Immunogenicity and Efficacy of Zika Virus Envelope Domain III in DNA, Protein, and ChAdOx1 Adenoviral-Vectored Vaccines

 , , ,
, , ,  and add
Show full author list
and add
Show full author list
Abstract
1. Introduction
2. Results
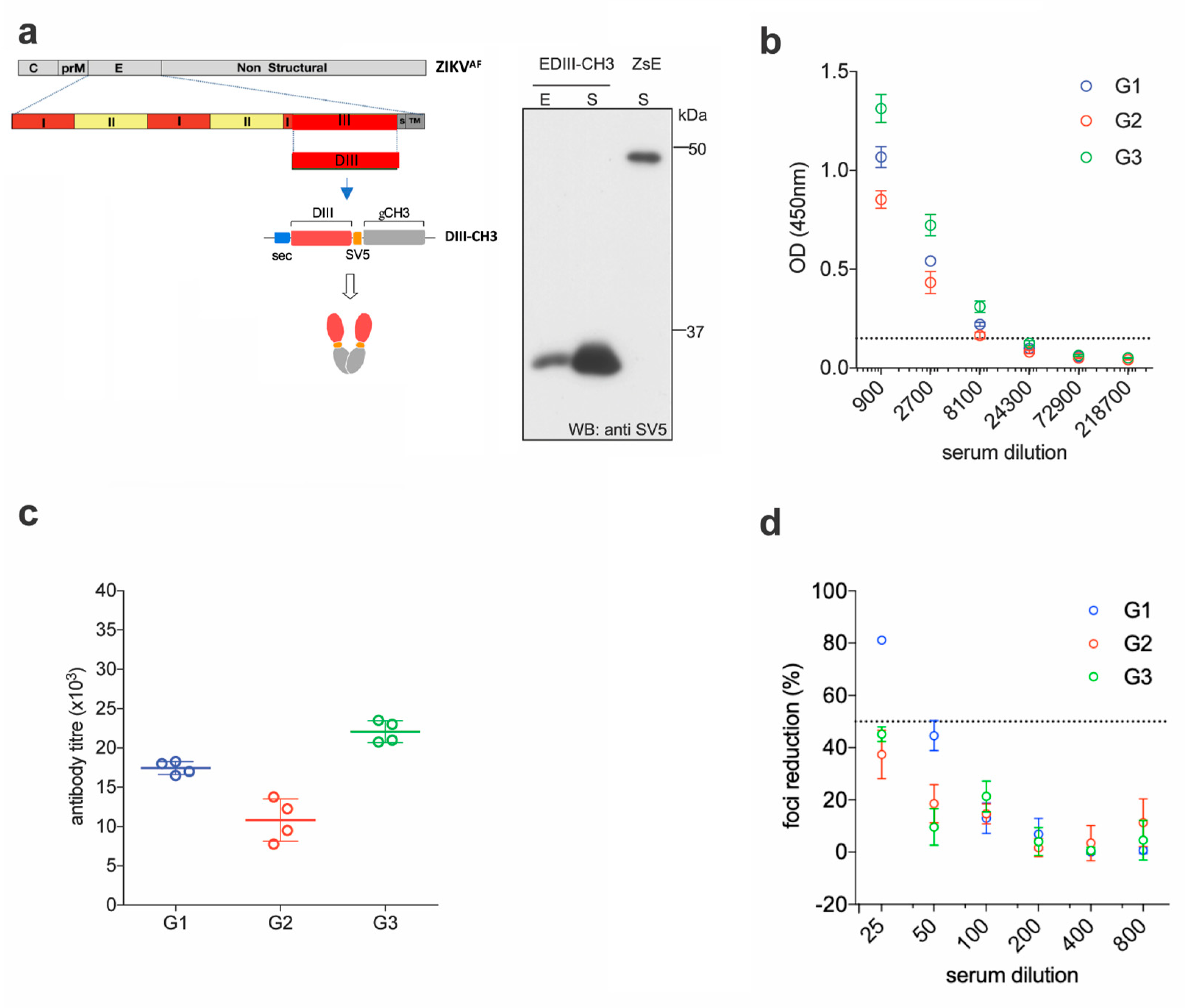
2.1. EDIII-CH3 DNA-Based Immunization Induces Poorly Neutralizing Antibody Responses
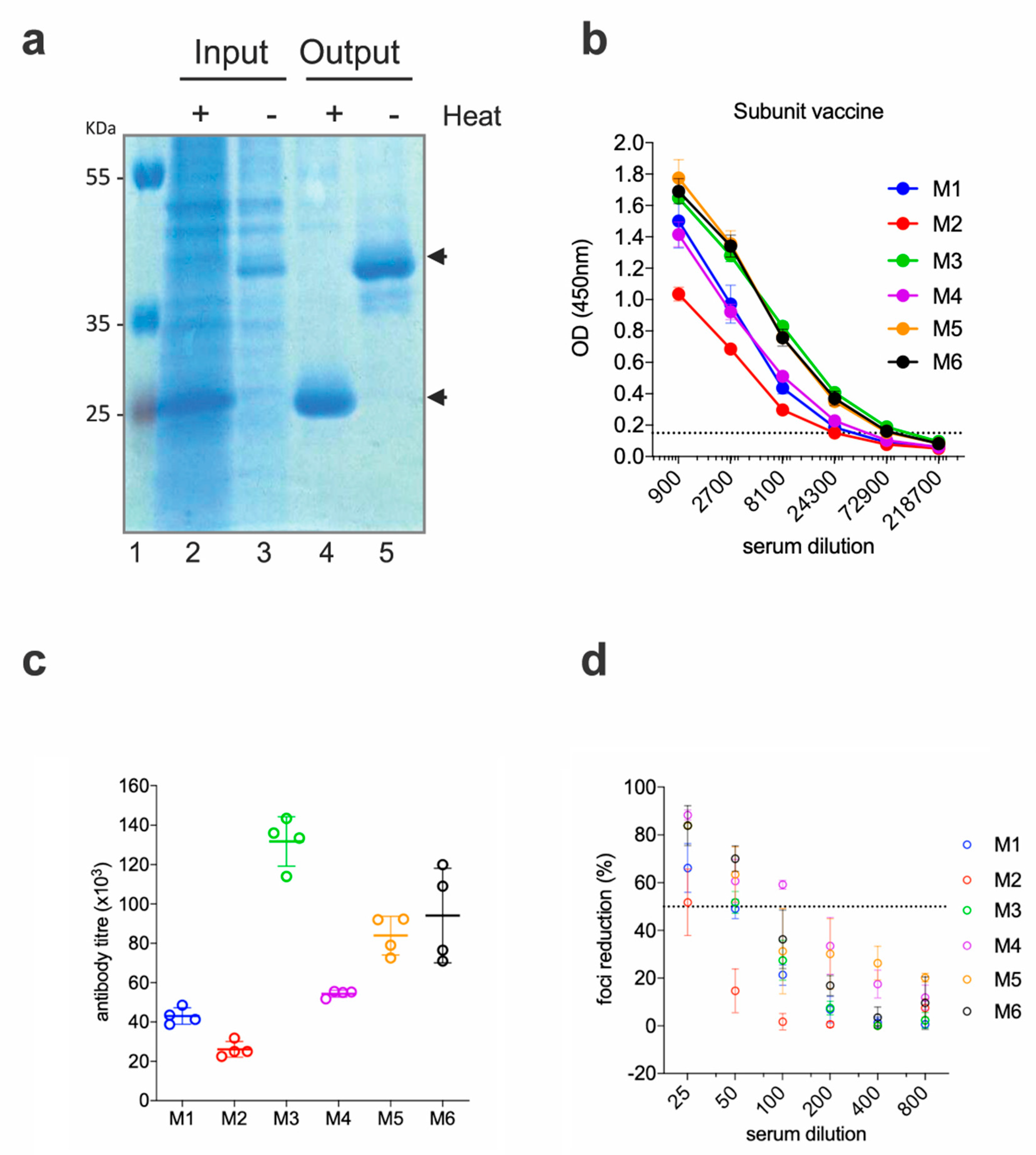
2.2. EDIII-CH3 Protein-Based Immunization Induce Poorly Neutralzsing Antibody Responses
2.3. Adenoviral Vaccine Design Carrying ZIKV EDIII
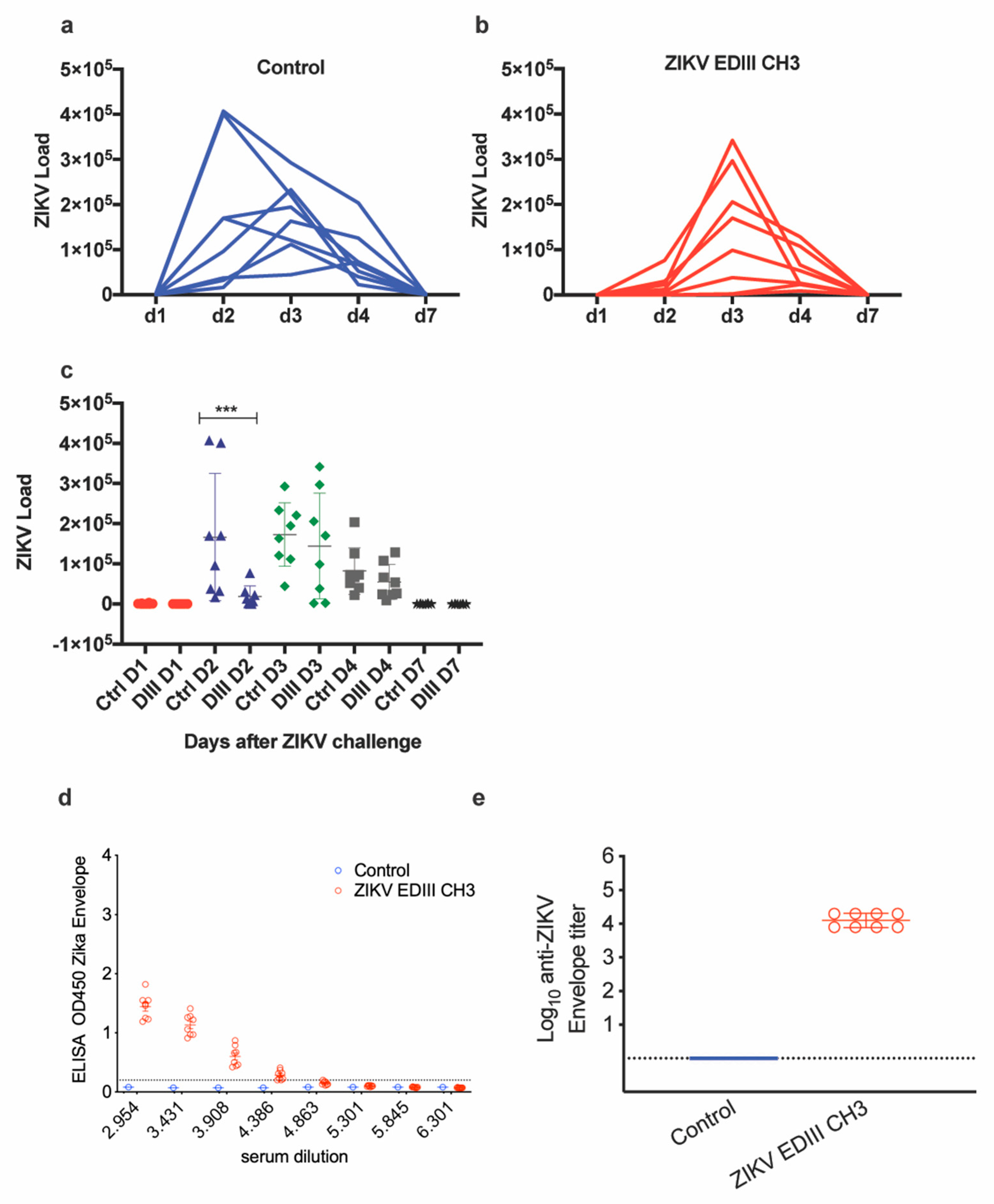
2.4. Immunogenicity and Efficacy of ChAdOx1-EDIII in A129 Mice
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Vaccines
4.3. Animal Ethics
4.3.1. DNA-Based Vaccine
4.3.2. Protein-Based Vaccine
4.3.3. Adenovirus-Based Vaccine
4.3.4. Pre-Challenge Bleed
4.3.5. Enzyme-Linked Immunosorbent Assay (ELISA) to Quantify Whole IgG
4.3.6. Challenge Virus
4.3.7. Clinical Measurements
4.3.8. Sample Collection
4.4. PCR Quantification of Viral Load
4.4.1. Protein-Based Vaccine
4.4.2. Adenovirus-Based Vaccine
4.4.3. Histology
4.4.4. Contributions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holbrook, M.R. Historical Perspectives on Flavivirus Research. Viruses 2017, 9, 97. [Google Scholar] [CrossRef]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Monath, T.P.; Vasconcelos, P.F. Yellow fever. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, Z.Y.; Han, J.F.; Jiang, T.; Li, X.F.; Qin, C.F. Genomic characterization and phylogenetic analysis of Zika virus circulating in the Americas. Infect. Genet. Evol. 2016, 43, 43–49. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Lim, E.X.; Zhang, S.; Fibriansah, G.; Ng, T.S.; Ooi, J.S.; Shi, J.; Lok, S.M. Structure of the thermally stable Zika virus. Nature 2016, 533, 425–428. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Ogata, S.; Clements, D.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Conformational changes of the flavivirus E glycoprotein. Structure 2004, 12, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.; Holbrook, M.; Shope, R.E.; Barrett, A.D.; Watowich, S.J. Biophysical characterization and vector-specific antagonist activity of domain III of the tick-borne flavivirus envelope protein. J. Virol. 2001, 75, 4002–4007. [Google Scholar] [CrossRef] [PubMed]
