Innate Immune Receptors and Defense Against Primary Pathogenic Fungi


Abstract
:1. Introduction
1.1. Fungi
1.2. Fungal Pathogen-Associated Cell Wall Products
2. Innate Immunity
3. Innate Immune Receptors
3.1. C-Type Lectin Receptors
3.2. Toll-Like Receptors
4. Innate Receptors for Pathogenic Fungi
4.1. Blastomyces spp.
4.1.1. BAD1 Receptors
4.1.2. CLR
4.1.3. TLR
4.1.4. Human Innate Immunity
4.2. Coccidioides spp.
4.2.1. CLR
4.2.2. TLR
4.2.3. Human Innate Immunity
4.3. Cryptococcus spp.
4.3.1. Capsule Receptors
4.3.2. CLR
4.3.3. TLR
4.3.4. Human Innate Immunity
4.4. Histoplasma spp.
CLR
4.5. Paracoccidioides spp.
4.5.1. CLR
4.5.2. TLR
4.5.3. Galectin
4.5.4. Humans
5. Evaluating the Data
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Blackwell, M. The fungi: 1, 2, 3... 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Salazar, F.; Brown, G.D. Antifungal Innate Immunity: A Perspective from the Last 10 Years. J. Innate Immun. 2018, 10, 373–397. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, T.N.; Fierer, J. Coccidioides immitis and posadasii; A review of their biology, genomics, pathogenesis, and host immunity. Virulence 2018, 9, 1426–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gow, N.A.R.; Latge, J.P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Hopke, A.; Brown, A.J.P.; Hall, R.A.; Wheeler, R.T. Dynamic Fungal Cell Wall Architecture in Stress Adaptation and Immune Evasion. Trends Microbiol. 2018, 26, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Coelho, C.; Cordero, R.J.B.; Dragotakes, Q.; Jung, E.; Vij, R.; Wear, M.P. The capsule of Cryptococcus neoformans. Virulence 2019, 10, 822–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Kwon-Chung, K.J. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol. Cell Biol. 1994, 14, 4912–4919. [Google Scholar] [CrossRef]
- Brown, G.D.; Herre, J.; Williams, D.L.; Willment, J.A.; Marshall, S.J.; Gordon, S. Dectin-1 mediates the biological effects of b-glucans. J. Exp. Med. 2003, 197, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Yang, X.L.; Yudate, T.; Chung, J.S.; Wu, J.; Luby-Phelps, K.; Kimberly, R.P.; Underhill, D.; Cruz, P.D., Jr.; Ariizumi, K. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J. Biol. Chem. 2006, 281, 38854–38866. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.L.; Zhao, X.Q.; Jiang, C.; You, Y.; Chen, X.P.; Jiang, Y.Y.; Jia, X.M.; Lin, X. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.; Malireddi, R.K.; Lenardon, M.D.; Koberle, M.; Vautier, S.; MacCallum, D.M.; Biedermann, T.; Schaller, M.; Netea, M.G.; Kanneganti, T.D.; et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog. 2014, 10, e1004050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, C.A.; Salvage-Jones, J.A.; Li, X.; Hitchens, K.; Butcher, S.; Murray, R.Z.; Beckhouse, A.G.; Lo, Y.L.S.; Manzanero, S.; Cobbold, C.; et al. The Macrophage-Inducible C-Type Lectin, Mincle, Is an Essential Component of the Innate Immune Response to Candida albicans. J. Immunol. 2008, 180, 7404–7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouault, T.; El Abed-El Behi, M.; Martinez-Esparza, M.; Breuilh, L.; Trinel, P.A.; Chamaillard, M.; Trottein, F.; Poulain, D. Specific Recognition of Candida albicans by Macrophages Requires Galectin-3 to Discriminate Saccharomyces cerevisiae and Needs Association with TLR2 for Signaling. J. Immunol. 2006, 177, 4679–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 2008, 283, 20590–20599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.-A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Biondo, C.; Malara, A.; Costa, A.; Signorino, G.; Cardile, F.; Midiri, A.; Galbo, R.; Papasergi, S.; Domina, M.; Pugliese, M.; et al. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur. J. Immunol. 2012, 42, 2632–2643. [Google Scholar] [CrossRef]
