Development of an IL-17A DNA Vaccine to Treat Systemic Lupus Erythematosus in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
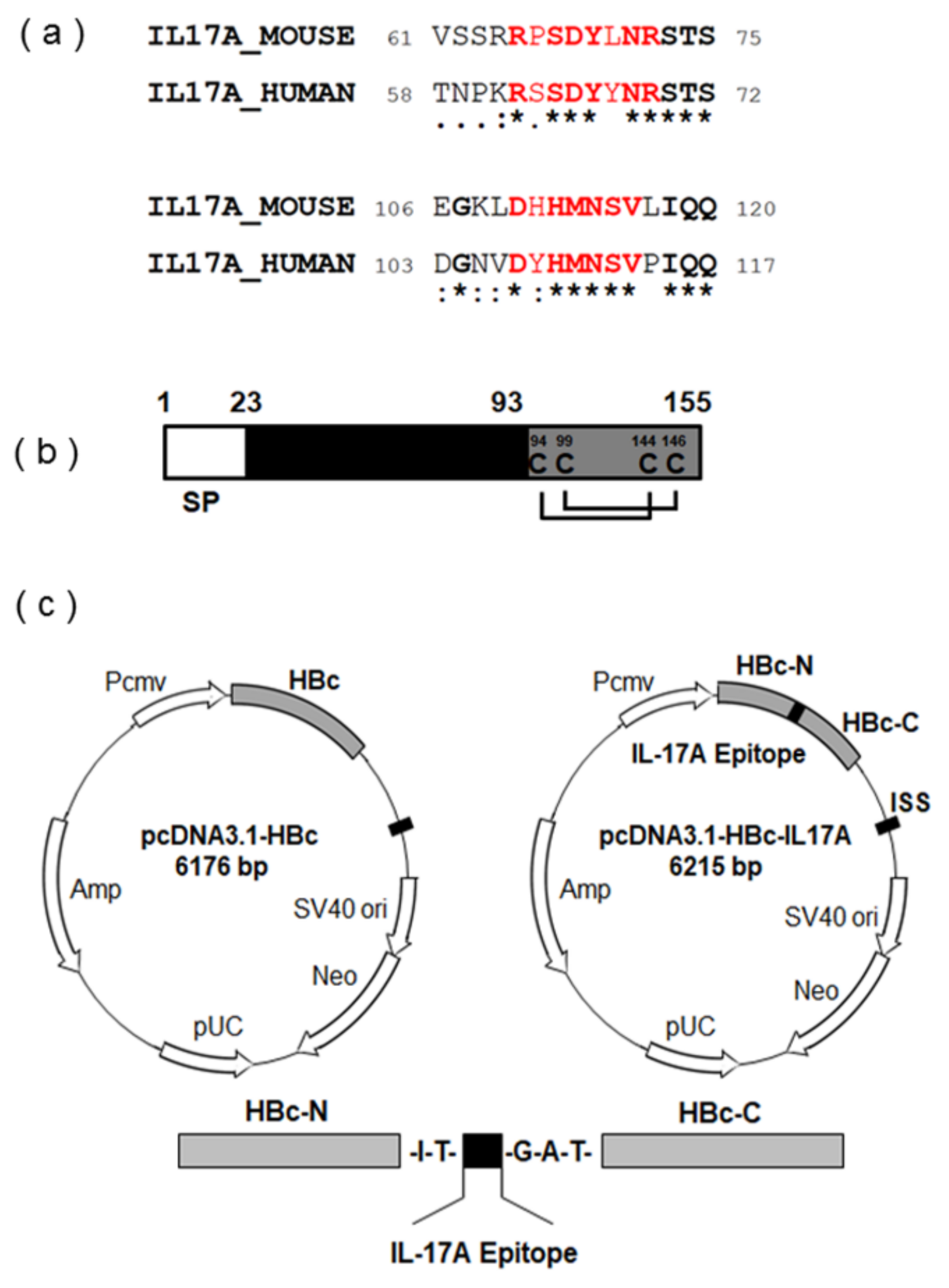
2.1. Vaccine Design and Synthesis
2.2. Mice
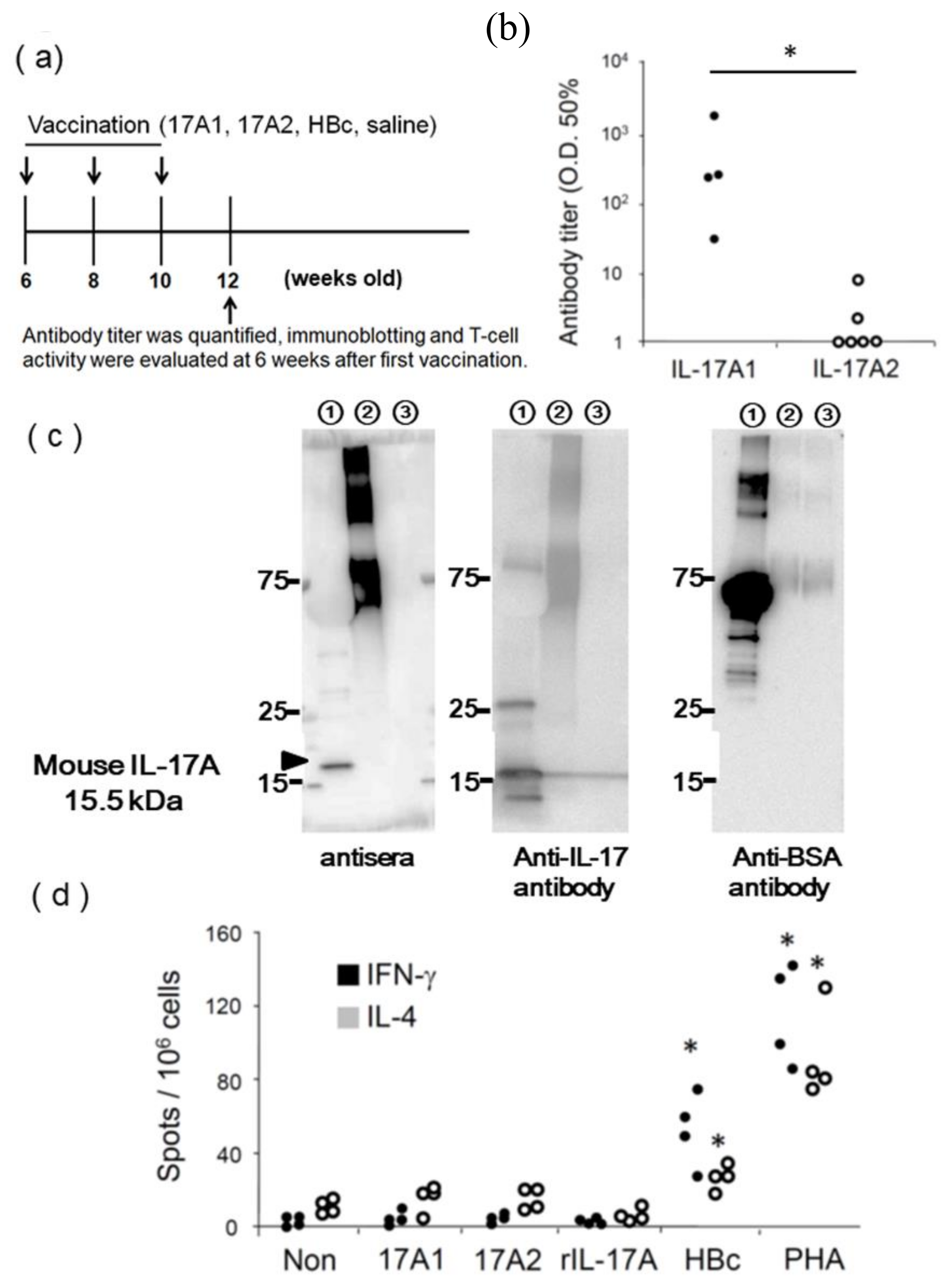
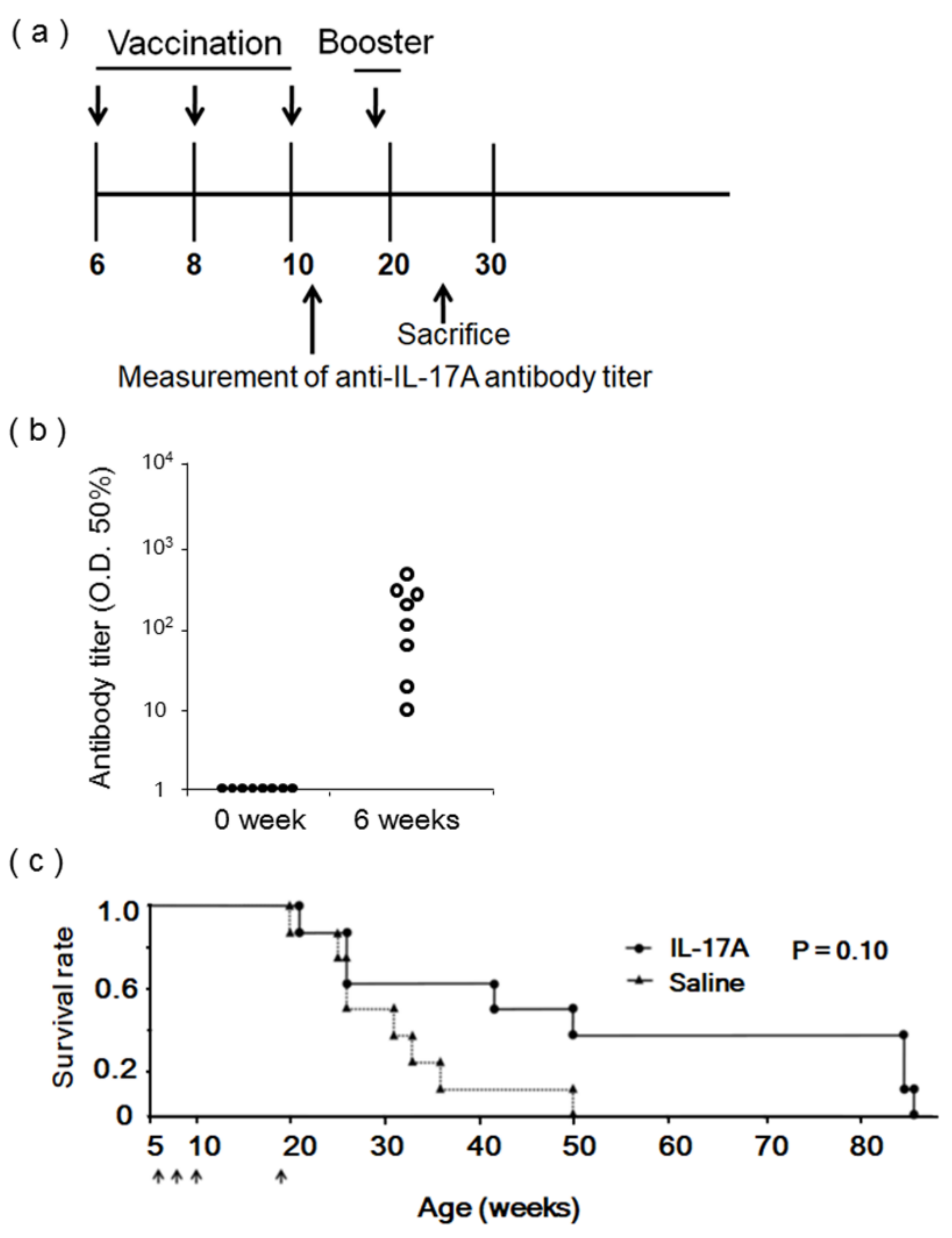
2.3. DNA Immunization
2.4. Anti-IL-17A Antibody Titer Assay
2.5. Immunoblotting
2.6. Measurement of Serum Cytokines
2.7. Histological Examination
2.8. ELISpot Assay
2.9. Statistical Analysis
3. Results
3.1. Screening of Appropriate Antigen Sequence for IL-17A DNA Vaccine
3.2. Evaluation of IL-17A1 Vaccine in NZBWF1 Mice
3.3. Evaluation of IL-17A1 DNA Vaccine in MRL/lpr Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borchers, A.T.; Naguwa, S.M.; Shoenfeld, Y.; Gershwin, M.E. The geoepidemiology of systemic lupus erythematosus. Autoimmun. Rev. 2010, 9, A277–A287. [Google Scholar] [CrossRef] [PubMed]
- Su, D.L.; Lu, Z.M.; Shen, M.N.; Li, X.; Sun, L.Y. Roles of pro- and anti-inflammatory cytokines in the pathogenesis of SLE. J. Biomed. Biotechnol. 2012, 2012, 347141. [Google Scholar] [CrossRef]
- Li, D.; Guo, B.; Wu, H.; Tan, L.; Chang, C.; Lu, Q. Interleukin-17 in systemic lupus erythematosus: A comprehensive review. Autoimmunity 2015, 48, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Baeten, D.L.; Josien, R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin. Immunol. 