- Barba-Spaeth, G.; Dejnirattisai, W.; Rouvinski, A.; Vaney, M.C.; Medits, I.; Sharma, A.; Simon-Loriere, E.; Sakuntabhai, A.; Cao-Lormeau, V.M.; Haouz, A.; et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 2016, 536, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Fremont, D.H.; Kuhn, R.J.; Diamond, M.S. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: Implications for vaccine development. Cell Host Microbe 2008, 4, 229–238. [Google Scholar] [CrossRef]
- Dai, L.; Song, J.; Lu, X.; Deng, Y.Q.; Musyoki, A.M.; Cheng, H.; Zhang, Y.; Yuan, Y.; Song, H.; Haywood, J.; et al. Structures of the Zika Virus Envelope Protein and Its Complex with a Flavivirus Broadly Protective Antibody. Cell Host Microbe 2016, 19, 696–704. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Oliphant, T.; Johnson, S.; Burke, S.; Diamond, M.S.; Fremont, D.H. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 2005, 437, 764–769. [Google Scholar] [CrossRef]
- Cockburn, J.J.; Navarro Sanchez, M.E.; Fretes, N.; Urvoas, A.; Staropoli, I.; Kikuti, C.M.; Coffey, L.L.; Arenzana Seisdedos, F.; Bedouelle, H.; Rey, F.A. Mechanism of dengue virus broad cross-neutralization by a monoclonal antibody. Structure 2012, 20, 303–314. [Google Scholar] [CrossRef]
- Zhao, H.; Fernandez, E.; Dowd, K.A.; Speer, S.D.; Platt, D.J.; Gorman, M.J.; Govero, J.; Nelson, C.A.; Pierson, T.C.; Diamond, M.S.; et al. Structural Basis of Zika Virus-Specific Antibody Protection. Cell 2016, 166, 1016–1027. [Google Scholar] [CrossRef]
- Dowd, K.A.; Pierson, T.C. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology 2011, 411, 306–315. [Google Scholar] [CrossRef]
- Beltramello, M.; Williams, K.L.; Simmons, C.P.; Macagno, A.; Simonelli, L.; Quyen, N.T.; Sukupolvi-Petty, S.; Navarro-Sanchez, E.; Young, P.R.; de Silva, A.M.; et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 2010, 8, 271–283. [Google Scholar] [CrossRef]
