A Pool of Eight Virally Vectored African Swine Fever Antigens Protect Pigs against Fatal Disease

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Viruses and Cells
2.3. Recombinant Vectors
2.4. Swine Leucocyte Antigen (SLA) Genotyping
2.5. Animal Experiments and Ethics Statement
2.5.1. Experiment 1
2.5.2. Experiment 2
2.6. Interferon Gamma (IFNγ) ELISpot
2.7. Fixed-Cell Assays
2.7.1. Immunoperoxidase Assay (IPA) against the Whole Virus
2.7.2. Immunofluorescence Assay (IFA) against Individual Proteins
2.8. Indirect ELISA
2.9. Statistical Analysis
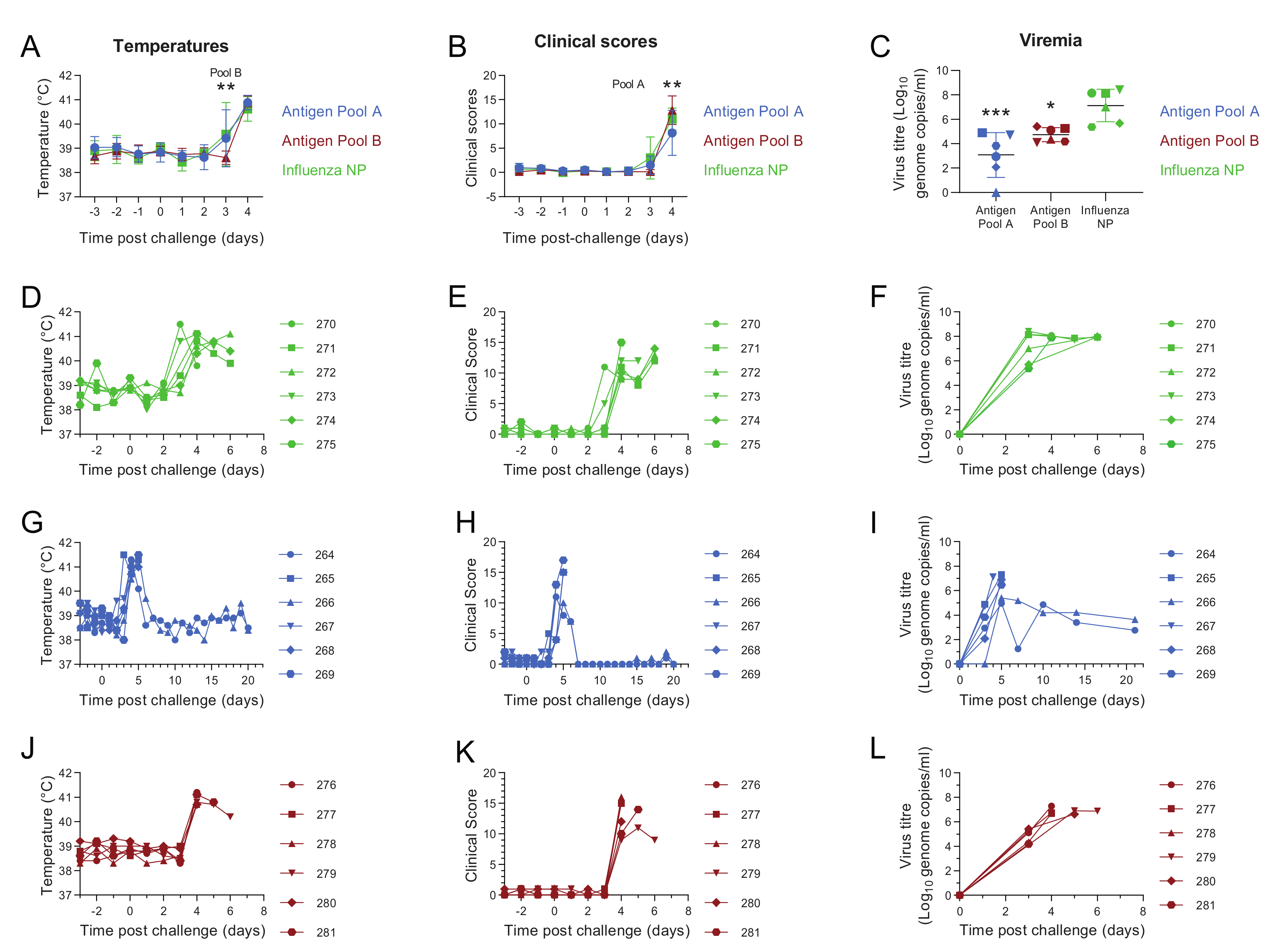
3. Results
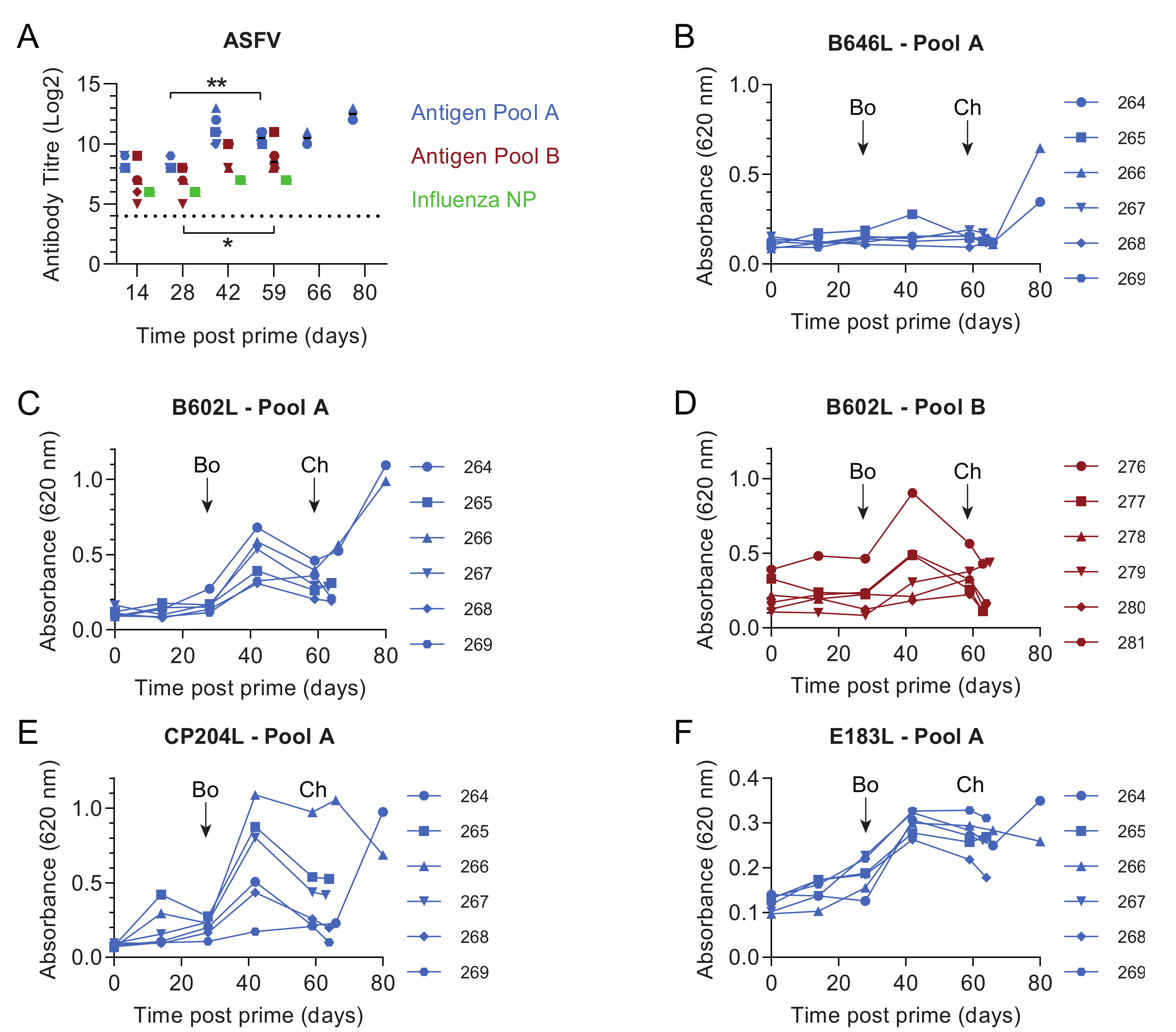
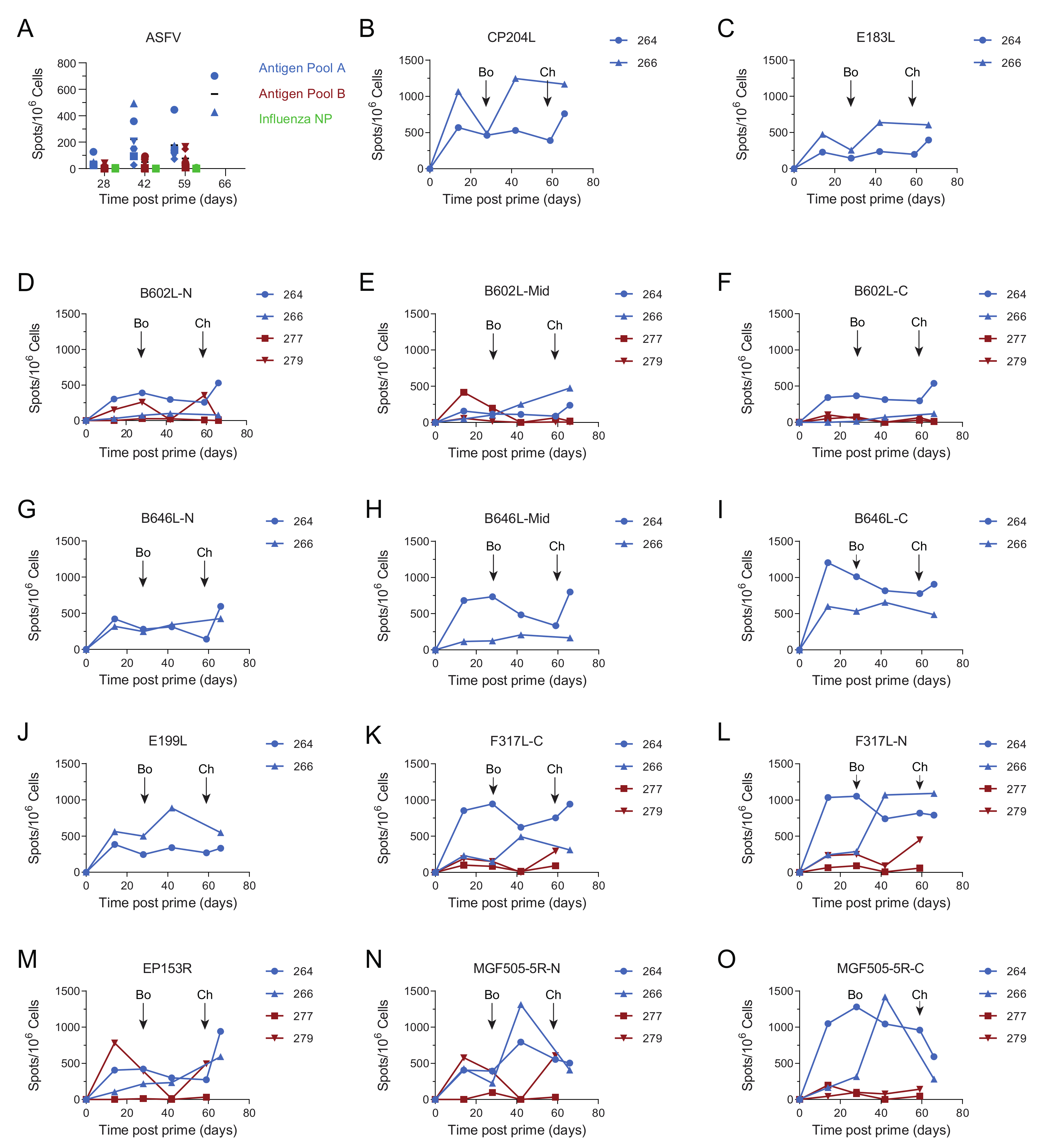
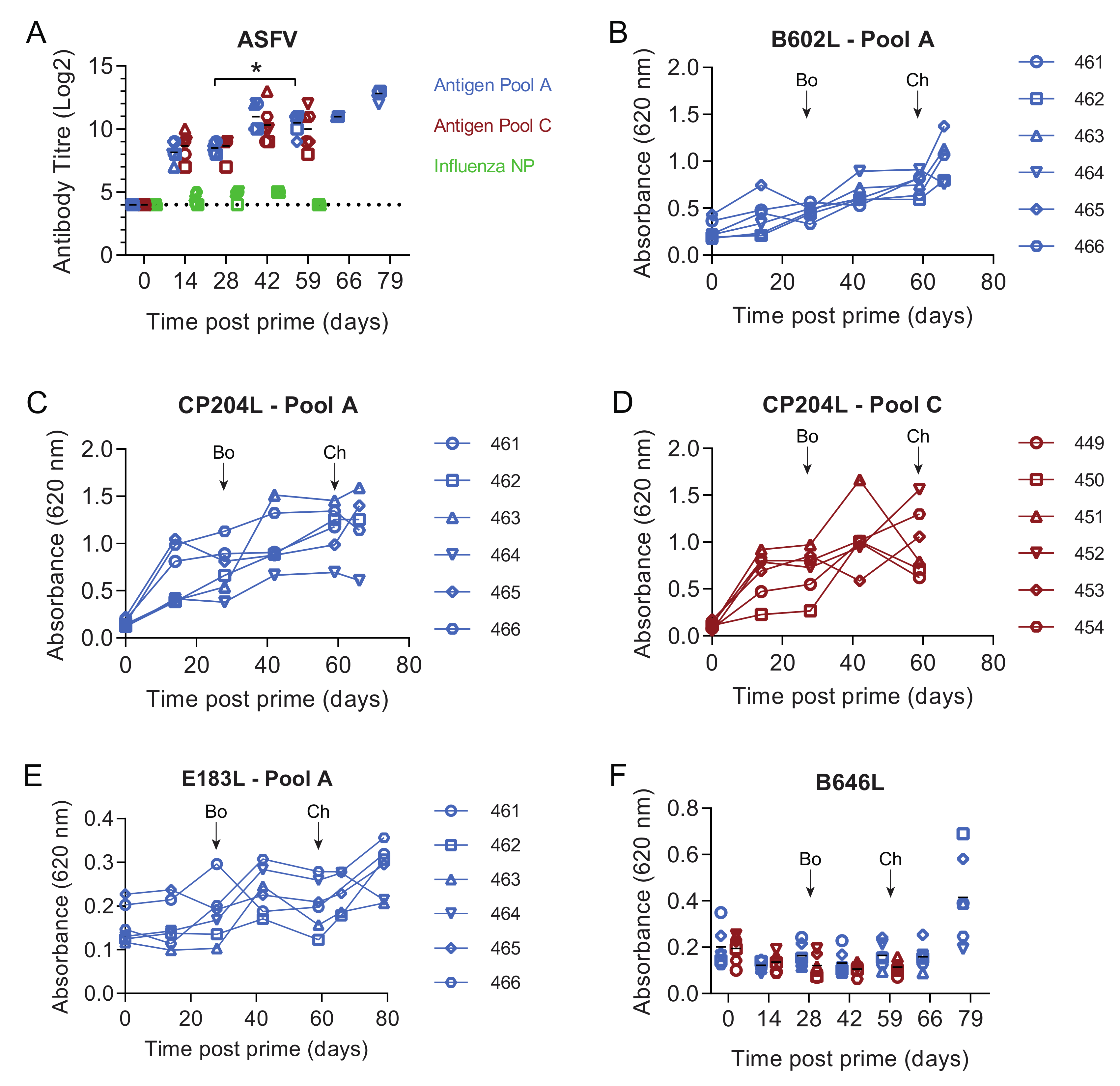
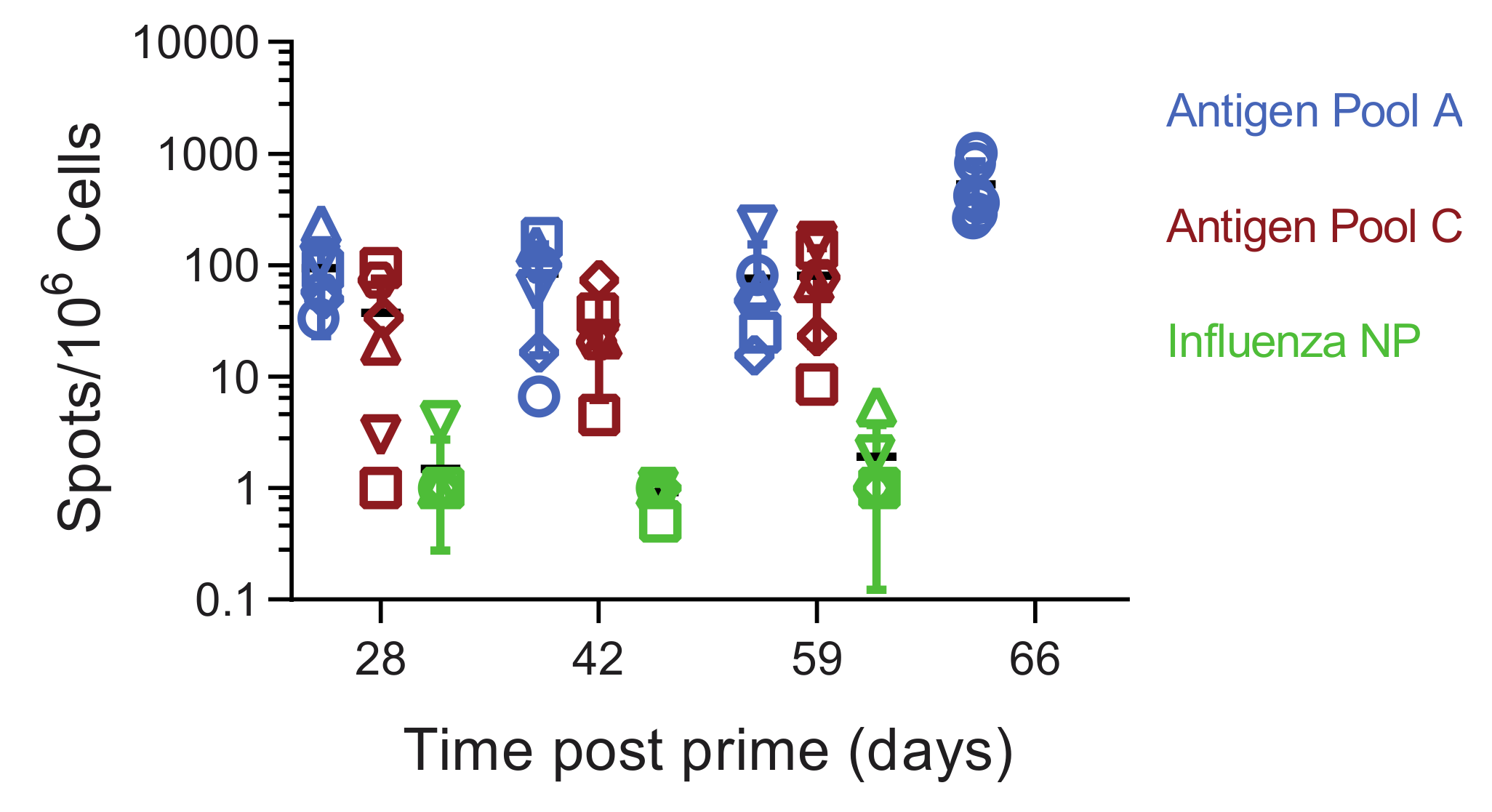
3.1. Pools of Eight Viral Vectors Induce ASFV-Specific and Antigen-Specific Responses in Swine
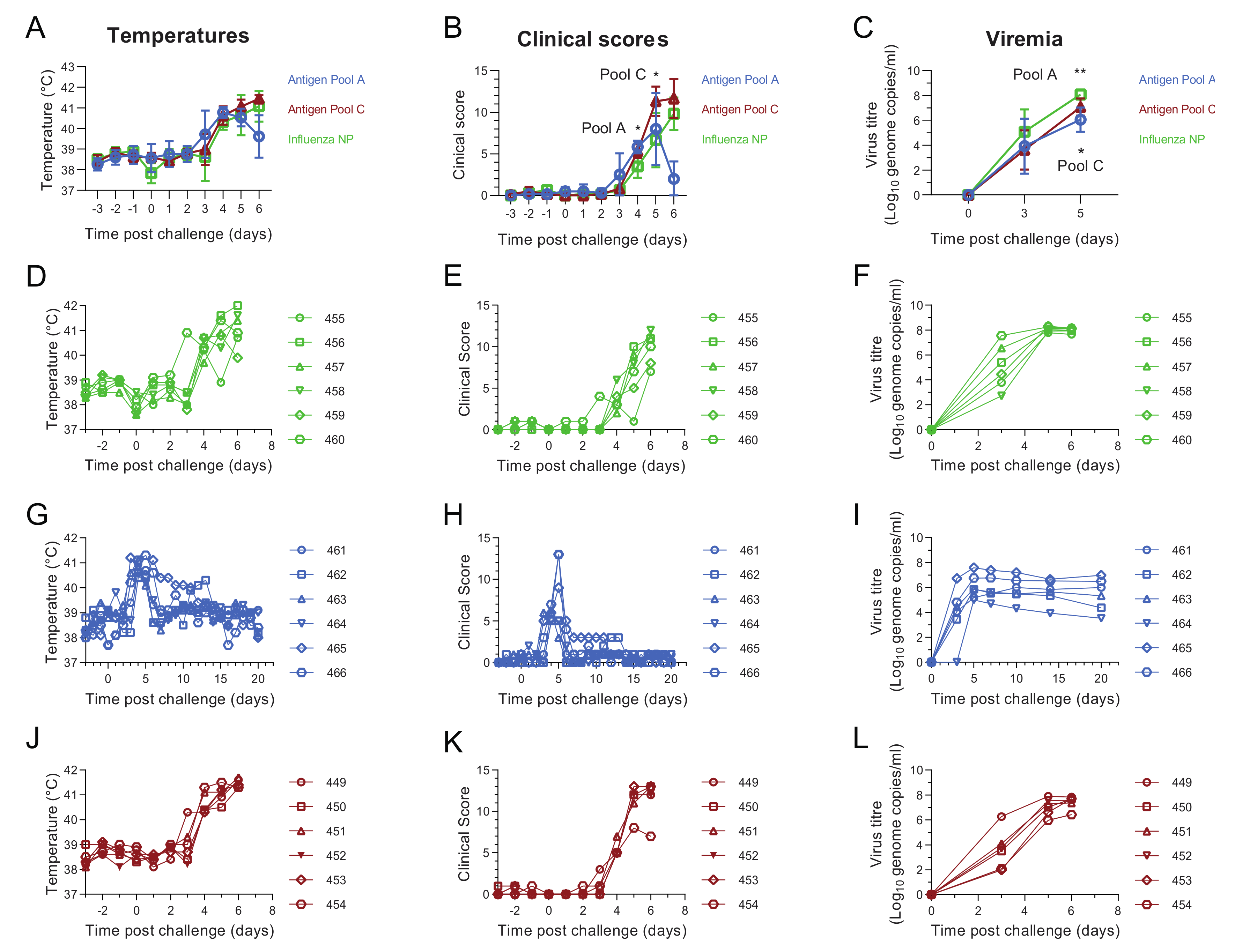
3.2. Comparative Evaluation of Temperatures, Clinical Signs and Viremia Levels in Experiment 1
3.3. Evaluation of Macroscopic Lesions and Viral Load in the Tissues in Experiment 1
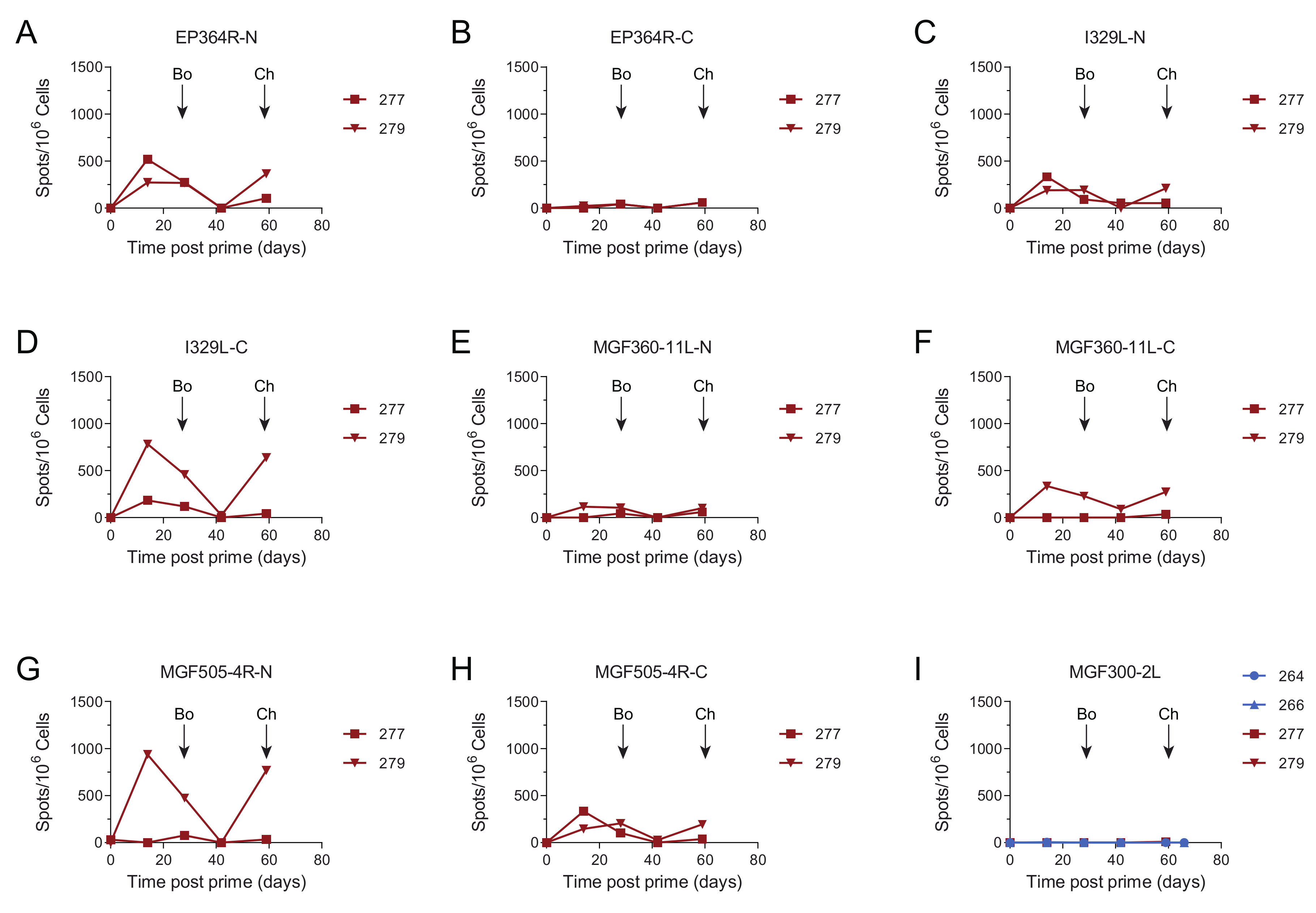
3.4. Immune Responses after Challenge in Surviving Animals in Experiment 1
3.5. Immune Responses in Experiment 2
3.6. Antigen Pool A Protected 100% of Pigs from Fatal Disease
3.7. Evaluation of Macroscopic Lesions in Experiment 2
3.8. Antibody and Cellular Responses after Challenge in Pigs Immunised with Antigen Pool A in Experiment 2
3.9. Clinical Outcomes Did Not Correlate with Inferred SLA Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manso Ribeiro, J. Déclaration sur la vaccination contre la Peste Porcine Africaine à la XXXe Session Générale de l’Office International des Epizooties. Bull. Off. Int. Épiz. 1962, 58, 1031–1040. [Google Scholar]
- Leitao, A.; Cartaxeiro, C.; Coelho, R.; Cruz, B.; Parkhouse, R.M.; Portugal, F.; Vigario, J.D.; Martins, C.L. The non-haemadsorbing African swine fever virus isolate ASFV/NH/P68 provides a model for defining the protective anti-virus immune response. J. Gen. Virol. 2001, 82, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cordon, P.J.; Chapman, D.; Jabbar, T.; Reis, A.L.; Goatley, L.; Netherton, C.L.; Taylor, G.; Montoya, M.; Dixon, L. Different routes and doses influence protection in pigs immunised with the naturally attenuated African swine fever virus isolate OURT88/3. Antiviral. Res. 2017, 138, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, P.L.; Lacasta, A.; Lopez, E.; Bosch, L.; Collado, J.; Pina-Pedrero, S.; Correa-Fiz, F.; Accensi, F.; Navas, M.J.; Vidal, E.; et al. BA71 Delta CD2: A new recombinant live attenuated African swine fever virus with cross-protective capabilities. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- O’Donnell, V.; Risatti, G.R.; Holinka, L.G.; Krug, P.W.; Carlson, J.; Velazquez-Salinas, L.; Azzinaro, P.A.; Gladue, D.P.; Borca, M.V. Simultaneous Deletion of the 9GL and UK Genes from the African Swine Fever Virus Georgia 2007 Isolate Offers Increased Safety and Protection against Homologous Challenge. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Reis, A.L.; Abrams, C.C.; Goatley, L.C.; Netherton, C.; Chapman, D.G.; Sanchez-Cordon, P.; Dixon, L.K. Deletion of African swine fever virus interferon inhibitors from the genome of a virulent isolate reduces virulence in domestic pigs and induces a protective response. Vaccine 2016, 34, 4698–4705. [Google Scholar] [CrossRef]
