6306. [Google Scholar] [CrossRef]
- Edlmayr, J.; Niespodziana, K.; Popow-Kraupp, T.; Krzyzanek, V.; Focke-Tejkl, M.; Blaas, D.; Grote, M.; Valenta, R. Antibodies induced with recombinant VP1 from human rhinovirus exhibit cross-neutralisation. Eur. Respir. J. 2011, 37, 44–52. [Google Scholar] [CrossRef]
- Watters, K.; Palmenberg, A.C. CDHR3 extracellular domains EC1-3 mediate rhinovirus C interaction with cells and as recombinant derivatives, are inhibitory to virus infection. PLoS Pathog. 2018, 14, e1007477. [Google Scholar] [CrossRef]
- Douglas, R.G., Jr.; Rossen, R.D.; Butler, W.T.; Couch, R.B. Rhinovirus neutralizing antibody in tears, parotid saliva, nasal secretions and serum. J. Immunol. 1967, 99, 297–303. [Google Scholar]
- Schild, G.C.; Hobson, D. Neutralizing Antibody Levels in Human Sera with Two Strains of Common Cold Virus. Br. J. Exp. Pathol. 1962, 43, 288. [Google Scholar]
- Dong, Y.; Liu, Y.; Jiang, W.; Smith, T.J.; Xu, Z.; Rossmann, M.G. Antibody-induced uncoating of human rhinovirus B14. Proc. Natl. Acad. Sci. USA 2017, 114, 8017–8022. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.H.; Kolatkar, P.R.; Oliveira, M.A.; Cheng, R.H.; Greve, J.M.; McClelland, A.; Baker, T.S.; Rossmann, M.G. Structure of a human rhinovirus complexed with its receptor molecule. In Regulation of Gene Expression in Animal Viruses; Springer: Heidelberg, Germany, 1993; pp. 1–12. [Google Scholar]
- Charles, C.H.; Luo, G.X.; Kohlstaedt, L.A.; Morantte, I.G.; Gorfain, E.; Cao, L.; Williams, J.H.; Fang, F. Prevention of human rhinovirus infection by multivalent fab molecules directed against ICAM-1. Antimicrob. Agents Chemother. 2003, 47, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yu, M. Viral receptor blockage by multivalent recombinant antibody fusion proteins: Inhibiting human rhinovirus (HRV) infection with CFY196. J. Antimicrob. Chemother. 2004, 53, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Kohlstaedt, L.A.; Charles, C.H.; Gorfain, E.; Morantte, I.; Williams, J.H.; Fang, F. Humanization of an anti-ICAM-1 antibody with over 50-fold affinity and functional improvement. J. Immunol. Methods 2003, 275, 31–40. [Google Scholar] [CrossRef]
- McClelland, A.; DeBear, J.; Yost, S.C.; Meyer, A.M.; Marlor, C.W.; Greve, J.M. Identification of monoclonal antibody epitopes and critical residues for rhinovirus binding in domain 1 of intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 7993–7997. [Google Scholar] [CrossRef]
- Traub, S.; Nikonova, A.; Carruthers, A.; Dunmore, R.; Vousden, K.A.; Gogsadze, L.; Hao, W.; Zhu, Q.; Bernard, K.; Zhu, J.; et al. An anti-human ICAM-1 antibody inhibits rhinovirus-induced exacerbations of lung inflammation. PLoS Pathog. 2013, 9, e1003520. [Google Scholar] [CrossRef] [PubMed]
- Greve, J.M.; Forte, C.P.; Marlor, C.W.; Meyer, A.M.; Hoover-Litty, H.; Wunderlich, D.; McClelland, A. Mechanisms of receptor-mediated rhinovirus neutralization defined by two soluble forms of ICAM-1. J. Virol. 1991, 65, 6015–6023. [Google Scholar] [CrossRef]
- Crump, C.E.; Arruda, E.; Hayden, F.G. Comparative antirhinoviral activities of soluble intercellular adhesion molecule-1 (sICAM-1) and chimeric ICAM-1/immunoglobulin A molecule. Antimicrob. Agents Chemother. 1994, 38, 1425–1427. [Google Scholar] [CrossRef]
- Martin, S.; Casasnovas, J.M.; Staunton, D.E.; Springer, T.A. Efficient neutralization and disruption of rhinovirus by chimeric ICAM-1/immunoglobulin molecules. J. Virol. 1993, 67, 3561–3568. [Google Scholar] [CrossRef]
- Huguenel, E.D.; Cohn, D.; Dockum, D.P.; Greve, J.M.; Fournel, M.A.; Hammond, L.; Irwin, R.; Mahoney, J.; McClelland, A.; Muchmore, E.; et al. Prevention of rhinovirus infection in chimpanzees by soluble intercellular adhesion molecule-1. Am. J. Respir. Crit. Care Med. 1997, 155, 1206–1210. [Google Scholar] [CrossRef]
- Turner, R.B.; Wecker, M.T.; Pohl, G.; Witek, T.J.; McNally, E.; St George, R.; Winther, B.; Hayden, F.G. Efficacy of tremacamra, a soluble intercellular adhesion molecule 1, for experimental rhinovirus infection: A randomized clinical trial. JAMA 1999, 281, 1797–1804. [Google Scholar] [CrossRef]
- Marlovits, T.C.; Abrahamsberg, C.; Blaas, D. Very-low-density lipoprotein receptor fragment shed from HeLa cells inhibits human rhinovirus infection. J. Virol. 1998, 72, 10246–10250. [Google Scholar] [CrossRef] [PubMed]