- Throsby, M.; Geuijen, C.; Goudsmit, J.; Bakker, A.Q.; Korimbocus, J.; Kramer, R.A.; Clijsters-van der Horst, M.; de Jong, M.; Jongeneelen, M.; Thijsse, S.; et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J. Virol. 2006, 80, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.; Engle, M.; Nybakken, G.E.; Doane, C.; Johnson, S.; Huang, L.; Gorlatov, S.; Mehlhop, E.; Marri, A.; Chung, K.M.; et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005, 11, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–13100. [Google Scholar] [CrossRef]
- Fahimi, H.; Mohammadipour, M.; Haddad Kashani, H.; Parvini, F.; Sadeghizadeh, M. Dengue viruses and promising envelope protein domain III-based vaccines. Appl. Microbiol. Biotechnol. 2018, 102, 2977–2996. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Recombinant Protein-Based Dengue Vaccines. Front. Immunol. 2018, 9, 1919. [Google Scholar] [CrossRef]
- Chua, A.J.; Vituret, C.; Tan, M.L.; Gonzalez, G.; Boulanger, P.; Ng, M.L.; Hong, S.S. A novel platform for virus-like particle-display of flaviviral envelope domain III: Induction of Dengue and West Nile virus neutralizing antibodies. Virol. J. 2013, 10, 129. [Google Scholar] [CrossRef]
- Mota, J.; Acosta, M.; Argotte, R.; Figueroa, R.; Mendez, A.; Ramos, C. Induction of protective antibodies against dengue virus by tetravalent DNA immunization of mice with domain III of the envelope protein. Vaccine 2005, 23, 3469–3476. [Google Scholar] [CrossRef]
- Poggianella, M.; Slon Campos, J.L.; Chan, K.R.; Tan, H.C.; Bestagno, M.; Ooi, E.E.; Burrone, O.R. Dengue E Protein Domain III-Based DNA Immunisation Induces Strong Antibody Responses to All Four Viral Serotypes. PLoS Negl. Trop. Dis. 2015, 9, e0003947. [Google Scholar] [CrossRef]
- Ramanathan, M.P.; Kuo, Y.C.; Selling, B.H.; Li, Q.; Sardesai, N.Y.; Kim, J.J.; Weiner, D.B. Development of a novel DNA SynCon tetravalent dengue vaccine that elicits immune responses against four serotypes. Vaccine 2009, 27, 6444–6453. [Google Scholar] [CrossRef]
- Khanam, S.; Pilankatta, R.; Khanna, N.; Swaminathan, S. An adenovirus type 5 (AdV5) vector encoding an envelope domain III-based tetravalent antigen elicits immune responses against all four dengue viruses in the presence of prior AdV5 immunity. Vaccine 2009, 27, 6011–6021. [Google Scholar] [CrossRef]