- Reis, A.L.; Goatley, L.C.; Jabbar, T.; Sanchez-Cordon, P.J.; Netherton, C.L.; Chapman, D.G.; Dixon, L.K. Deletion of the African swine fever virus gene DP148R does not reduce virus replication in culture but reduces virus virulence in pigs and induces high levels of protection against challenge. J. Virol. 2017. [Google Scholar] [CrossRef]
- Ramirez-Medina, E.; Vuono, E.; O’Donnell, V.; Holinka, L.G.; Silva, E.; Rai, A.; Pruitt, S.; Carrillo, C.; Gladue, D.P.; Borca, M.V. Differential Effect of the Deletion of African Swine Fever Virus Virulence-Associated Genes in the Induction of Attenuation of the Highly Virulent Georgia Strain. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Borca, M.V.; Ramirez-Medina, E.; Silva, E.; Vuono, E.; Rai, A.; Pruitt, S.; Holinka, L.G.; Velazquez-Salinas, L.; Zhu, J.; Gladue, D.P. Development of a Highly Effective African Swine Fever Virus Vaccine by Deletion of the I177L Gene Results in Sterile Immunity against the Current Epidemic Eurasia Strain. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, D.; He, X.; Liu, R.; Wang, Z.; Zhang, X.; Li, F.; Shan, D.; Chen, H.; Zhang, J.; et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci. China Life Sci. 2020, 10. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Richt, J.A. Subunit Vaccine Approaches for African Swine Fever Virus. Vaccines 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Neilan, J.G.; Zsak, L.; Lu, Z.; Burrage, T.G.; Kutish, G.F.; Rock, D.L. Neutralizing antibodies to African swine fever virus proteins p30, p54, and p72 are not sufficient for antibody-mediated protection. Virology 2004, 319, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Barderas, M.G.; Rodriguez, F.; Gomez-Puertas, P.; Aviles, M.; Beitia, F.; Alonso, C.; Escribano, J.M. Antigenic and immunogenic properties of a chimera of two immunodominant African swine fever virus proteins. Arch. Virol. 2001, 146, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gonzalvo, F.; Rodriguez, F.; Escribano, J.M. Functional and immunological properties of the baculovirus-expressed hemagglutinin of African swine fever virus. Virology 1996, 218, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Argilaguet, J.M.; Perez-Martin, E.; Nofrarias, M.; Gallardo, C.; Accensi, F.; Lacasta, A.; Mora, M.; Ballester, M.; Galindo-Cardiel, I.; Lopez-Soria, S.; et al. DNA vaccination partially protects against African swine fever virus lethal challenge in the absence of antibodies. PLoS ONE 2012, 7, e40942. [Google Scholar] [CrossRef] [PubMed]