- Miyazato, A.; Nakamura, K.; Yamamoto, N.; Mora-Montes, H.M.; Tanaka, M.; Abe, Y.; Tanno, D.; Inden, K.; Gang, X.; Ishii, K.; et al. Toll-like receptor 9-dependent activation of myeloid dendritic cells by Deoxynucleic acids from Candida albicans. Infect. Immun. 2009, 77, 3056–3064. [Google Scholar] [CrossRef] [Green Version]
- Kasperkovitz, P.V.; Cardenas, M.L.; Vyas, J.M. TLR9 is actively recruited to Aspergillus fumigatus phagosomes and requires the N-terminal proteolytic cleavage domain for proper intracellular trafficking. J. Immunol. 2010, 185, 7614–7622. [Google Scholar] [CrossRef] [Green Version]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef]
- Willment, J.A.; Lin, H.-H.; Reid, D.M.; Taylor, P.R.; Williams, D.L.; Wong, S.Y.C.; Gordon, S.; Brown, G.D. Dectin-1 Expression and Function Are Enhanced on Alternatively Activated and GM-CSF-Treated Macrophages and Are Negatively Regulated by IL-10, Dexamethasone, and Lipopolysaccharide. J. Immunol. 2003, 171, 4569–4573. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Reyes, C.N.; Becker, C.A.; Katsumoto, T.R.; Ma, J.; Wolf, A.J.; Bose, N.; Chan, A.S.H.; Magee, A.S.; Danielson, M.E.; et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011, 472, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glocker, E.-O.; Hennigs, A.; Nabavi, M.; Schäffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A HomozygousCARD9Mutation in a Family with Susceptibility to Fungal Infections. N. Engl. J. Med. 2009, 361, 1727–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeekens, S.P.; van de Veerdonk, F.L.; Kullberg, B.J.; Netea, M.G. Genetic susceptibility to Candida infections. EMBO Mol. Med. 2013, 5, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.K.; Mansell, A. Toll-like receptors: The swiss army knife of immunity and vaccine development. Clin. Transl. Immunol. 2016, 5, e85. [Google Scholar] [CrossRef]
- Saccente, M.; Woods, G.L. Clinical and Laboratory Update on Blastomycosis. Clin. Microbiol. Rev. 2010, 23, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Friedman; Schwartz. Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. J. Fungi 2019, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Castillo, C.G.; Kauffman, C.A.; Miceli, M.H. Blastomycosis. Infect. Dis. Clin. North. Am. 2016, 30, 247–264. [Google Scholar] [CrossRef]
- Finkel-Jimenez, B.; Wüthrich, M.; Brandhorst, T.; Klein, B.S. The WI-1 adhesin blocks phagocyte TNF-a production, imparting pathogenicity on Blastomyces dermatitidis. J. Immunol. 2001, 166, 2665–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, J.A.; Gauthier, G.M.; Klein, B.S. Turning on virulence: Mechanisms that underpin the morphologic transition and pathogenicity of Blastomyces. Virulence 2019, 10, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandhorst, T.T.; Wüthrich, M.; Finkel-Jimenez, B.; Warner, T.; Klein, B.S. Exploiting Type 3 Complement Receptor for TNF-α Suppression, Immune Evasion, and Progressive Pulmonary Fungal Infection. J. Immunol. 2004, 173, 7444–7453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuthrich, M.; Gern, B.; Hung, C.Y.; Ersland, K.; Rocco, N.; Pick-Jacobs, J.; Galles, K.; Filutowicz, H.; Warner, T.; Evans, M.; et al. Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J. Clin. Investig. 2011, 121, 554–568. [Google Scholar] [CrossRef]
- Wang, H.; LeBert, V.; Hung, C.-Y.; Galles, K.; Saijo, S.; Lin, X.; Cole, G.T.; Klein, B.S.; Wüthrich, M. C-type lectin receptors differentially induce th17 cells and vaccine immunity to the endemic mycosis of North America. J. Immunol. 2014, 192, 1107–1119. [Google Scholar] [CrossRef]