- Shao, Q.; Herrlinger, S.; Zhu, Y.N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.P.; Brindley, M.A.; Chen, J.F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.; Guimaraes, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Pedraza, A.; Bestagno, M.; Mancardi, S.; Sanchez, R.; Burrone, O. Mammalian cell expression of dimeric small immune proteins (SIP). Protein. Eng. 1997, 10, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Slon Campos, J.L.; Poggianella, M.; Marchese, S.; Bestagno, M.; Burrone, O.R. Secretion of dengue virus envelope protein ectodomain from mammalian cells is dependent on domain II serotype and affects the immune response upon DNA vaccination. J. Gen. Virol. 2015, 96, 3265–3279. [Google Scholar] [CrossRef]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A Susceptible Mouse Model for Zika Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef]
- Lopez-Camacho, C.; Abbink, P.; Larocca, R.A.; Dejnirattisai, W.; Boyd, M.; Badamchi-Zadeh, A.; Wallace, Z.R.; Doig, J.; Velazquez, R.S.; Neto, R.D.L.; et al. Rational Zika vaccine design via the modulation of antigen membrane anchors in chimpanzee adenoviral vectors. Nat. Commun. 2018, 9, 2441. [Google Scholar] [CrossRef]
- Larocca, R.A.; Abbink, P.; Peron, J.P.; Zanotto, P.M.; Iampietro, M.J.; Badamchi-Zadeh, A.; Boyd, M.; Ng’ang’a, D.; Kirilova, M.; Nityanandam, R.; et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536, 474–478. [Google Scholar] [CrossRef]
- Slon Campos, J.L.; Poggianella, M.; Marchese, S.; Mossenta, M.; Rana, J.; Arnoldi, F.; Bestagno, M.; Burrone, O.R. DNA-immunisation with dengue virus E protein domains I/II, but not domain III, enhances Zika, West Nile and Yellow Fever virus infection. PLoS ONE 2017, 12, e0181734. [Google Scholar] [CrossRef]
- Diamond, M.S.; Pierson, T.C.; Fremont, D.H. The structural immunology of antibody protection against West Nile virus. Immunol. Rev. 2008, 225, 212–225. [Google Scholar] [CrossRef]
- Oliphant, T.; Nybakken, G.E.; Engle, M.; Xu, Q.; Nelson, C.A.; Sukupolvi-Petty, S.; Marri, A.; Lachmi, B.E.; Olshevsky, U.; Fremont, D.H.; et al. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J. Virol. 2006, 80, 12149–12159. [Google Scholar] [CrossRef]