- Lacasta, A.; Ballester, M.; Monteagudo, P.L.; Rodriguez, J.M.; Salas, M.L.; Accensi, F.; Pina-Pedrero, S.; Bensaid, A.; Argilaguet, J.; Lopez-Soria, S.; et al. Expression library immunization can confer protection against lethal challenge with African swine fever virus. J. Virol. 2014, 88, 13322–13332. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, S.Y.; Perez-Nunez, D.; Morozov, I.; Sanchez, E.G.; Gaudreault, N.N.; Trujillo, J.D.; Mur, L.; Nogal, M.; Madden, D.; Urbaniak, K.; et al. DNA-Protein Vaccination Strategy Does Not Protect from Challenge with African Swine Fever Virus Armenia 2007 Strain. Vaccines 2019, 7. [Google Scholar] [CrossRef]
- Lokhandwala, S.; Waghela, S.D.; Bray, J.; Martin, C.L.; Sangewar, N.; Charendoff, C.; Shetti, R.; Ashley, C.; Chen, C.H.; Berghman, L.R.; et al. Induction of Robust Immune Responses in Swine by Using a Cocktail of Adenovirus-Vectored African Swine Fever Virus Antigens. Clin. Vaccine Immun. 2016, 23, 888–900. [Google Scholar] [CrossRef]
- Lokhandwala, S.; Waghela, S.D.; Bray, J.; Sangewar, N.; Charendoff, C.; Martin, C.L.; Hassan, W.S.; Koynarski, T.; Gabbert, L.; Burrage, T.G.; et al. Adenovirus-vectored novel African Swine Fever Virus antigens elicit robust immune responses in swine. PLoS ONE 2017, 12, e0177007. [Google Scholar] [CrossRef]
- Lopera-Madrid, J.; Osorio, J.E.; He, Y.; Xiang, Z.; Adams, L.G.; Laughlin, R.C.; Mwangi, W.; Subramanya, S.; Neilan, J.; Brake, D.; et al. Safety and immunogenicity of mammalian cell derived and Modified Vaccinia Ankara vectored African swine fever subunit antigens in swine. Vet. Immunol. Immunopathol. 2017, 185, 20–33. [Google Scholar] [CrossRef]
- Netherton, C.L.; Goatley, L.C.; Reis, A.L.; Portugal, R.; Nash, R.H.; Morgan, S.B.; Gault, L.; Nieto, R.; Norlin, V.; Gallardo, C.; et al. Identification and Immunogenicity of African Swine Fever Virus Antigens. Front. Immunol. 2019, 10, 1318. [Google Scholar] [CrossRef] [PubMed]
- Lokhandwala, S.; Petrovan, V.; Popescu, L.; Sangewar, N.; Elijah, C.; Stoian, A.; Olcha, M.; Ennen, L.; Bray, J.; Bishop, R.P.; et al. Adenovirus-vectored African Swine Fever Virus antigen cocktails are immunogenic but not protective against intranasal challenge with Georgia 2007/1 isolate. Vet. Microbiol. 2019, 235, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Cadenas-Fernandez, E.; Sanchez-Vizcaino, J.M.; Kosowska, A.; Rivera, B.; Mayoral-Alegre, F.; Rodriguez-Bertos, A.; Yao, J.; Bray, J.; Lokhandwala, S.; Mwangi, W.; et al. Adenovirus-vectored African Swine Fever Virus Antigens Cocktail Is Not Protective against Virulent Arm07 Isolate in Eurasian Wild Boar. Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Onisk, D.V.; Borca, M.V.; Kutish, G.; Kramer, E.; Irusta, P.; Rock, D.L. Passively transferred African swine fever virus antibodies protect swine against lethal infection. Virology 1994, 198, 350–354. [Google Scholar] [CrossRef]
- Burmakina, G.; Malogolovkin, A.; Tulman, E.R.; Zsak, L.; Delhon, G.; Diel, D.G.; Shobogorov, N.M.; Morgunov, Y.P.; Morgunov, S.Y.; Kutish, G.F.; et al. African swine fever virus serotype-specific proteins are significant protective antigens for African swine fever. J. Gen. Virol. 2016, 97, 1670–1675. [Google Scholar] [CrossRef]
- Oura, C.A.; Denyer, M.S.; Takamatsu, H.; Parkhouse, R.M. In vivo depletion of CD8+ T lymphocytes abrogates protective immunity to African swine fever virus. J. Gen. Virol. 2005, 86, 2445–2450. [Google Scholar] [CrossRef]
