Downregulation of A20 Expression Increases the Immune Response and Apoptosis and Reduces Virus Production in Cells Infected by the Human Respiratory Syncytial Virus

 , and
, and
Abstract
1. Introduction
2. Material and Methods
2.1. Cells and Virus
2.2. Viral Infections and Plaque Assays
2.3. Quantitative RT-PCR
2.4. Western Blots
2.5. siRNA Silencing
2.6. A20 Knockout A549 Cells
2.7. Overexpression Assays
2.8. Apoptosis Assay
2.9. Stranded mRNA-seq Library Preparation and Sequencing
2.10. Data Analysis
2.11. Functional Annotation of Candidate Genes
2.12. Statistical Analysis
3. Results
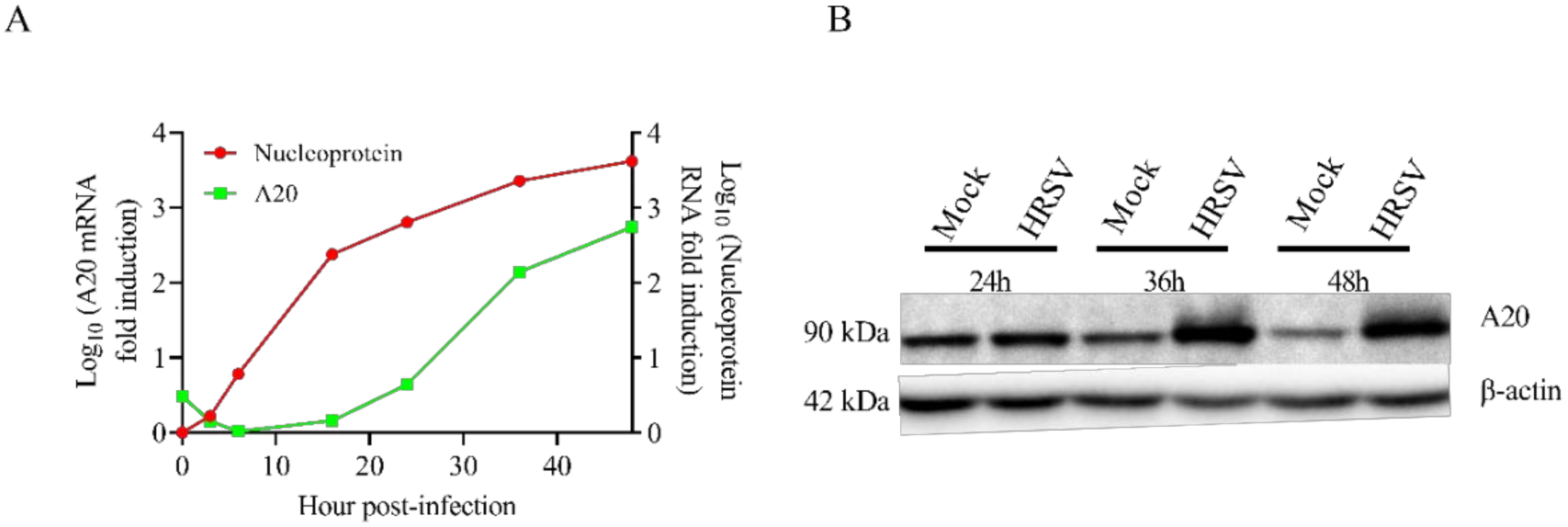
3.1. HRSV Infection Induces High Levels of A20 Expression
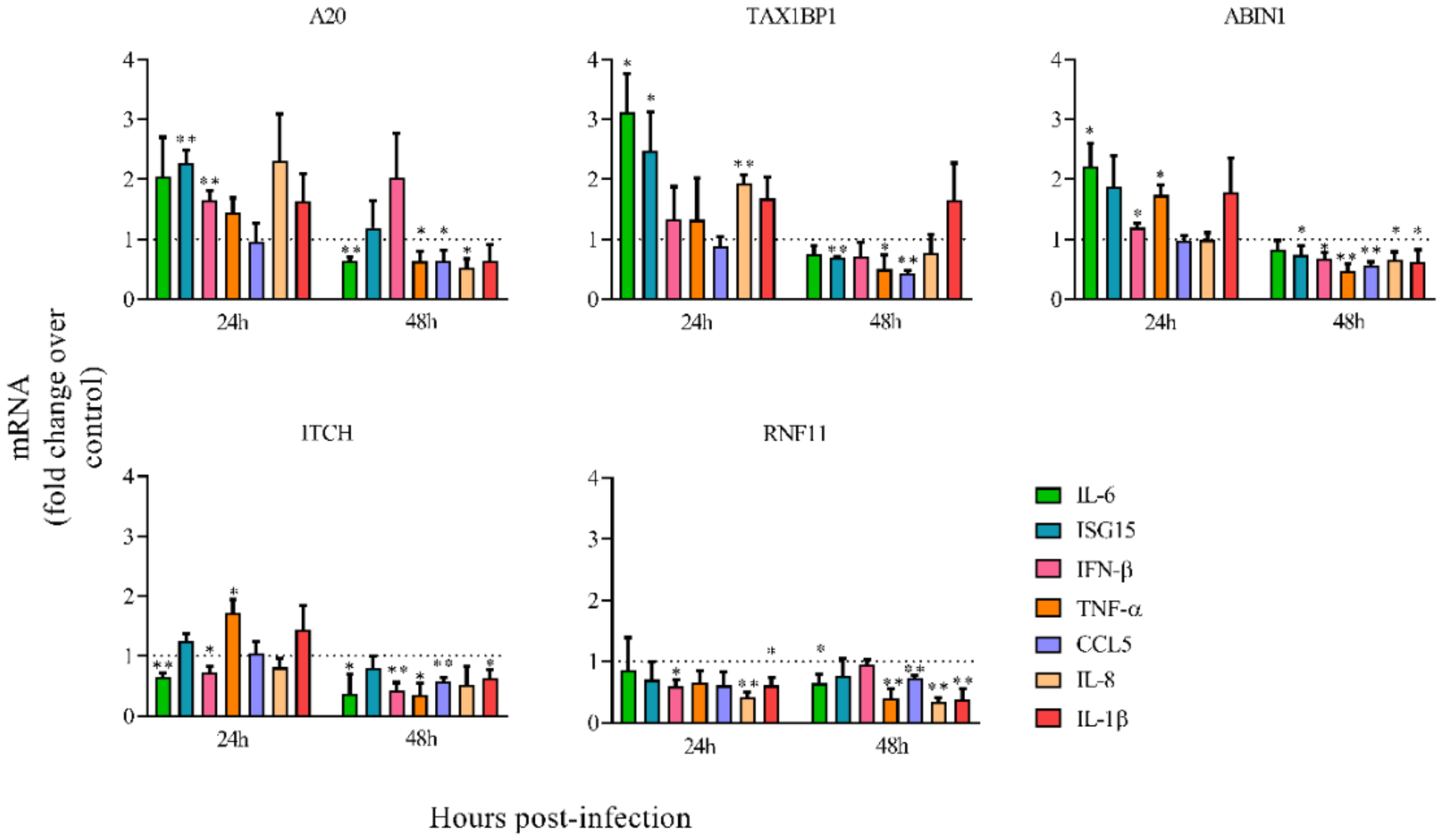
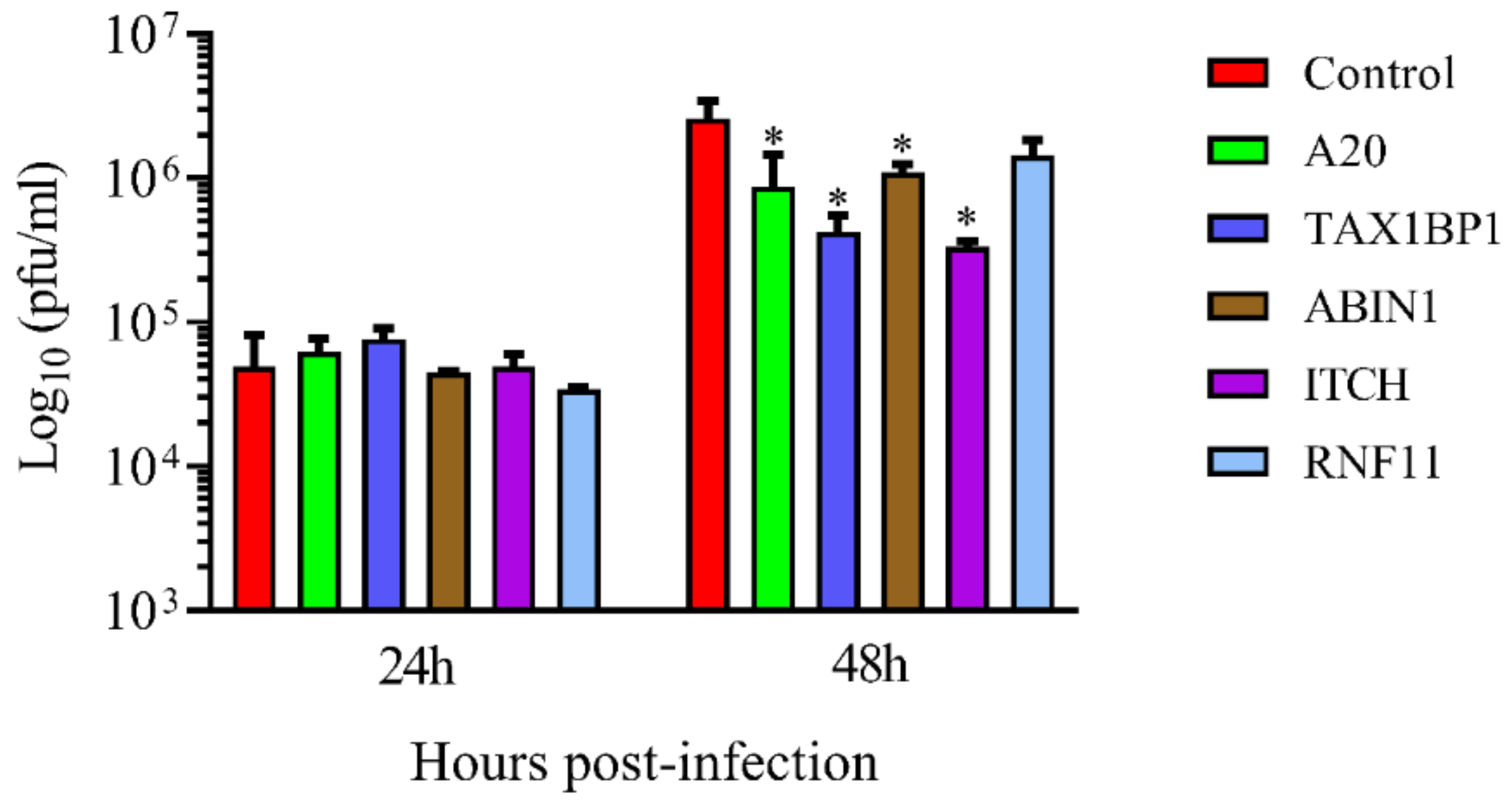
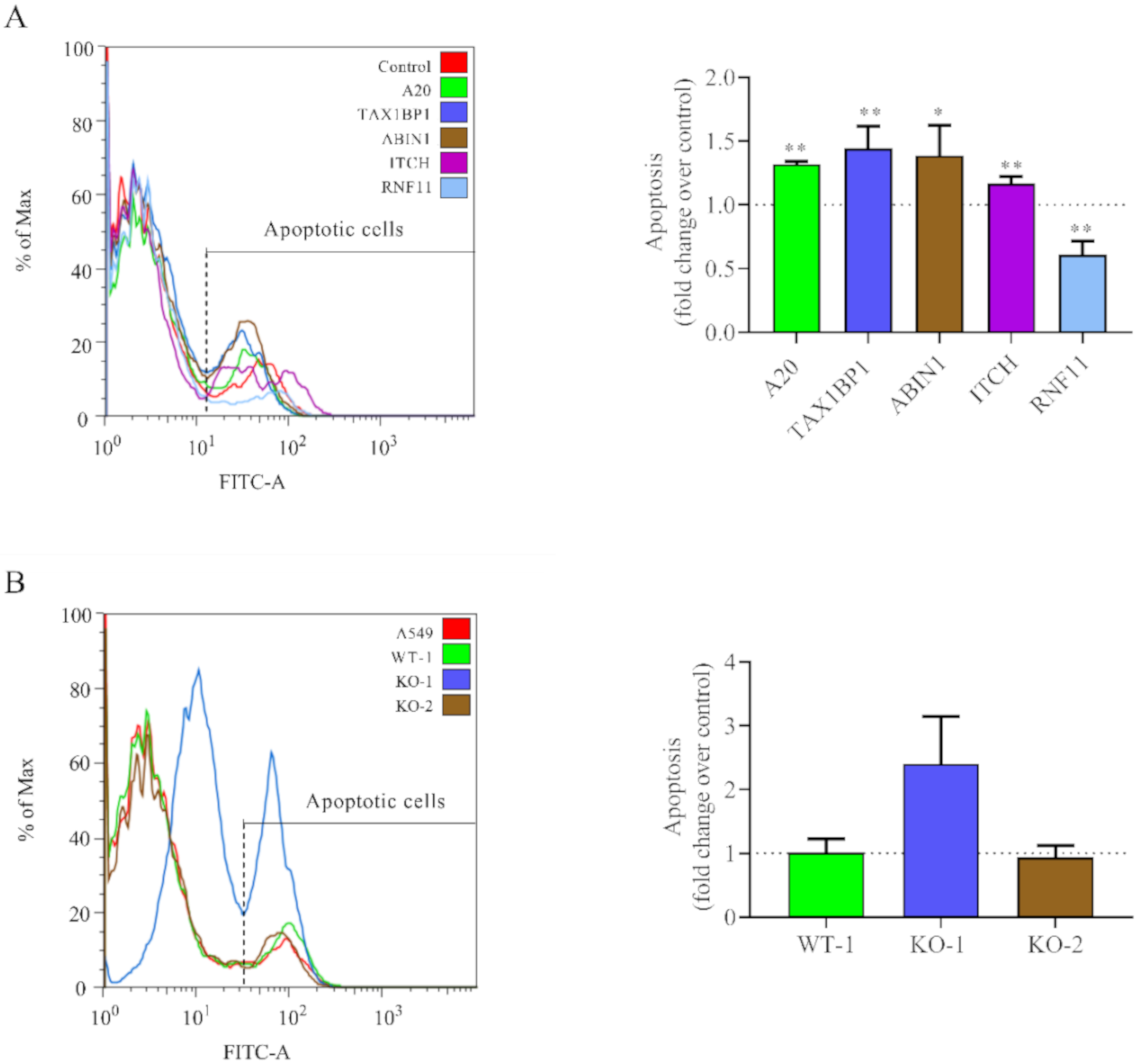
3.2. Downregulation of A20, TAX1BP1, or ABIN1 Enhances Intracellular Immune Response and Decreases HRSV Virus Production
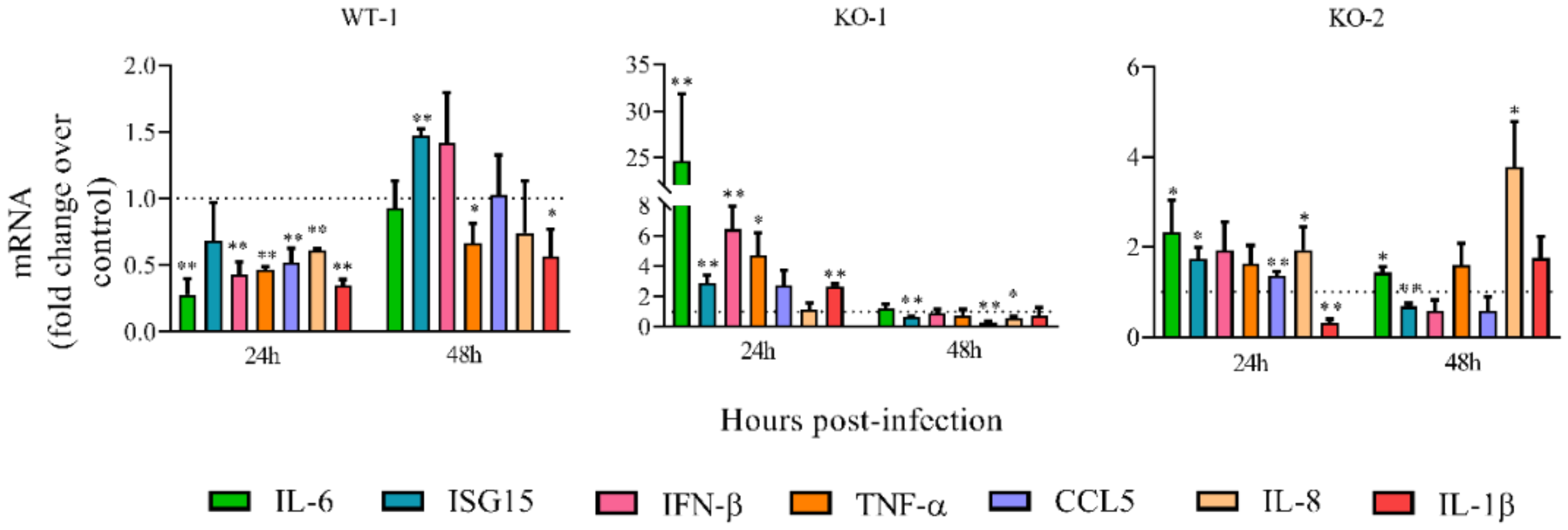
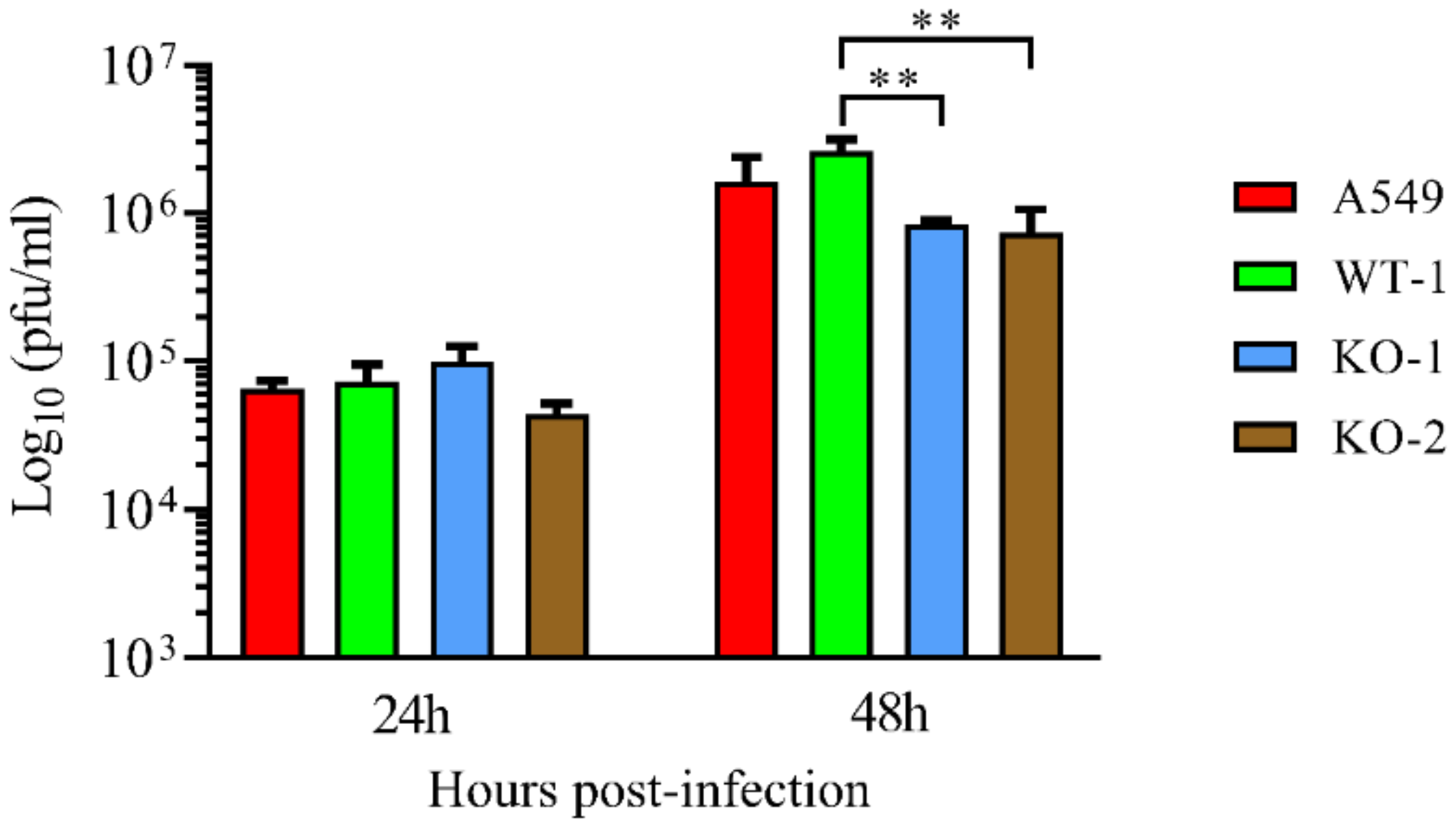
3.3. HRSV Production is Impaired in A20 Knockout Cells While the Antiviral/Inflammatory Response Is Augmented in These Cells
3.4. Immune Response and Infection-Related Signaling Pathways Are Upregulated in A20 Knockout Cells Following HRSV Infection
3.5. Downregulation of A20 and A20-Interacting Proteins Increases Cell Apoptosis in HRSV-Infected Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E. Respiratory Syncytial Virus Infection: An Illness for All Ages. Clin. Chest. Med. 2017, 38, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Everard, M.L. The relationship between respiratory syncytial virus infections and the development of wheezing and asthma in children. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Groskreutz, D.J.; Look, D.C. Recognizing the importance of respiratory syncytial virus in chronic obstructive pulmonary disease. COPD 2009, 6, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S.; Boyapalle, S. Epidemiologic, experimental, and clinical links between respiratory syncytial virus infection and asthma. Clin. Microbiol. Rev. 2008, 21, 495–504. [Google Scholar] [CrossRef]
- Graham, B.S.; Johnson, T.R.; Peebles, R.S. Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology 2000, 48, 237–247. [Google Scholar] [CrossRef]
- Openshaw, P.J.; Tregoning, J.S. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin. Microbiol. Rev. 2005, 18, 541–555. [Google Scholar] [CrossRef]
- Tripp, R.A. Pathogenesis of respiratory syncytial virus infection. Viral. Immunol. 2004, 17, 165–181. [Google Scholar] [CrossRef]
- Bennett, B.L.; Garofalo, R.P.; Cron, S.G.; Hosakote, Y.M.; Atmar, R.L.; Macias, C.G.; Piedra, P.A. Immunopathogenesis of respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2007, 195, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, E.G.; Schlegel, C.; Garofalo, R.P.; Mehta, R.; Scheffler, M.; Mei, M.; Piedra, P.A. Robust Cytokine and Chemokine Response in Nasopharyngeal Secretions: Association With Decreased Severity in Children With Physician Diagnosed Bronchiolitis. J. Infect. Dis. 2016, 214, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Bohmwald, K.; Galvez, N.M.S.; Canedo-Marroquin, G.; Pizarro-Ortega, M.S.; Andrade-Parra, C.; Gomez-Santander, F.; Kalergis, A.M. Contribution of Cytokines to Tissue Damage During Human Respiratory Syncytial Virus Infection. Front. Immunol. 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lopez, C.B. The innate immune response to RSV: Advances in our understanding of critical viral and host factors. Vaccine 2017, 35, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res. 2011, 21, 22–39. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479, 52–65. [Google Scholar] [CrossRef]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef]