- Marlovits, T.C.; Zechmeister, T.; Gruenberger, M.; Ronacher, B.; Schwihla, H.; Blaas, D. Recombinant soluble low density lipoprotein receptor fragment inhibits minor group rhinovirus infection in vitro. FASEB J. 1998, 12, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Nicodemou, A.; Petsch, M.; Konecsni, T.; Kremser, L.; Kenndler, E.; Casasnovas, J.M.; Blaas, D. Rhinovirus-stabilizing activity of artificial VLDL-receptor variants defines a new mechanism for virus neutralization by soluble receptors. FEBS Lett. 2005, 579, 5507–5511. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, G.; Russell, S.M.; Clarke, B.E.; Speller, S.A.; Trowbridge, M.; Vadolas, J. Neutralization epitopes of human rhinovirus type 2. J. Gen. Virol. 1990, 71 Pt 6, 1275–1282. [Google Scholar] [CrossRef]
- Barnett, P.V.; Rowlands, D.J.; Parry, N.R. Characterization of monoclonal antibodies raised against a synthetic peptide capable of inducing a neutralizing response to human rhinovirus type 2. J. Gen. Virol. 1993, 74 Pt 7, 1295–1302. [Google Scholar] [CrossRef]
- Hastings, G.Z.; Speller, S.A.; Francis, M.J. Neutralizing antibodies to human rhinovirus produced in laboratory animals and humans that recognize a linear sequence from VP2. J. Gen. Virol. 1990, 71, 3055–3059. [Google Scholar] [CrossRef]
- McCray, J.; Werner, G. Different rhinovirus serotypes neutralized by antipeptide antibodies. Nature 1987, 329, 736–738. [Google Scholar] [CrossRef]
- Molins, M.A.; Contreras, M.À.; Fita, I.; Pons, M. Solution conformation of an immunogenic peptide from HRV2: Comparison with the conformation found in a complex with a Fab fragment of an anti-HRV2 neutralizing antibody. J. Pept. Sci. 1998, 4, 101–110. [Google Scholar] [CrossRef]
- Sherry, B.; Mosser, A.G.; Colonno, R.J.; Rueckert, R.R. Use of monoclonal antibodies to identify four neutralization immunogens on a common cold picornavirus, human rhinovirus 14. J. Virol. 1986, 57, 246–257. [Google Scholar] [CrossRef]
- Skern, T.; Neubauer, C.; Frasel, L.; Gründler, P.; Sommergruber, W.; Zorn, M.; Kuechler, E.; Blaas, D. A neutralizing epitope on human rhinovirus type 2 includes amino acid residues between 153 and 164 of virus capsid protein VP2. J. Gen. Virol. 1987, 68, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Tormo, J.; Blaas, D.; Parry, N.R.; Rowlands, D.; Stuart, D.; Fita, I. Crystal structure of a human rhinovirus neutralizing antibody complexed with a peptide derived from viral capsid protein VP2. EMBO J. 1994, 13, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Glanville, N.; McLean, G.R.; Guy, B.; Lecouturier, V.; Berry, C.; Girerd, Y.; Gregoire, C.; Walton, R.P.; Pearson, R.M.; Kebadze, T.; et al. Cross-serotype immunity induced by immunization with a conserved rhinovirus capsid protein. PLoS Pathog. 2013, 9, e1003669. [Google Scholar] [CrossRef] [PubMed]
- Hewat, E.A.; Marlovits, T.C.; Blaas, D. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J. Virol. 1998, 72, 4396–4402. [Google Scholar] [CrossRef] [PubMed]
- Katpally, U.; Fu, T.M.; Freed, D.C.; Casimiro, D.R.; Smith, T.J. Antibodies to the buried N terminus of rhinovirus VP4 exhibit cross-serotypic neutralization. J. Virol. 2009, 83, 7040–7048. [Google Scholar] [CrossRef]
- Panjwani, A.; Asfor, A.S.; Tuthill, T.J. The conserved N-terminus of human rhinovirus capsid protein VP4 contains membrane pore-forming activity and is a target for neutralizing antibodies. J. Gen. Virol. 2016, 97, 3238. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pazderova, P.; Waltl, E.E.; Niederberger-Leppin, V.; Flicker, S.; Valenta, R.; Niespodziana, K. ELISA-Based Assay for Studying Major and Minor Group Rhinovirus–Receptor Interactions. Vaccines 2020, 8, 315. https://doi.org/10.3390/vaccines8020315
Pazderova P, Waltl EE, Niederberger-Leppin V, Flicker S, Valenta R, Niespodziana K. ELISA-Based Assay for Studying Major and Minor Group Rhinovirus–Receptor Interactions. Vaccines. 2020; 8(2):315. https://doi.org/10.3390/vaccines8020315
Chicago/Turabian StylePazderova, Petra, Eva E. Waltl, Verena Niederberger-Leppin, Sabine Flicker, Rudolf Valenta, and Katarzyna Niespodziana. 2020. "ELISA-Based Assay for Studying Major and Minor Group Rhinovirus–Receptor Interactions" Vaccines 8, no. 2: 315. https://doi.org/10.3390/vaccines8020315
APA StylePazderova, P., Waltl, E. E., Niederberger-Leppin, V., Flicker, S., Valenta, R., & Niespodziana, K. (2020). ELISA-Based Assay for Studying Major and Minor Group Rhinovirus–Receptor Interactions. Vaccines, 8(2), 315. https://doi.org/10.3390/vaccines8020315