- King, K.; Chapman, D.; Argilaguet, J.M.; Fishbourne, E.; Hutet, E.; Cariolet, R.; Hutchings, G.; Oura, C.A.; Netherton, C.L.; Moffat, K.; et al. Protection of European domestic pigs from virulent African isolates of African swine fever virus by experimental immunisation. Vaccine 2011, 29, 4593–4600. [Google Scholar] [CrossRef]
- Lacasta, A.; Monteagudo, P.L.; Jimenez-Marin, A.; Accensi, F.; Ballester, M.; Argilaguet, J.; Galindo-Cardiel, I.; Segales, J.; Salas, M.L.; Dominguez, J.; et al. Live attenuated African swine fever viruses as ideal tools to dissect the mechanisms involved in viral pathogenesis and immune protection. Vet. Res. 2015, 46, 135. [Google Scholar] [CrossRef]
- Jancovich, J.K.; Chapman, D.; Hansen, D.T.; Robida, M.D.; Loskutov, A.; Craciunescu, F.; Borovkov, A.; Kibler, K.; Goatley, L.; King, K.; et al. Immunisation of pigs by DNA prime and recombinant vaccinia virus boost to identify and rank African swine fever virus immunogenic and protective proteins. J. Virol. 2018, 92, e02219-17. [Google Scholar] [CrossRef]
- Netherton, C.; Rouiller, I.; Wileman, T. The subcellular distribution of multigene family 110 proteins of African swine fever virus is determined by differences in C-terminal KDEL endoplasmic reticulum retention motifs. J. Virol. 2004, 78, 3710–3721. [Google Scholar] [CrossRef]
- Enjuanes, L.; Carrascosa, A.L.; Moreno, M.A.; Viñuela, E. Titration of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Boinas, F.S.; Hutchings, G.H.; Dixon, L.K.; Wilkinson, P.J. Characterization of pathogenic and non-pathogenic African swine fever virus isolates from Ornithodoros erraticus inhabiting pig premises in Portugal. J. Gen. Virol. 2004, 85, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Reid, S.M.; Hutchings, G.H.; Grierson, S.S.; Wilkinson, P.J.; Dixon, L.K.; Bastos, A.D.; Drew, T.W. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J. Virol. Methods 2003, 107, 53–61. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Guzman, E.; Turner, A.; Gilbert, S.C.; Prentice, H.; Hope, J.C.; Charleston, B. Differential effects of viral vectors on migratory afferent lymph dendritic cells in vitro predict enhanced immunogenicity in vivo. J. Virol. 2011, 85, 9385–9394. [Google Scholar] [CrossRef]
- Chapman, D.A.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Morris, S.J.; Turner, A.V.; Green, N.; Warimwe, G.M. Laboratory-Scale Production of Replication-Deficient Adenovirus Vectored Vaccines. Methods Mol. Biol. 2016, 1349, 121–135. [Google Scholar] [CrossRef]
- Pavot, V.; Sebastian, S.; Turner, A.V.; Matthews, J.; Gilbert, S.C. Generation and Production of Modified Vaccinia Virus Ankara (MVA) as a Vaccine Vector. Methods Mol. Biol. Clifton N.J. 2017, 1581, 97–119. [Google Scholar] [CrossRef]
- Ho, C.S.; Lunney, J.K.; Ando, A.; Rogel-Gaillard, C.; Lee, J.H.; Schook, L.B.; Smith, D.M. Nomenclature for factors of the SLA system, update 2008. Tissue Antigens 2009, 73, 307–315. [Google Scholar] [CrossRef]