- Saitoh, T.; Yamamoto, M.; Miyagishi, M.; Taira, K.; Nakanishi, M.; Fujita, T.; Akira, S.; Yamamoto, N.; Yamaoka, S. A20 is a negative regulator of IFN regulatory factor 3 signaling. J. Immunol. 2005, 174, 1507–1512. [Google Scholar] [CrossRef]
- Lin, R.; Yang, L.; Nakhaei, P.; Sun, Q.; Sharif-Askari, E.; Julkunen, I.; Hiscott, J. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J. Biol. Chem. 2006, 281, 2095–2103. [Google Scholar] [CrossRef]
- Evans, P.C.; Ovaa, H.; Hamon, M.; Kilshaw, P.J.; Hamm, S.; Bauer, S.; Ploegh, H.L.; Smith, T.S. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem. J. 2004, 378, 727–734. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Mele, A.; Cervantes, J.R.; Chien, V.; Friedman, D.; Ferran, C. Single nucleotide polymorphisms at the TNFAIP3/A20 locus and susceptibility/resistance to inflammatory and autoimmune diseases. Adv. Exp. Med. Biol. 2014, 809, 163–183. [Google Scholar] [PubMed]
- Arguello, M.; Paz, S.; Ferran, C.; Moll, H.P.; Hiscott, J. Anti-viral tetris: Modulation of the innate anti-viral immune response by A20. Adv. Exp. Med. Biol. 2014, 809, 49–64. [Google Scholar] [PubMed]
- De, A.; Dainichi, T.; Rathinam, C.V.; Ghosh, S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep. 2014, 15, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Roose, K.; Bogaert, P.; Sze, M.; Saelens, X.; Pasparakis, M.; Carpentier, I.; van Loo, G.; Beyaert, R. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog. 2012, 8, e1002570. [Google Scholar] [CrossRef]