- Wang, H.; Lee, T.J.; Fites, S.J.; Merkhofer, R.; Zarnowski, R.; Brandhorst, T.; Galles, K.; Klein, B.; Wuthrich, M. Ligation of Dectin-2 with a novel microbial ligand promotes adjuvant activity for vaccination. PLoS Pathog. 2017, 13, e1006568. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, M.; Lerksuthirat, T.; Klein, B.; Wuthrich, M. The C-Type Lectin Receptor MCL Mediates Vaccine-Induced Immunity against Infection with Blastomyces dermatitidis. Infect. Immun. 2015, 84, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; LeBert, V.; Li, M.; Lerksuthirat, T.; Galles, K.; Klein, B.; Wuthrich, M. Mannose Receptor Is Required for Optimal Induction of Vaccine-Induced T-Helper Type 17 Cells and Resistance to Blastomyces dermatitidis Infection. J. Infect. Dis. 2016, 213, 1762–1766. [Google Scholar] [CrossRef]
- Roy, M.; Benedict, K.; Deak, E.; Kirby, M.A.; McNiel, J.T.; Sickler, C.J.; Eckardt, E.; Marx, R.K.; Heffernan, R.T.; Meece, J.K.; et al. A Large Community Outbreak of Blastomycosis in Wisconsin With Geographic and Ethnic Clustering. Clin. Infect. Dis. 2013, 57, 655–662. [Google Scholar] [CrossRef]
- Merkhofer, R.M., Jr.; O’Neill, M.B.; Xiong, D.; Hernandez-Santos, N.; Dobson, H.; Fites, J.S.; Shockey, A.C.; Wuethrich, M.; Pepperell, C.S.; Klein, B.S. Investigation of Genetic Susceptibility to Blastomycosis Reveals Interleukin-6 as a Potential Susceptibility Locus. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.; Barker, B.M.; Hoover, S.; Nix, D.E.; Ampel, N.M.; Frelinger, J.A.; Orbach, M.J.; Galgiani, J.N. Recent advances in our understanding of the environmental, epidemiological, immunological, and clinical dimensions of coccidioidomycosis. Clin. Microbiol. Rev. 2013, 26, 505–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, L.; Nix, D.; Wright, M.; Lindberg, E.; Fagan, T.; Lieberman, D.; Stoffer, T.P.; Ampel, N.M.; Galgiani, J.N. Coccidioidomycosis as a common cause of community-acquired pneumonia. Emerg. Inf. Dis. 2006, 12, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Viriyakosol, S.; Fierer, J.; Brown, G.D.; Kirkland, T.N. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on TLR2 and dectin-1. Infect. Immun. 2005, 73, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viriyakosol, S.; Jimenez, M.d.P.; Gurney, M.A.; Ashbaugh, M.E.; Fierer, J. Dectin-1 Is Required for Resistance to Coccidioidomycosis in Mice. mBio 2013, 4, e005970-0512. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, T.; Fierer, J. Inbred mouse strains differ in resistance to lethal Coccidioides immitis infection. Antimicrob. Agents Chemother. 1983, 24, 921–924. [Google Scholar] [CrossRef] [Green Version]
- Del Pilar Jiménez-A, M.; Viriyakosol, S.; Walls, L.; Datta, S.K.; Kirkland, T.; Heinsbroek, S.E.M.; Brown, G.; Fierer, J. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a). Genes Immun. 2008, 9, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Fierer, J.; Walls, L.; Wright, F.; Kirkland, T.N. Genes influencing resistance to Coccidioides immitis and the interleukin-10 response map to chromosomes 4 and 6 in mice. Infect. Immun. 1999, 67, 2916–2919. [Google Scholar] [CrossRef] [Green Version]