- Ho, C.S.; Lunney, J.K.; Lee, J.H.; Franzo-Romain, M.H.; Martens, G.W.; Rowland, R.R.; Smith, D.M. Molecular characterization of swine leucocyte antigen class II genes in outbred pig populations. Anim. Genet. 2010, 41, 428–432. [Google Scholar] [CrossRef]
- Smith, D.M.; Lunney, J.K.; Ho, C.S.; Martens, G.W.; Ando, A.; Lee, J.H.; Schook, L.; Renard, C.; Chardon, P. Nomenclature for factors of the swine leukocyte antigen class II system, 2005. Tissue Antigens 2005, 66, 623–639. [Google Scholar] [CrossRef]
- Smith, D.M.; Lunney, J.K.; Martens, G.W.; Ando, A.; Lee, J.H.; Ho, C.S.; Schook, L.; Renard, C.; Chardon, P. Nomenclature for factors of the SLA class-I system, 2004. Tissue Antigens 2005, 65, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Maccari, G.; Robinson, J.; Ballingall, K.; Guethlein, L.A.; Grimholt, U.; Kaufman, J.; Ho, C.S.; de Groot, N.G.; Flicek, P.; Bontrop, R.E.; et al. IPD-MHC 2.0: an improved inter-species database for the study of the major histocompatibility complex. Nucleic. Acids. Res. 2017, 45, d860–d864. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Cardiel, I.; Ballester, M.; Solanes, D.; Nofrarias, M.; Lopez-Soria, S.; Argilaguet, J.M.; Lacasta, A.; Accensi, F.; Rodriguez, F.; Segales, J. Standardization of pathological investigations in the framework of experimental ASFV infections. Virus Res. 2013, 173, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Soler, A.; Nieto, R.; Carrascosa, A.L.; De Mia, G.M.; Bishop, R.P.; Martins, C.; Fasina, F.O.; Couacy-Hymman, E.; Heath, L.; et al. Comparative evaluation of novel African swine fever virus (ASF) antibody detection techniques derived from specific ASF viral genotypes with the OIE internationally prescribed serological tests. Vet. Microbiol. 2013, 162, 32–43. [Google Scholar] [CrossRef]
- Reis, A.L.; Parkhouse, R.M.; Penedos, A.R.; Martins, C.; Leitao, A. Systematic analysis of longitudinal serological responses of pigs infected experimentally with African swine fever virus. J. Gen. Virol. 2007, 88, 2426–2434. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Hemmink, J.D.; Graham, S.P.; Tchilian, E.; Charleston, B.; Hammer, S.E.; Ho, C.S.; Hammond, J.A. The MHC homozygous inbred Babraham pig as a resource for veterinary and translational medicine. Hla. 2018, 1, 40–43. [Google Scholar] [CrossRef]
- Manso Ribeiro, J.; Nunes Petisca, J.L.; Lopes Frazao, F.; Sobral, M. Vaccination contre la Peste Porcine Africaine. Bull. Off. Int. Épiz. 1963, 60, 921–937. [Google Scholar]
- Zsak, L.; Onisk, D.V.; Afonso, C.L.; Rock, D.L. Virulent African swine fever virus isolates are neutralized by swine immune serum and by monoclonal antibodies recognizing a 72-kDa viral protein. Virology 1993, 196, 596–602. [Google Scholar] [CrossRef]
- Borca, M.V.; Irusta, P.; Carrillo, C.; Afonso, C.L.; Burrage, T.; Rock, D.L. African swine fever virus structural protein p72 contains a conformational neutralizing epitope. Virology 1994, 201, 413–418. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Liu, S.; Luo, Y.; Wang, Y.; Li, S.; Zhao, Z.; Bi, Y.; Sun, J.; Peng, R.; Song, H.; Zhu, D.; et al. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843. [Google Scholar] [CrossRef]