- Maelfait, J.; Roose, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Schuijs, M.J.; Willart, M.; Ibanez, L.I.; Hammad, H.; Lambrecht, B.; et al. A20 Deficiency in Lung Epithelial Cells Protects against Influenza A Virus Infection. PLoS Pathog. 2016, 12, e1005410. [Google Scholar] [CrossRef]
- Martinez, I.; Lombardia, L.; Garcia-Barreno, B.; Dominguez, O.; Melero, J.A. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J. Gen. Virol. 2007, 88, 570–581. [Google Scholar] [CrossRef]
- Mbiguino, A.; Menezes, J. Purification of human respiratory syncytial virus: Superiority of sucrose gradient over percoll, renografin, and metrizamide gradients. J. Virol. Methods 1991, 31, 161–170. [Google Scholar] [CrossRef]
- Gonzalez-Sanz, R.; Mata, M.; Bermejo-Martin, J.; Alvarez, A.; Cortijo, J.; Melero, J.A.; Martinez, I. ISG15 Is Upregulated in Respiratory Syncytial Virus Infection and Reduces Virus Growth through Protein ISGylation. J. Virol. 2016, 90, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barreno, B.; Palomo, C.; Penas, C.; Delgado, T.; Perez-Brena, P.; Melero, J.A. Marked differences in the antigenic structure of human respiratory syncytial virus F and G glycoproteins. J. Virol. 1989, 63, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kweon, J.; Kim, A.; Chon, J.K.; Yoo, J.Y.; Kim, H.J.; Kim, S.; Lee, C.; Jeong, E.; Chung, E.; et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013, 31, 251–258. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, N.S.; Liebl, D.J.; Harhaj, E.W. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007, 26, 3910–3922. [Google Scholar] [CrossRef] [PubMed]
- Zetoune, F.S.; Murthy, A.R.; Shao, Z.; Hlaing, T.; Zeidler, M.G.; Li, Y.; Vincenz, C. A20 inhibits NF-kappa B activation downstream of multiple Map3 kinases and interacts with the I kappa B signalosome. Cytokine 2001, 15, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef]