- Yang, M.; Lai, H.; Sun, H.; Chen, Q. Virus-like particles that display Zika virus envelope protein domain III induce potent neutralizing immune responses in mice. Sci. Rep. 2017, 7, 7679. [Google Scholar] [CrossRef]
- Yang, M.; Dent, M.; Lai, H.; Sun, H.; Chen, Q. Immunization of Zika virus envelope protein domain III induces specific and neutralizing immune responses against Zika virus. Vaccine 2017, 35, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Miranda, G.; Lim, S.M.; Mohsen, M.O.; Pobelov, I.V.; Roesti, E.S.; Heath, M.D.; Skinner, M.A.; Kramer, M.F.; Martina, B.E.E.; Bachmann, M.F. Zika Virus-Derived E-DIII Protein Displayed on Immunologically Optimized VLPs Induces Neutralizing Antibodies without Causing Enhancement of Dengue Virus Infection. Vaccines (Basel) 2019, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, R.; Gao, F.; Li, M.; Liu, J.; Wang, J.; Hong, W.; Zhao, L.; Wen, Y.; Yin, C.; et al. Delineating antibody recognition against Zika virus during natural infection. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, S.; Du, L.; Wang, C.; Zou, P.; Hong, B.; Yuan, M.; Ren, X.; Tai, W.; Kong, Y.; et al. Neutralization of Zika virus by germline-like human monoclonal antibodies targeting cryptic epitopes on envelope domain III. Emerg. Microbes Infect. 2017, 6, e89. [Google Scholar] [CrossRef] [PubMed]
- Sapparapu, G.; Fernandez, E.; Kose, N.; Bin, C.; Fox, J.M.; Bombardi, R.G.; Zhao, H.; Nelson, C.A.; Bryan, A.L.; Barnes, T.; et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016, 540, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.C.; Wirchnianski, A.S.; Govero, J.; Vergnolle, O.; Dowd, K.A.; Pierson, T.C.; Kielian, M.; Girvin, M.E.; Diamond, M.S.; Lai, J.R. Engineered Dengue Virus Domain III Proteins Elicit Cross-Neutralizing Antibody Responses in Mice. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Kim, Y.C.; Lopez-Camacho, C.; Nettleship, J.E.; Rahman, N.; Hill, M.L.; Silva-Reyes, L.; Ortiz-Martinez, G.; Figueroa-Aguilar, G.; Mar, M.A.; Vivanco-Cid, H.; et al. Optimization of Zika virus envelope protein production for ELISA and correlation of antibody titers with virus neutralization in Mexican patients from an arbovirus endemic region. Virol J. 2018, 15, 193. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine | Animal ID | Histology ID | Culled by Day | Diffusely Scattered Nuclear Debris | Lymphocytic Perivascular Cuffing | Diffusely Scattered PMNs | Degenerating Neurons-Hippocampus | Patchy, Meningeal Infiltration by Inflammatory Cells | Poorly Defined Areas of White Pulp with Large Mononuclear Cells | EMH +/− Apoptosis | Mature PMNs in Red Pulp Sinuses |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vehicle | 31,371 | 732/17 | 7 | Mild | Mild | WNL | WNL | Moderate | Mild | Moderate | Mild |
| 33,333 | 733/17 | 7 | Mild | Minimal | Minimal | WNL | Minimal | Mild | Moderate | Mild | |
| 31,110 | 734/17 | 7 | Minimal | Minimal | WNL | Minimal | Minimal | Mild | Moderate | Minimal | |
| 13,509 | 735/17 | 7 | Minimal | Minimal | WNL | WNL | Mild | WNL | Moderate | Mild | |
| 31,127 | 736/17 | 6 | WNL | Minimal | WNL | WNL | Minimal | WNL | Moderate | Mild | |
| 13,035 | 737/17 | 7 | Moderate | Moderate | Minimal | Moderate | Moderate | Mild | Moderate | Minimal | |
| ChAdOx1 EDIII | 31,764 | 714/17 | 8 | Moderate | Moderate | WNL | Marked | Moderate | WNL | Moderate | WNL |
| 12,300 | 715/17 | 8 | Mild | Mild | WNL | Moderate | Moderate | WNL | Moderate | Minimal | |
| 31,303 | 716/17 | 8 | Mild | Moderate | WNL | Minimal | Moderate | Minimal | Moderate | Mild | |
| 31,131 | 717/17 | 8 | Mild | Mild | Minimal | WNL | Moderate | Minimal | Moderate | Mild | |
| 13,219 | 718/17 | 7 | Minimal | Moderate | WNL | Minimal | Moderate | WNL | Moderate | Minimal | |
| 31,398 | 719/17 | 8 | Minimal | Moderate | WNL | Minimal | Moderate | Minimal | Severe | Mild | |
| Reference | 13,657 | 690/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Minimal | WNL |
| 12,304 | 691/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 13,545 | 692/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 31,609 | 693/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 15,214 | 694/17 | 21 | WNL | WNL | WNL | WNL | Minimal | WNL | Mild | WNL | |
| 13,448 | 695/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Minimal | WNL | |