2014, 154, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Chen, Y.M.; Wen, M.C.; Hsieh, T.Y.; Hung, W.T.; Lan, J.L. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus 2012, 21, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Abdel Galil, S.M.; Ezzeldin, N.; El-Boshy, M.E. The role of serum IL-17 and IL-6 as biomarkers of disease activity and predictors of remission in patients with lupus nephritis. Cytokine 2015, 76, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Apostolidis, S.A.; Rosetti, F.; Keszei, M.; Wang, N.; Terhorst, C.; Mayadas, T.N.; Tsokos, G.C. Cutting edge: Protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J. Immunol. 2012, 188, 3567–3571. [Google Scholar] [CrossRef]
- Koriyama, H.; Nakagami, H.; Nakagami, F.; Osako, M.K.; Kyutoku, M.; Shimamura, M.; Kurinami, H.; Katsuya, T.; Rakugi, H.; Morishita, R. Long-Term Reduction of High Blood Pressure by Angiotensin II DNA Vaccine in Spontaneously Hypertensive Rats. Hypertension 2015, 66, 167–174. [Google Scholar] [CrossRef]
- Pang, Z.; Nakagami, H.; Osako, M.K.; Koriyama, H.; Nakagami, F.; Tomioka, H.; Shimamura, M.; Kurinami, H.; Takami, Y.; Morishita, R.; et al. Therapeutic vaccine against DPP4 improves glucose metabolism in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1256–E1263. [Google Scholar] [CrossRef]