- Andres, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G.A. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, C.; Windsor, M.; Wileman, T. A virally encoded chaperone specialized for folding of the major capsid protein of African swine fever virus. J. Virol. 2001, 75, 7221–7229. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, B.; Qian, N.; Zhang, F.; Tan, X.; Lei, J.; Xiang, Y. Structure of the African swine fever virus major capsid protein p72. Cell Res. 2019, 29, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Norley, S.G.; Wardley, R.C. Effector mechanisms in the pig. Antibody-dependent cellular cytolysis of African swine fever virus infected cells. Res. Vet. Sci. 1983, 35, 75–79. [Google Scholar] [CrossRef]
- Popescu, L.; Gaudreault, N.N.; Whitworth, K.M.; Murgia, M.V.; Nietfeld, J.C.; Mileham, A.; Samuel, M.; Wells, K.D.; Prather, R.S.; Rowland, R.R.R. Genetically edited pigs lacking CD163 show no resistance following infection with the African swine fever virus isolate, Georgia 2007/1. Virology 2017, 501, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, H.H.; Denyer, M.S.; Lacasta, A.; Stirling, C.M.; Argilaguet, J.M.; Netherton, C.L.; Oura, C.A.; Martins, C.; Rodriguez, F. Cellular immunity in ASFV responses. Virus. Res. 2013, 173, 110–121. [Google Scholar] [CrossRef]
- Mur, L.; Gallardo, C.; Soler, A.; Zimmermman, J.; Pelayo, V.; Nieto, R.; Sanchez-Vizcaino, J.M.; Arias, M. Potential use of oral fluid samples for serological diagnosis of African swine fever. Vet. Microbiol. 2013, 165, 135–139. [Google Scholar] [CrossRef]
- Gallardo, C.; Sanchez, E.G.; Perez-Nunez, D.; Nogal, M.; de Leon, P.; Carrascosa, A.L.; Nieto, R.; Soler, A.; Arias, M.L.; Revilla, Y. African swine fever virus (ASFV) protection mediated by NH/P68 and NH/P68 recombinant live-attenuated viruses. Vaccine 2018, 36, 2694–2704. [Google Scholar] [CrossRef]
- Luka, P.D.; Achenbach, J.E.; Mwiine, F.N.; Lamien, C.E.; Shamaki, D.; Unger, H.; Erume, J. Genetic Characterization of Circulating African Swine Fever Viruses in Nigeria (2007–2015). Trans. Emerg. Dis. 2017, 64, 1598–1609. [Google Scholar] [CrossRef]
- Kouakou, K.V.; Michaud, V.; Biego, H.G.; Gnabro, H.P.G.; Kouakou, A.V.; Mossoun, A.M.; Awuni, J.A.; Minoungou, G.L.; Aplogan, G.L.; Awoume, F.K.; et al. African and classical swine fever situation in Ivory-Coast and neighboring countries, 2008–2013. Acta Trop. 2017, 166, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.A.; Penrith, M.L.; Fasina, F.O.; Beltran-Alcrudo, D. The African swine fever epidemic in West Africa, 1996–2002. Trans. Emerg. Dis. 2018, 65, 64–76. [Google Scholar] [CrossRef]
- Wade, A.; Achenbach, J.E.; Gallardo, C.; Settypalli, T.B.K.; Souley, A.; Djonwe, G.; Loitsch, A.; Dauphin, G.; Ngang, J.J.E.; Boyomo, O.; et al. Genetic characterization of African swine fever virus in Cameroon, 2010–2018. J. Microbiol. 2019, 57, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Couacy-Hymann, E.; Kouakou, K.V.; Achenbach, J.E.; Kouadio, L.; Koffi, Y.M.; Godji, H.P.; Adje, K.E.; Oulai, J.; Pell-Minhiaud, H.J.; Lamien, C.E. Re-emergence of genotype I of African swine fever virus in Ivory Coast. Trans. Emerg. Dis. 2019, 66, 882–896. [Google Scholar] [CrossRef]