| ChAdOx1 Mock | 31,220 | 726/17 | 8 | Minimal | Moderate | WNL | Mild | Mild | Mild | Moderate | Mild |
| 13,690 | 727/17 | 8 | Mild | Moderate | WNL | Mild | Moderate | Minimal | Moderate | Moderate | |
| 34,144 | 728/17 | 8 | Minimal | Mild | Mild | Moderate | Moderate | Minimal | Severe | Mild | |
| 13,122 | 729/17 | 8 | WNL | Mild | WNL | Not present | Minimal | Not present | |||
| 12,189 | 730/17 | 7 | Minimal | Mild | WNL | WNL | Moderate | Minimal | Moderate | Mild | |
| 13,555 | 731/17 | 7 | Minimal | Minimal | Minimal | WNL | Mild | Mild | WNL | Mild | |
| Vehicle | 31,371 | 732/17 | 7 | Mild | Mild | WNL | WNL | Moderate | Mild | Moderate | Mild |
| 33,333 | 733/17 | 7 | Mild | Minimal | Minimal | WNL | Minimal | Mild | Moderate | Mild | |
| 31,110 | 734/17 | 7 | Minimal | Minimal | WNL | Minimal | Minimal | Mild | Moderate | Minimal | |
| 13,509 | 735/17 | 7 | Minimal | Minimal | WNL | WNL | Mild | WNL | Moderate | Mild | |
| 31,127 | 736/17 | 6 | WNL | Minimal | WNL | WNL | Minimal | WNL | Moderate | Mild | |
| 13,035 | 737/17 | 7 | Moderate | Moderate | Minimal | Moderate | Moderate | Mild | Moderate | Minimal | |
| ChAdOx1 EDIII | 31,764 | 714/17 | 8 | Moderate | Moderate | WNL | Marked | Moderate | WNL | Moderate | WNL |
| 12,300 | 715/17 | 8 | Mild | Mild | WNL | Moderate | Moderate | WNL | Moderate | Minimal | |
| 31,303 | 716/17 | 8 | Mild | Moderate | WNL | Minimal | Moderate | Minimal | Moderate | Mild | |
| 31,131 | 717/17 | 8 | Mild | Mild | Minimal | WNL | Moderate | Minimal | Moderate | Mild | |
| 13,219 | 718/17 | 7 | Minimal | Moderate | WNL | Minimal | Moderate | WNL | Moderate | Minimal | |
| 31,398 | 719/17 | 8 | Minimal | Moderate | WNL | Minimal | Moderate | Minimal | Severe | Mild | |
| Reference | 13,657 | 690/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Minimal | WNL |
| 12,304 | 691/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 13,545 | 692/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 31,609 | 693/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Mild | WNL | |
| 15,214 | 694/17 | 21 | WNL | WNL | WNL | WNL | Minimal | WNL | Mild | WNL | |
| 13,448 | 695/17 | 21 | WNL | WNL | WNL | WNL | WNL | WNL | Minimal | WNL | |