- Viriyakosol, S.; Jimenez Mdel, P.; Saijo, S.; Fierer, J. Neither dectin-2 nor the mannose receptor is required for resistance to Coccidioides immitis in mice. Infect. Immun. 2014, 82, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Yamakmoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Viriyakosol, S.; Walls, L.; Okamoto, S.; Raz, E.; Williams, D.L.; Fierer, J. Myeloid Differentiation Factor 88 and Interleukin-1R1 Signaling Contribute to Resistance to Coccidioides immitis. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionne, S.O.; Podany, A.B.; Ruiz, Y.W.; Ampel, N.M.; Galgiani, J.N.; Lake, D.F. Spherules Derived from Coccidioides posadasii Promote Human Dendritic Cell Maturation and Activation. Infect. Immun. 2006, 74, 2415–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampel, N.M.; Nelson, D.K.; Li, L.; Dionne, S.O.; Lake, D.F.; Simmons, K.A.; Pappagianis, D. The Mannose Receptor Mediates the Cellular Immune Response in Human Coccidioidomycosis. Infect. Immun. 2005, 73, 2554–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinh, D.C.; Masannat, F.; Dzioba, R.B.; Galgiani, J.N.; Holland, S.M. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-gamma receptor 1 deficiency. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 49, e626-5. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Schwartz, B.; Hsu, A.P.; Miranda, D.J.; Valdez, P.A.; Fink, D.; Lau, K.P.; Long-Priel, D.; Kuhns, D.B.; Uzel, G.; et al. Interleukin-12 Receptor 1 Deficiency Predisposing to Disseminated Coccidioidomycosis. Clin. Infect. Dis. 2011, 52, e99–e102. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, E.P.; Hsu, A.P.; Pechacek, J.; Bax, H.I.; Dias, D.L.; Paulson, M.L.; Chandrasekaran, P.; Rosen, L.B.; Carvalho, D.S.; Ding, L.; et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 2013, 131, 1624–1634. [Google Scholar] [CrossRef]
- Jones, J.; Fleming, P.; Ciesielski, C.; Hu, D.; Kaplan, J.; Ward, J. Coccidioidomycosis among persons with AIDS in the united states. J. Infect. Dis. 1995, 171, 961–966. [Google Scholar] [CrossRef]
- Kirkland, T.N.; Fierer, J. Coccidioidomycosis: A reemerging infectious disease. Emerg. Infect. Dis. 1996, 2. [Google Scholar] [CrossRef]
- Rosenstein, N.E.; Emery, K.W.; Werner, B.; Kao, A.; Johnson, R.H.; Rogers, D.; Vugia, D.; Reingold, A.L.; Talbot, R.; Plikaytis, B.D.; et al. Risk factors for severe pulmonary and disseminated coccidioidomycosis: Kern County, California, 1995–1996. Clin. Infect. Dis. 2001, 32, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, C.; Lucas, K.D.; Mohle-Boetani, J.C. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg. Infect. Dis. 2015, 21, 70–75. [Google Scholar] [CrossRef]
- Maziarz, E.K.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2016, 30, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Goldman, D.L.; Khine, H.; Abadi, J.; Lindenberg, D.J.; Pirofski, L.A.; Niang, R.; Casadevall, A. Serologic Evidence for Cryptococcus neoformans Infection in Early Childhood. Pediatrics 2001, 107, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.H.; Sorrell, T.C.; Allworth, A.M.; Heath, C.H.; McGregor, A.R.; Papanaoum, K.; Richards, M.J.; Gottlieb, T. Cryptococcal Disease of the CNS in Immunocompetent Hosts: Influence of Cryptococcal Variety on Clinical Manifestations and Outcome. Clin. Infect. Dis. 1995, 20, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Perfect, J.R.; Cloud, G.A.; Larsen, R.A.; Pankey, G.A.; Lancaster, D.J.; Henderson, H.; Kauffman, C.A.; Haas, D.W.; Saccente, M.; et al. Cryptococcosis in Human Immunodeficiency Virus-Negative Patients in the Era of Effective Azole Therapy. Clin. Infect. Dis. 2001, 33, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-Y.; Wang, S.-Y.; Shih, H.-P.; Tu, K.-H.; Huang, W.-C.; Ding, J.-Y.; Lin, C.-H.; Yeh, C.-F.; Ho, M.-W.; Chang, S.-C.; et al. Disseminated Cryptococcosis Due to Anti-Granulocyte-Macrophage Colony-Stimulating Factor Autoantibodies in the Absence of Pulmonary Alveolar Proteinosis. J. Clin. Immunol. 2017, 37, 143–152. [Google Scholar] [CrossRef]