- Angers, A.; Ramjaun, A.R.; McPherson, P.S. The HECT domain ligase itch ubiquitinates endophilin and localizes to the trans-Golgi network and endosomal system. J. Biol. Chem. 2004, 279, 11471–11479. [Google Scholar] [CrossRef] [PubMed]
- Web FASTQC, P.B.B. FastQC A Quality Control. Tool for High. Trhroughput Sequence Data. 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 February 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.; Trapnell, C.; Kelley, D. Cummerbund: Analysis, Exploration, Manipulation, and Visualization of Cufflinks High. Throughput Sequencing Data. R Packag. Version 2.14.0. 2013. Available online: https://bioconductor.org/packages/release/bioc/html/cummeRbund.html (accessed on 19 February 2020).
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Gao, L.; Coope, H.; Grant, S.; Ma, A.; Ley, S.C.; Harhaj, E.W. ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J. Biol. Chem. 2011, 286, 36592–36602. [Google Scholar] [CrossRef]
- De Valck, D.; Jin, D.Y.; Heyninck, K.; Van de Craen, M.; Contreras, R.; Fiers, W.; Jeang, K.T.; Beyaert, R. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 1999, 18, 4182–4190. [Google Scholar] [CrossRef]
- Choi, Y.B.; Shembade, N.; Parvatiyar, K.; Balachandran, S.; Harhaj, E.W. TAX1BP1 Restrains Virus-Induced Apoptosis by Facilitating Itch-Mediated Degradation of the Mitochondrial Adaptor MAVS. Mol. Cell Biol. 2017, 37, e00422-16. [Google Scholar] [CrossRef]
- Verstrepen, L.; Verhelst, K.; Carpentier, I.; Beyaert, R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem. Sci. 2011, 36, 347–354. [Google Scholar] [CrossRef]
- Oshima, S.; Turer, E.E.; Callahan, J.A.; Chai, S.; Advincula, R.; Barrera, J.; Shifrin, N.; Lee, B.; Benedict Yen, T.S.; Woo, T.; et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature 2009, 457, 906–909. [Google Scholar] [CrossRef]