- Murgia, M.V.; Mogler, M.; Certoma, A.; Green, D.; Monaghan, P.; Williams, D.T.; Rowland, R.R.R.; Gaudreault, N.N. Evaluation of an African swine fever (ASF) vaccine strategy incorporating priming with an alphavirus-expressed antigen followed by boosting with attenuated ASF virus. Arch. Virol. 2019, 164, 359–370. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | 1 | 2 | ||
|---|---|---|---|---|
| Antigen Pool | A | B | A | C |
| Genes | B602L | B602L | B602L | B646L |
| B646L | EP153R | B646L | CP204L | |
| CP204L | EP364R | CP204L | E199L | |
| E183L | F317L | E183L | F317L | |
| E199L | I329L | E199L | MGF505-5R | |
| EP153R | MGF360-11L | EP153R | ||
| F317L | MGF505-4R | F317L | ||
| MGF505-5R | MGF505-5R | MGF505-5R | ||
| rAd Dose (IU) | 5 × 109 | 5 × 109 | 1.5 × 1010 | 1.5 × 1010 |
| MVA Dose (pfu) | 7.5 × 107 | 7.5 × 107 | 2 × 108 | 2 × 108 |
| Protein | Antigen Pool A | Antigen Pool B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 264 | 265 | 266 | 267 | 268 | 269 | 276 | 277 | 278 | 279 | 280 | 281 | |
| E199L | No | No | No | No | No | No | - | - | - | - | - | - |
| EP153R | No | No | No | No | No | No | No | No | No | No | No | No |
| EP364R | - | - | - | - | - | - | No | No | No | No | No | Yes |
| F317L | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| I329L | - | - | - | - | - | - | Yes | Yes | Yes | No | No | Yes |
| MGF360-11L | - | - | - | - | - | - | No | No | No | No | No | No |
| MGF505-4R | - | - | - | - | - | - | No | No | No | No | No | No |
| MGF505-5R | No | No | No | No | No | No | No | No | No | No | No | No |
| Exp. Group | Pig ID | SLA I Allele Specificity | SLA II Allele Specificity | Inferred Haplotype | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SLA-1 | SLA-3 | SLA-2 | DRB1 | DQB1 | DQA | Class I | Class II | Complete | ||
| Bab | Reference | 14:02 | 04:03 | 11:04 | 05:01 | 08:01 | 01:03 | 55/55 | 6/6 | 55.6/55.6 |
| Pool A | 264 | 04:XX,07:XX | 04:XX | 02:XX,04:XX | 06:01,09:XX | 07:XX,08:XX | 01:XX,03:XX | 4/32 | 12b/14 | 4.14/32.12b |
| 265 | 04:XX,16:03 | 04:XX | 04:XX,06:XX | 02:XX,04:03-04 | 04:XX,07:XX | 02:XX,03:XX | 4/24 | 4/19a | 4.4/24.19a | |
| 266 | 04:XX,16:03 | 04:XX | 04:XX,06:XX | 01:XX,04:03-04 | 01:XX,07:XX | 01:XX,03:XX | 4/24 | 1/19a | 4.1/24.19a | |
| 267 | 08:XX,14:01 | 04:XX,06:01 | 06:XX,12:XX | 04:XX,06:XX | 02:02,07:XX | 01:XX,02:XX | 22/62 | 12a/15b | 22.15b/62.12a | |
| 268 | 04:XX,08:XX | 04:XX,06:01 | 04:XX,12:XX | 01:XX,04:XX | 01:XX,02:02 | 01:XX,02:XX | 4/22 | 1/15b | 4.1/22.15b | |
| 269 | 08:XX,09:XX,15:XX | 06:01,07:XX | 05:XX,12:XX | 04:XX,10:XX | 02:02,06:XX | 01:XX,02:XX | 22/28 | 15b/23 | 22.15b/28.23 | |
| Pool B | 276 | 08:XX,16:03 | 04:XX,06:01 | 06:XX,12:XX | 04:XX,04:03-04 | 02:02,07:XX | 02:XX,03:XX | 22/24 | 15b/19a | 22.15b/24.19a |
| 277 | Blank/undetected | 05:XX | 06:XX | 09:XX,11:XX | 04:XX,08:XX | 02:XX,03:XX | 33/33 | 14/26 | 33.14/33.26 | |
| 278 | 04:XX,14:02 | 04:XX | 04:XX,11:04-05 | 01:XX,05:XX | 01:XX,08:XX | 01:XX | 4/55 | 1/6 | 4.1/55.6 | |
| 279 | 11:XX,12:XX,13:01 | 04:XX,05:XX | 04:XX,10:XX | 04:03-04,09:XX | 03:02-03,08:XX | 02:XX,03:XX | 35/43 | 13/14 | 35.13/43.14 | |