- Browne, S.K.; Burbelo, P.D.; Chetchotisakd, P.; Suputtamongkol, Y.; Kiertiburanakul, S.; Shaw, P.A.; Kirk, J.L.; Jutivorakool, K.; Zaman, R.; Ding, L.; et al. Adult-onset immunodeficiency in Thailand and Taiwan. N. Engl. J. Med. 2012, 367, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon-Chung, K.J.; Fraser, J.A.; Doering, T.L.; Wang, Z.A.; Janbon, G.; Idnurm, A.; Bahn, Y.S. Cryptococcus neoformans and Cryptococcus gattii, the Etiologic Agents of Cryptococcosis. Cold Spring Harb. Perspect. Med. 2014, 4, a019760. [Google Scholar] [CrossRef]
- Decote-Ricardo, D.; LaRocque-de-Freitas, I.F.; Rocha, J.D.B.; Nascimento, D.O.; Nunes, M.P.; Morrot, A.; Freire-de-Lima, L.; Previato, J.O.; Mendonça-Previato, L.; Freire-de-Lima, C.G. Immunomodulatory Role of Capsular Polysaccharides Constituents of Cryptococcus neoformans. Front. Med. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huston, S.M.; Ngamskulrungroj, P.; Xiang, R.F.; Ogbomo, H.; Stack, D.; Li, S.S.; Timm-Mccann, M.; Kyei, S.K.; Oykhman, P.; Kwon-Chung, K.J.; et al. Cryptococcus gattii Capsule Blocks Surface Recognition Required for Dendritic Cell Maturation Independent of Internalization and Antigen Processing. J. Immunol. 2016, 196, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Syme, R.M.; Spurrell, J.C.L.; Amankwah, E.K.; Green, F.H.Y.; Mody, C.H. Primary Dendritic Cells Phagocytose Cryptococcus neoformans via Mannose Receptors and Fc Receptor II for Presentation to T Lymphocytes. Infect. Immun. 2002, 70, 5972–5981. [Google Scholar] [CrossRef] [Green Version]
- Surawut, S.; Ondee, T.; Taratummarat, S.; Palaga, T.; Pisitkun, P.; Chindamporn, A.; Leelahavanichkul, A. The role of macrophages in the susceptibility of Fc gamma receptor IIb deficient mice to Cryptococcus neoformans. Sci. Rep. 2017, 7, 40006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Nakamura, Y.; Sato, K.; Takahashi, Y.; Nomura, T.; Miyasaka, T.; Ishii, K.; Hara, H.; Yamamoto, N.; Kanno, E.; et al. Defect of CARD9 leads to impaired accumulation of gamma interferon-producing memory phenotype T cells in lungs and increased susceptibility to pulmonary infection with Cryptococcus neoformans. Infect. Immun. 2014, 82, 1606–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.M.; Wuthrich, M.; Wang, H.; Klein, B.; Hull, C.M. Characterization of C-type lectins reveals an unexpectedly limited interaction between Cryptococcus neoformans spores and Dectin-1. PLoS ONE 2017, 12, e0173866. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kinjo, T.; Saijo, S.; Miyazato, A.; Adachi, Y.; Ohno, N.; Fujita, J.; Kaku, M.; Iwakura, Y.; Kawakami, K. Dectin-1 Is Not Required for the Host Defense to Cryptococcus neoformans. Microbiol. Immunol. 2007, 51, 1115–1119. [Google Scholar] [CrossRef]
- Nakamura, Y.; Sato, K.; Yamamoto, H.; Matsumura, K.; Matsumoto, I.; Nomura, T.; Miyasaka, T.; Ishii, K.; Kanno, E.; Tachi, M.; et al. Dectin-2 Deficiency Promotes Th2 Response and Mucin Production in the Lungs after Pulmonary Infection with Cryptococcus neoformans. Infect. Immun. 2015, 83, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-R.; Li, F.; Han, H.; Xu, X.; Li, N.; Wang, S.; Xu, J.-F.; Jia, X.-M. Dectin-3 Recognizes Glucuronoxylomannan of Cryptococcus neoformans Serotype AD and Cryptococcus gattii Serotype B to Initiate Host Defense Against Cryptococcosis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Biondo, C.; Midiri, A.; Messina, L.; Tomasello, F.; Garufi, G.; Catania, M.R.; Bombaci, M.; Beninati, C.; Teti, G.; Mancuso, G. MyD88 and TLR2, but not TLR4, are required for host defense against Cryptococcus neoformans. Eur. J. Immunol. 2005, 35, 870–878. [Google Scholar] [CrossRef]