- Pérez de Lema, G.; Maier, H.; Franz, T.J.; Escribese, M.; Chilla, S.; Segerer, S.; Camarasa, N.; Schmid, H.; Banas, B.; Kalaydjiev, S.; et al. Chemokine receptor Ccr2 deficiency reduces renal disease and prolongs survival in MRL/lpr lupus-prone mice. J. Am. Soc. Nephrol. 2005, 16, 3592–3601. [Google Scholar] [CrossRef]
- Liu, S.; Song, X.; Chrunyk, B.A.; Shanker, S.; Hoth, L.R.; Marr, E.S.; Griffor, M.C. Crystal structures of interleukin 17A and its complex with IL-17 receptor A. Nat. Commun. 2013, 4, 1888. [Google Scholar] [CrossRef] [PubMed]
- Pumpens, P.; Grens, E. HBV core particles as a carrier for B cell/T cell epitopes. Intervirology 2001, 44, 98–114. [Google Scholar] [CrossRef]
- Seavey, M.M.; Lu, L.D.; Stump, K.L.; Wallace, N.H.; Ruggeri, B.A. Novel, orally active, proteasome inhibitor, delanzomib (CEP-18770), ameliorates disease symptoms and glomerulonephritis in two preclinical mouse models of SLE. Int. Immunopharmacol. 2012, 12, 257–270. [Google Scholar] [CrossRef]
- Venegas-Pont, M.; Manigrasso, M.B.; Grifoni, S.C.; LaMarca, B.B.; Maric, C.; Racusen, L.C.; Glover, P.H.; Jones, A.V.; Drummond, H.A.; Ryan, M.J. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 2010, 56, 643–649. [Google Scholar] [CrossRef]
- Hayashi, T.; Shimoyama, N.; Mizuno, T. Destruction of salivary and lacrimal glands by Th1-polarized reaction in a model of secondary Sjogren’s syndrome in lupus-prone female NZB × NZWF1 mice. Inflammation 2012, 35, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Amarilyo, G.; Lourenco, E.V.; Shi, F.D.; La Cava, A. IL-17 promotes murine lupus. J. Immunol. 