- Monick, M.M.; Cameron, K.; Staber, J.; Powers, L.S.; Yarovinsky, T.O.; Koland, J.G.; Hunninghake, G.W. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 2005, 280, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, D.R.; Milligan, L.; Stark, J.M. Induction of CD95 (Fas) and apoptosis in respiratory epithelial cell cultures following respiratory syncytial virus infection. Virology 1999, 257, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vicente, M.; Medrano, L.M.; Resino, S.; Garcia-Sastre, A.; Martinez, I. TRIM25 in the Regulation of the Antiviral Innate Immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef]
- Bosanac, I.; Wertz, I.E.; Pan, B.; Yu, C.; Kusam, S.; Lam, C.; Phu, L.; Phung, Q.; Maurer, B.; Arnott, D.; et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol. Cell 2010, 40, 548–557. [Google Scholar] [CrossRef]
- Verhelst, K.; Carpentier, I.; Kreike, M.; Meloni, L.; Verstrepen, L.; Kensche, T.; Dikic, I.; Beyaert, R. A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J. 2012, 31, 3845–3855. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Barber, G.N.; Harhaj, E.W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 2010, 285, 14999–15009. [Google Scholar] [CrossRef]
- Shembade, N.; Parvatiyar, K.; Harhaj, N.S.; Harhaj, E.W. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J. 2009, 28, 513–522. [Google Scholar] [CrossRef]
- Wagner, S.; Carpentier, I.; Rogov, V.; Kreike, M.; Ikeda, F.; Lohr, F.; Wu, C.J.; Ashwell, J.D.; Dotsch, V.; Dikic, I.; et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene 2008, 27, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Oyama, M.; Kozuka-Hata, H.; Inoue, J. Involvement of A20 in the molecular switch that activates the non-canonical NF-small ka, CyrillicB pathway. Sci. Rep. 2013, 3, 2568. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Boldogh, S.; Garofalo, R.; Jamaluddin, M.; Brasier, A.R. Respiratory syncytial virus influences NF-kappaB-dependent gene expression through a novel pathway involving MAP3K14/NIK expression and nuclear complex formation with NF-kappaB2. J. Virol. 2005, 79, 8948–8959. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, K.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I{middle dot}NF- kappa B-inducing kinase signaling pathway. J. Biol. Chem. 2008, 283, 23169–23178. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vicente, M.; Resino, S.; Martinez, I. siRNA-Mediated Simultaneous Regulation of the Cellular Innate Immune Response and Human Respiratory Syncytial Virus Replication. Biomolecules 2019, 9, E165. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a virus NS1 protein induced A20 contributes to viral replication by suppressing interferon-induced antiviral response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Lee, J.; Chan, S.T.; Kim, J.Y.; Ou, J.J. Hepatitis C Virus Induces the Ubiquitin-Editing Enzyme A20 via Depletion of the Transcription Factor Upstream Stimulatory Factor 1 To Support Its Replication. mBio 2019, 10, e01660-19. [Google Scholar] [CrossRef]
- Song, X.; Yao, Z.; Yang, J.; Zhang, Z.; Deng, Y.; Li, M.; Ma, C.; Yang, L.; Gao, X.; Li, W.; et al. HCV core protein binds to gC1qR to induce A20 expression and inhibit cytokine production through MAPKs and NF-kappaB signaling pathways. Oncotarget 2016, 7, 33796–33808. [Google Scholar]
- Gu, S.Y.; Kim, Y.E.; Kwon, K.M.; Han, T.H.; Ahn, J.H. Biphasic regulation of A20 gene expression during human cytomegalovirus infection. Virol. J. 2014, 11, 124. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef]