- Yauch, L.E.; Mansour, M.K.; Shoham, S.; Rottman, J.B.; Levitz, S.M. Involvement of CD14, Toll-Like Receptors 2 and 4, and MyD88 in the Host Response to the Fungal Pathogen Cryptococcus neoformans In Vivo. Infect. Immun. 2004, 72, 5373–5382. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Miyagi, K.; Koguchi, Y.; Kinjo, Y.; Uezu, K.; Kinjo, T.; Akamine, M.; Fujita, J.; Kawamura, I.; Mitsuyama, M.; et al. Limited contribution of Toll-like receptor 2 and 4 to the host response to a fungal infectious pathogen, Cryptococcus neoformans. FEMS Immunol. Med Microbiol. 2006, 47, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Lee, C.K.; Akalin, A.; Finberg, R.W.; Levitz, S.M. Contributions of the MyD88-Dependent Receptors IL-18R, IL-1R, and TLR9 to Host Defenses following Pulmonary Challenge with Cryptococcus neoformans. PLoS ONE 2011, 6, e26232. [Google Scholar] [CrossRef]
- Kawakami, K.; Koguchi, Y.; Qureshi, M.H.; Miyazato, A.; Yara, S.; Kinjo, Y.; Iwakura, Y.; Takeda, K.; Akira, S.; Kurimoto, M.; et al. IL-18 Contributes to Host Resistance Against Infection with Cryptococcus neoformans in Mice with Defective IL-12 Synthesis Through Induction of IFN-γ Production by NK Cells. J. Immunol. 2000, 165, 941–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.-P.; Wu, J.-Q.; Zhu, L.-P.; Wang, X.; Xu, B.; Wang, R.-Y.; Ou, X.-T.; Weng, X.-H. Association of Fcγ Receptor IIB Polymorphism with Cryptococcal Meningitis in HIV-Uninfected Chinese Patients. PLoS ONE 2012, 7, e42439. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, S.; Gohil, S.; Kuniholm, M.H.; Schultz, H.; Dufaud, C.; Armour, K.L.; Badri, S.; Mailliard, R.B.; Pirofski, L.-A. Fc Gamma Receptor 3A Polymorphism and Risk for HIV-Associated Cryptococcal Disease. mBio 2013, 4, e00573-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohatgi, S.; Pirofski, L.A. Host immunity to Cryptococcus neoformans. Future Microbiol. 2015, 10, 565–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, J.R.; Hube, B.; Puccia, R.; Casadevall, A.; Perfect, J.R. Fungi that Infect Humans. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Kauffman, C.A. Histoplasmosis: A Clinical and Laboratory Update. Clin. Microbiol. Rev. 2007, 20, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.L.; Gootee, L.; Morris, R.; Bullock, W.E. Digestion of Histoplasma capsulatum yeasts by human macrophages. J. Immunol. 1992, 149, 574–580. [Google Scholar]
- Bullock, W.E. Role of the adherence-promoting receptors, CR3, LFA-1, and p150,95, in binding of Histoplasma capsulatum by human macrophages. J. Exp. Med. 1987, 165, 195–210. [Google Scholar] [CrossRef]
- Lin, J.-S.; Huang, J.-H.; Hung, L.-Y.; Wu, S.-Y.; Wu-Hsieh, B.A. Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with Histoplasma yeast. J. Leukoc. Biol. 2010, 88, 95–106. [Google Scholar] [CrossRef]
- Huang, J.H.; Lin, C.Y.; Wu, S.Y.; Chen, W.Y.; Chu, C.L.; Brown, G.D.; Chuu, C.P.; Wu-Hsieh, B.A. CR3 and Dectin-1 Collaborate in Macrophage Cytokine Response through Association on Lipid Rafts and Activation of Syk-JNK-AP-1 Pathway. PLoS Pathog. 2015, 11, e1004985. [Google Scholar] [CrossRef] [Green Version]