2014, 193, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Pisitkun, P.; Ha, H.L.; Wang, H.; Claudio, E.; Tivy, C.C.; Zhou, H.; Mayadas, T.N.; Illei, G.G.; Siebenlist, U. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 2012, 37, 1104–1115. [Google Scholar] [CrossRef]
- Röhn, T.A.; Jennings, G.T.; Hernandez, M.; Grest, P.; Beck, M.; Zou, Y.; Kopf, M.; Bachmann, M.F. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur. J. Immunol. 2006, 36, 2857–2867. [Google Scholar] [CrossRef]
- Guan, Q.; Weiss, C.R.; Qing, G.; Ma, Y.; Peng, Z. An IL-17 peptide-based and virus-like particle vaccine enhances the bioactivity of IL-17 in vitro and in vivo. Immunotherapy 2012, 4, 1799–1807. [Google Scholar] [CrossRef]
- Tabarkiewicz, J.; Pogoda, K.; Karczmarczyk, A.; Pozarowski, P.; Giannopoulos, K. The Role of IL-17 and Th17 Lymphocytes in Autoimmune Diseases. Arch. Immunol. Ther. Exp. 2015, 63, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Titov, A.A.; Morel, L. An update on lupus animal models. Curr. Opin. Rheumatol. 2017, 29, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Cather, J.C.; Jarratt, M.; Meng, X.; Guana, A.; Nyirady, J. Efficacy of secukinumab on moderate-to-severe plaque psoriasis affecting different body regions: A pooled analysis of four