| ChAdOx1 Mock | 31,220 | 726/17 | 8 | Minimal | Moderate | WNL | Mild | Mild | Mild | Moderate | Mild |
| 13,690 | 727/17 | 8 | Mild | Moderate | WNL | Mild | Moderate | Minimal | Moderate | Moderate | |
| 34,144 | 728/17 | 8 | Minimal | Mild | Mild | Moderate | Moderate | Minimal | Severe | Mild | |
| 13,122 | 729/17 | 8 | WNL | Mild | WNL | Not present | Minimal | Not present | |||
| 12,189 | 730/17 | 7 | Minimal | Mild | WNL | WNL | Moderate | Minimal | Moderate | Mild | |
| 13,555 | 731/17 | 7 | Minimal | Minimal | Minimal | WNL | Mild | Mild | WNL | Mild | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Camacho, C.; De Lorenzo, G.; Slon-Campos, J.L.; Dowall, S.; Abbink, P.; Larocca, R.A.; Kim, Y.C.; Poggianella, M.; Graham, V.; Findlay-Wilson, S.; et al. Immunogenicity and Efficacy of Zika Virus Envelope Domain III in DNA, Protein, and ChAdOx1 Adenoviral-Vectored Vaccines. Vaccines 2020, 8, 307. https://doi.org/10.3390/vaccines8020307
López-Camacho C, De Lorenzo G, Slon-Campos JL, Dowall S, Abbink P, Larocca RA, Kim YC, Poggianella M, Graham V, Findlay-Wilson S, et al. Immunogenicity and Efficacy of Zika Virus Envelope Domain III in DNA, Protein, and ChAdOx1 Adenoviral-Vectored Vaccines. Vaccines. 2020; 8(2):307. https://doi.org/10.3390/vaccines8020307
Chicago/Turabian StyleLópez-Camacho, César, Giuditta De Lorenzo, Jose Luis Slon-Campos, Stuart Dowall, Peter Abbink, Rafael A. Larocca, Young Chan Kim, Monica Poggianella, Victoria Graham, Stephen Findlay-Wilson, and et al. 2020. "Immunogenicity and Efficacy of Zika Virus Envelope Domain III in DNA, Protein, and ChAdOx1 Adenoviral-Vectored Vaccines" Vaccines 8, no. 2: 307. https://doi.org/10.3390/vaccines8020307
APA StyleLópez-Camacho, C., De Lorenzo, G., Slon-Campos, J. L., Dowall, S., Abbink, P., Larocca, R. A., Kim, Y. C., Poggianella, M., Graham, V., Findlay-Wilson, S., Rayner, E., Carmichael, J., Dejnirattisai, W., Boyd, M., Hewson, R., Mongkolsapaya, J., Screaton, G. R., Barouch, D. H., Burrone, O. R., ... Reyes-Sandoval, A. (2020). Immunogenicity and Efficacy of Zika Virus Envelope Domain III in DNA, Protein, and ChAdOx1 Adenoviral-Vectored Vaccines. Vaccines, 8(2), 307. https://doi.org/10.3390/vaccines8020307