- Rappleye, C.A.; Eissenberg, L.G.; Goldman, W.E. Histoplasma capsulatum-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfoot, A.L.; Shen, Q.; Wüthrich, M.; Klein, B.S.; Rappleye, C.A. The Eng1 β-Glucanase Enhances Histoplasma Virulence by Reducing β-Glucan Exposure. mBio 2016, 7, e013880-1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayfield, J.A.; Rine, J. The genetic basis of variation in susceptibility to infection with Histoplasma capsulatum in the mouse. Genes Immun. 2007, 8, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayfield, J.A.; Fontana, M.F.; Rine, J. Genetic control of immune cell types in fungal disease. Proc. Natl. Acad. Sci. USA 2010, 107, 22202–22206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.H.; Huang, J.H.; Lin, H.C.; Chen, W.Y.; Lee, Y.H.; Hsu, L.C.; Netea, M.G.; Ting, J.P.; Wu-Hsieh, B.A. Dectin-2 is a primary receptor for NLRP3 inflammasome activation in dendritic cell response to Histoplasma capsulatum. PLoS Pathog. 2017, 13, e1006485. [Google Scholar] [CrossRef] [PubMed]
- Coady, A.; Sil, A. MyD88-dependent signaling drives host survival and early cytokine production during Histoplasma capsulatum infection. Infect. Immun. 2015, 83, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Van Prooyen, N.; Henderson, C.A.; Hocking Murray, D.; Sil, A. CD103+ Conventional Dendritic Cells Are Critical for TLR7/9-Dependent Host Defense against Histoplasma capsulatum, an Endemic Fungal Pathogen of Humans. PLoS Pathog. 2016, 12, e1005749. [Google Scholar] [CrossRef]
- Queiroz-Telles, F.; Fahal, A.H.; Falci, D.R.; Caceres, D.H.; Chiller, T.; Pasqualotto, A.C. Neglected endemic mycoses. Lancet Infect. Dis. 2017, 17, e367–e377. [Google Scholar] [CrossRef]
- Loures, F.V.; Araujo, E.F.; Feriotti, C.; Bazan, S.B.; Costa, T.A.; Brown, G.D.; Calich, V.L.G. Dectin-1 Induces M1 Macrophages and Prominent Expansion of CD8+IL-17+ Cells in Pulmonary Paracoccidioidomycosis. J. Infect. Dis. 2014, 210, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Feriotti, C.; Loures, F.V.; Frank De Araújo, E.; Costa, T.A.D.; Calich, V.L.G. Mannosyl-Recognizing Receptors Induce an M1-Like Phenotype in Macrophages of Susceptible Mice but an M2-Like Phenotype in Mice Resistant to a Fungal Infection. PLoS ONE 2013, 8, e54845. [Google Scholar] [CrossRef] [Green Version]
- Tavares, A.H.; Derengowski, L.S.; Ferreira, K.S.; Silva, S.S.; Macedo, C.; Bocca, A.L.; Passos, G.A.; Almeida, S.R.; Silva-Pereira, I. Murine Dendritic Cells Transcriptional Modulation upon Paracoccidioides brasiliensis Infection. PLoS Negl. Trop. Dis. 2012, 6, e1459. [Google Scholar] [CrossRef] [PubMed]
- Loures, F.V.; Pina, A.; Felonato, M.; Calich, V.L.G. TLR2 Is a Negative Regulator of Th17 Cells and Tissue Pathology in a Pulmonary Model of Fungal Infection. J. Immunol. 2009, 183, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loures, F.V.; Araujo, E.F.; Feriotti, C.; Bazan, S.B.; Calich, V.L. TLR-4 cooperates with Dectin-1 and mannose receptor to expand Th17 and Tc17 cells induced by Paracoccidioides brasiliensis stimulated dendritic cells. Front. Microbiol. 2015, 6, 261. [Google Scholar] [CrossRef] [PubMed]
- Loures, F.V.; Pina, A.; Felonato, M.; Feriotti, C.; De Araújo, E.F.; Calich, V.L.G. MyD88 Signaling Is Required for Efficient Innate and Adaptive Immune Responses to Paracoccidioides brasiliensis Infection. Infect. Immun. 2011, 79, 2470–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Yáñez, A.; Gozalbo, D.; Gil, M.L. MyD88 is dispensable for resistance to Paracoccidioides brasiliensis in a murine model of blood-borne disseminated infection. FEMS Immunol. Med. Microbiol. 2008, 54, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Menino, J.F.; Saraiva, M.; Gomes-Alves, A.G.; Lobo-Silva, D.; Sturme, M.; Gomes-Rezende, J.; Saraiva, A.L.; Goldman, G.H.; Cunha, C.; Carvalho, A.; et al. TLR9 Activation Dampens the Early Inflammatory Response to Paracoccidioides brasiliensis, Impacting Host Survival. PLoS Negl. Trop. Dis. 2013, 7, e2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calich, V.L.; Burger, E.; Kashino, S.S.; Fazioli, R.A.; Singer-Vermes, L.M. Resistance to Paracoccidioides brasiliensis in mice is controlled by a single dominant autosomal gene. Infect. Immun. 