phase 3 studies. Dermatol. Ther. 2016, 6, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Paust, H.J.; Krebs, C.F.; Turner, J.E.; Kaffke, A.; Bennstein, S.B.; Koyro, T.; Peters, A.; Velden, J.; Hünemörder, S.; et al. Function of the Th17/Interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol. 2015, 67, 475–487. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koriyama, H.; Ikeda, Y.; Nakagami, H.; Shimamura, M.; Yoshida, S.; Rakugi, H.; Morishita, R. Development of an IL-17A DNA Vaccine to Treat Systemic Lupus Erythematosus in Mice. Vaccines 2020, 8, 83. https://doi.org/10.3390/vaccines8010083
Koriyama H, Ikeda Y, Nakagami H, Shimamura M, Yoshida S, Rakugi H, Morishita R. Development of an IL-17A DNA Vaccine to Treat Systemic Lupus Erythematosus in Mice. Vaccines. 2020; 8(1):83. https://doi.org/10.3390/vaccines8010083
Chicago/Turabian StyleKoriyama, Hiroshi, Yuka Ikeda, Hironori Nakagami, Munehisa Shimamura, Shota Yoshida, Hiromi Rakugi, and Ryuichi Morishita. 2020. "Development of an IL-17A DNA Vaccine to Treat Systemic Lupus Erythematosus in Mice" Vaccines 8, no. 1: 83. https://doi.org/10.3390/vaccines8010083
APA StyleKoriyama, H., Ikeda, Y., Nakagami, H., Shimamura, M., Yoshida, S., Rakugi, H., & Morishita, R. (2020). Development of an IL-17A DNA Vaccine to Treat Systemic Lupus Erythematosus in Mice. Vaccines, 8(1), 83. https://doi.org/10.3390/vaccines8010083