| 280 | 11:XX,12:XX,13:01 | 04:XX,05:XX | 04:XX,10:XX | 04:03-04,09:XX | 03:02-03,08:XX | 02:XX,03:XX | 35/43 | 13/14 | 35.13/43.14 | |
| 281 | 08:XX,09:XX,15:XX | 06:01,07:XX | 05:XX,12:XX | 04:XX,10:XX | 02:02,06:XX | 01:XX,02:XX | 22/28 | 15b/23 | 22.15b/28.23 | |
| Pool A | 461 | 07:03,08:XX | 06:01 | 05:XX,12:XX | 04:XX | 02:02 | 02:XX | 21/22 | 15b/15b | 21.15b/22.15b |
| 462 | 07:03,11:XX | 04:XX,06:01 | 04:XX,05:XX | 04:XX,09:XX | 02:02,08:XX | 02:XX,03:XX | 21/43 | 14/15b | 21.15b/43.14 | |
| 463 | 04:XX | 04:XX | 04:XX | 02:XX,09:XX | 04:XX,08:XX | 02:XX,03:XX | 4/4 | 4/14 | 4.4/4.14 | |
| 464 | Blank/undetected | 05:XX,07:XX | 05:XX,06:XX | 10:XX,11:XX | 06:XX | 01:XX | */33 | 23/26 | * 23/33.26 | |
| 465 | 07:03,08:XX | 06:01 | 05:XX,12:XX | 04:XX | 02:02 | 02:XX | 21/22 | 15b/15b | 21.15b/22.15b | |
| 466 | 12:XX,13:01,16:03 | 04:XX,05:XX | 06:XX,10:XX | 04:03-04,10:XX | 06:XX,07:XX | 01:XX,03:XX | 24/35 | 19a/23 | 24.19a/35.23 | |
| Pool C | 449 | 07:03,08:XX | 06:01 | 05:XX,12:XX | 04:XX | 02:02 | 02:XX | 21/22 | 15b/15b | 21.15b/22.15b |
| 450 | 11:XX,14:01 | 04:XX | 04:XX,06:XX | 06:XX,09:XX | 07:XX,08:XX | 01:XX,03:XX | 43/62 | 12a/14 | 43.14/62.12a | |
| 451 | 04:XX,07:03 | 04:XX,06:XX | 04:XX,05:XX | 04:XX,09:XX | 02:02,08:XX | 02:XX,03:XX | 4/21 | 14/15b | 4.14/21.15b | |
| 452 | 08:XX,11:XX | 04:XX,07:XX | 04:XX,05:XX | 04:03-04,10:XX | 06:XX,07:XX | 01:XX,03:XX | 7/43 | 19a/23 | 7.23/43.19a | |
| 453 | 08:XX,12:XX,13:01 | 05:XX,0601 | 10:XX,12:XX | 04:XX,04:03-04 | 02:02,03:02-03 | 02:XX | 22/35 | 13/15b | 22.15b/35.13 | |
| 454 | 08:XX,16:03 | 04:XX,07:XX | 05:XX,06:XX | 04:03-04,10:XX | 06:XX,07:XX | 01:XX,03:XX | 7/24 | 19a/23 | 7.23/24.19a | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goatley, L.C.; Reis, A.L.; Portugal, R.; Goldswain, H.; Shimmon, G.L.; Hargreaves, Z.; Ho, C.-S.; Montoya, M.; Sánchez-Cordón, P.J.; Taylor, G.; et al. A Pool of Eight Virally Vectored African Swine Fever Antigens Protect Pigs against Fatal Disease. Vaccines 2020, 8, 234. https://doi.org/10.3390/vaccines8020234
Goatley LC, Reis AL, Portugal R, Goldswain H, Shimmon GL, Hargreaves Z, Ho C-S, Montoya M, Sánchez-Cordón PJ, Taylor G, et al. A Pool of Eight Virally Vectored African Swine Fever Antigens Protect Pigs against Fatal Disease. Vaccines. 2020; 8(2):234. https://doi.org/10.3390/vaccines8020234
Chicago/Turabian StyleGoatley, Lynnette C., Ana Luisa Reis, Raquel Portugal, Hannah Goldswain, Gareth L. Shimmon, Zoe Hargreaves, Chak-Sum Ho, María Montoya, Pedro J. Sánchez-Cordón, Geraldine Taylor, and et al. 2020. "A Pool of Eight Virally Vectored African Swine Fever Antigens Protect Pigs against Fatal Disease" Vaccines 8, no. 2: 234. https://doi.org/10.3390/vaccines8020234
APA StyleGoatley, L. C., Reis, A. L., Portugal, R., Goldswain, H., Shimmon, G. L., Hargreaves, Z., Ho, C.-S., Montoya, M., Sánchez-Cordón, P. J., Taylor, G., Dixon, L. K., & Netherton, C. L. (2020). A Pool of Eight Virally Vectored African Swine Fever Antigens Protect Pigs against Fatal Disease. Vaccines, 8(2), 234. https://doi.org/10.3390/vaccines8020234