1987, 55, 1919–1923. [Google Scholar] [CrossRef] [Green Version]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, O.; Rezende, C.P.; Moreno, P.; Freitas Fernandes, F.; Oliveira Brito, P.K.M.; Martinez, R.; Coelho, C.; Roque-Barreira, M.C.; Casadevall, A.; Almeida, F. Galectin-3 Inhibits Paracoccidioides brasiliensis Growth and Impacts Paracoccidioidomycosis through Multiple Mechanisms. mSphere 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Preite, N.W.; Feriotti, C.; Souza de Lima, D.; da Silva, B.B.; Condino-Neto, A.; Pontillo, A.; Calich, V.L.G.; Loures, F.V. The Syk-Coupled C-Type Lectin Receptors Dectin-2 and Dectin-3 Are Involved in Paracoccidioides brasiliensis Recognition by Human Plasmacytoid Dendritic Cells. Front. Immunol. 2018, 9, 464. [Google Scholar] [CrossRef]
- Balderramas, H.A.; Penitenti, M.; Rodrigues, D.R.; Bachiega, T.F.; Fernandes, R.K.; Ikoma, M.R.; Dias-Melicio, L.A.; Oliveira, S.L.; Soares, A.M. Human neutrophils produce IL-12, IL-10, PGE2 and LTB4 in response to Paracoccidioides brasiliensis. Involvement of TLR2, mannose receptor and dectin-1. Cytokine 2014, 67, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Goldani, L.Z.; Sugar, A.M. Paracoccidioidomycosis and AIDS: An Overview. Clin. Infect. Dis. 1995, 21, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Alves Pereira Neto, T.; Costa Pereira, A.A.; Costa Hanemann, J.A.; Coelho, L.F.L.; Malaquias, L.C.C. DC-SIGN and VDR polymorphisms are associated with chronic form of paracoccidioidomycosis with oral manifestations. Mycoses 2019, 62, 186–192. [Google Scholar] [CrossRef] [PubMed]
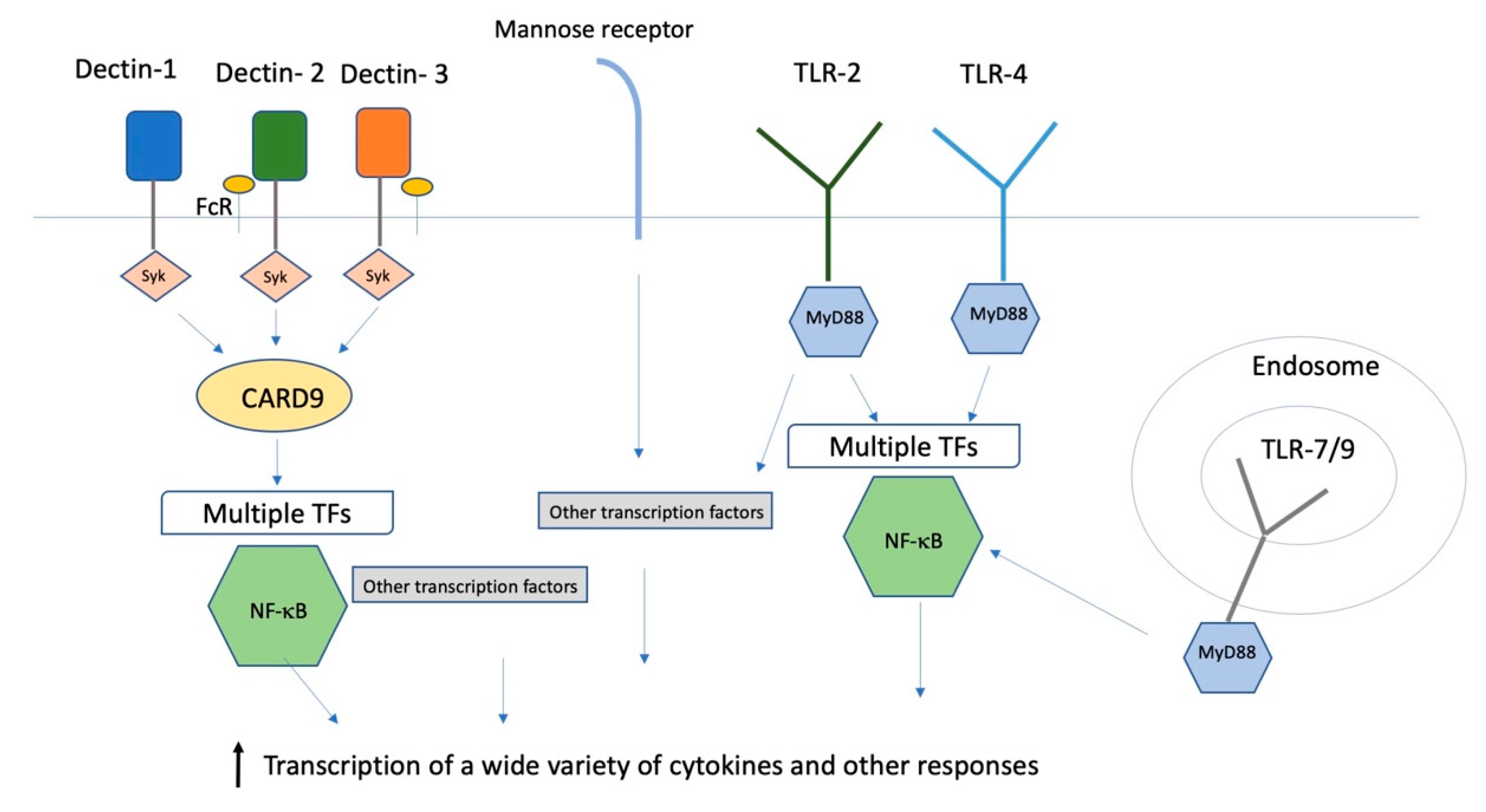

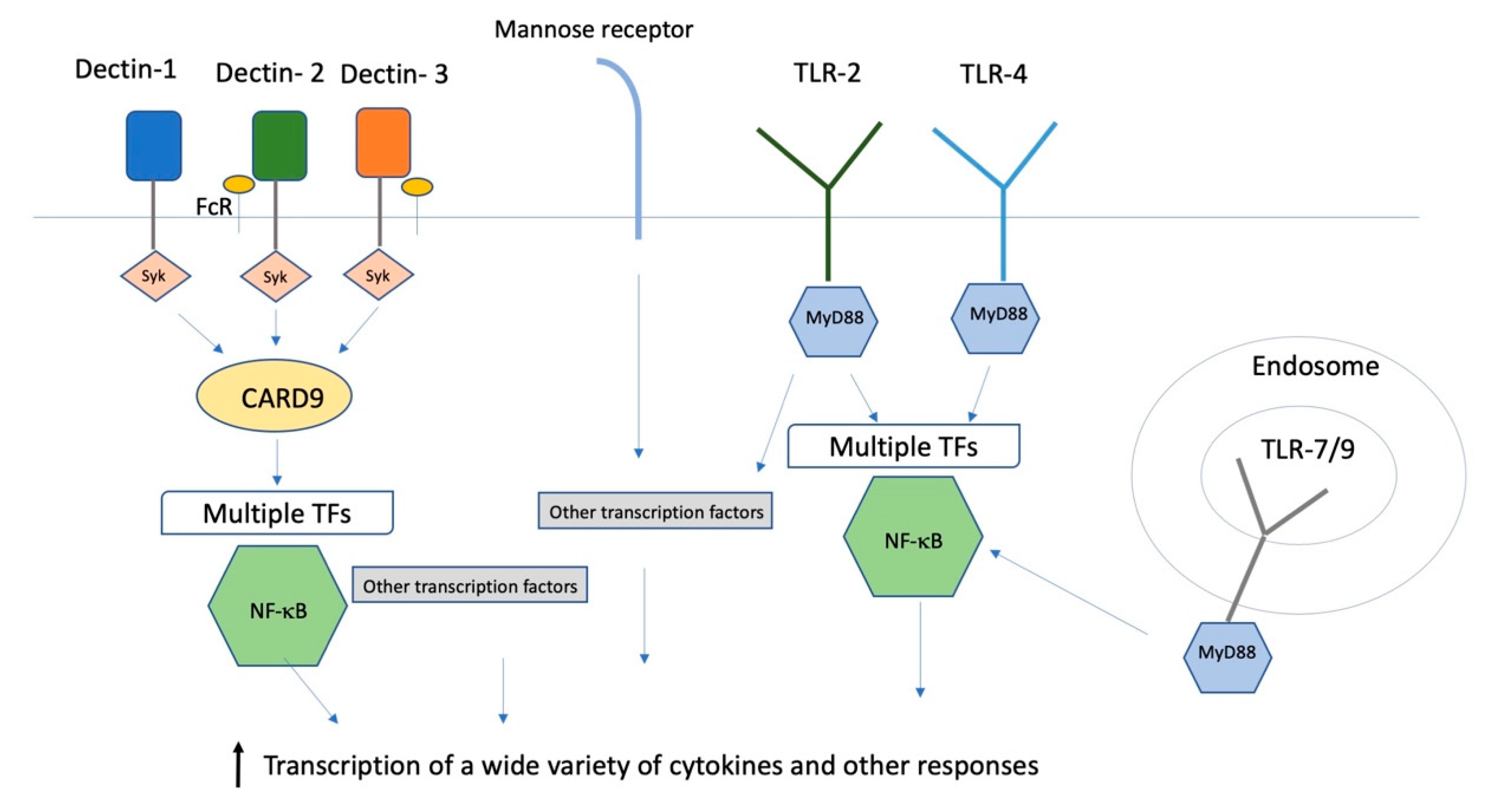{kind=link}
| Pattern Recognition Receptor | Ligand | Reference | |
|---|---|---|---|
| CLR a | Dectin-1 | β-(1,3)-glucan | [8] |
| Dectin-2 | High mannose | [9] | |
| Dectin-2/Dectin-3 | α-mannan | [10] | |
| Mannose receptor | N-linked mannan | [11] | |
| Mannose receptor | Chitin | [12] | |
| Mincle b | α-mannan | [13] | |
| Galectin-3 | β-(1,2)-mannoside | [14] | |
| DC-SIGN | N-linked mannan | [15] | |
| Others | NOD-2 c | Chitin | [12] |
| TLR d | TLR-2 | Phospholipomannan | [16] |
| TLR-2 | β-(1,6)-glucan | [11] | |
| TLR-4 | O-linked mannans | [11] | |
| TLR-7 | ssRNAe | [17] | |
| TLR-9 | CpG DNA | [18] | |
| TLR-9 | Chitin | [12] |
| Organism | Innate Resistance | Acquired Response |
|---|---|---|
| Blastomyces spp. | CR3, CARD9-dependent | CARD9-dependent Dectin-2, Dectin-3, MR |
| Coccidioides spp. | Dectin-1 MyD88-dependent IL-1R | CARD9-dependent Dectin-2 |
| Cryptococcus spp. | CARD9-dependent Dectin-3, FcRγ MyD88-dependent TLR-2, IL-18R | ? |
| Histoplasma spp. | MyD88-dependent TLR-7/9 a,b | CARD9-dependent Dectin-2 |
| Paracoccidioides spp. | Resistance - Dectin-1 MyD88-probably TLR-2 Galectin | ? |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirkland, T.N.; Fierer, J. Innate Immune Receptors and Defense Against Primary Pathogenic Fungi. Vaccines 2020, 8, 303. https://doi.org/10.3390/vaccines8020303
Kirkland TN, Fierer J. Innate Immune Receptors and Defense Against Primary Pathogenic Fungi. Vaccines. 2020; 8(2):303. https://doi.org/10.3390/vaccines8020303
Chicago/Turabian StyleKirkland, Theo N., and Joshua Fierer. 2020. "Innate Immune Receptors and Defense Against Primary Pathogenic Fungi" Vaccines 8, no. 2: 303. https://doi.org/10.3390/vaccines8020303
APA StyleKirkland, T. N., & Fierer, J. (2020). Innate Immune Receptors and Defense Against Primary Pathogenic Fungi. Vaccines, 8(2), 303. https://doi.org/10.3390/vaccines8020303