- Bitko, V.; Shulyayeva, O.; Mazumder, B.; Musiyenko, A.; Ramaswamy, M.; Look, D.C.; Barik, S. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 2007, 81, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, J.; Zeng, R.; Yang, J.; Liu, J.; Zhang, Z.; Song, X.; Yao, Z.; Ma, C.; Li, W.; et al. Respiratory Syncytial Virus Replication Is Promoted by Autophagy-Mediated Inhibition of Apoptosis. J. Virol. 2018, 92, e02193-17. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.C.C.; Maubach, G.; Naumann, M. NF-kappaB-regulated ubiquitin-editing enzyme A20 paves the way for infection persistency. Cell Cycle 2018, 17, 3–4. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Gene Symbol | Benjamini (p-Value) |
|---|---|---|
| GO:0006955 Immune response | CCL26, CXCL2, CXCL5, CXCL8, CD70, CD74, SAMHD1, TNFRSF1B, TNFRSF21, B2M, CTSS, CIITA, C1R, C3, GBP2, IFITM2, IL1RAP, IL7R, HLA-C, PDCD1LG2, TLR4, TRIM22, TNFSF10, TNFSF13B, TNFSF14, TNFSF9 | 0.017 |
| GO:0001666 Response to hypoxia | CD24, CD38, CASP1, DPP4, EGLN1, EDNRA, HIF1A, LOXL2, MMP2, MUC1, PLAT, PLOD2, SLC2A8, SOD2, TGFBR2 | 0.031 |
| GO:0051607 Defense response to virus | MX2, SAMHD1, APOBEC3G, GBP1, GBP3, IFI16, IFI44L, IFIT1, IFITM1, IFITM2, IL33, SERINC5, TLR3, TRIM22 | 0.055 |
| GO:0006954 Inflammatory response | CCL17, CCL26, CXCL2, CXCL5, CXCL8, SMAD1, TNFRSF1B, TNFRSF21, AOX1, CHST2, CASP4, CIITA, C3, ITGB2, IFI16, IL1RAP, NR1H4, PTX3, RPS6KA5, THEMIS2, TLR3, TLR4 | 0.060 |
| GO:0060333 Interferon-gamma-mediated signaling pathway | IFI30, B2M, CIITA, GBP1, GBP2, IFNGR1, HLA-C, MT2A, TRIM22 | 0.069 |
| GO:0007155 Cell adhesion | ADAM12, CD24, EDIL3, EPHA4, KIAA1462, KITLG, CDH2, CDH6, CEACAM1, COL4A6, COL5A1, EFNB2, FAP, FN1, LGALS3BP, ISLR, IGFBP7, ITGB2, LSAMP, LOXL2, RGMB, SCN1B, TNC, THEMIS2 | 0.084 |
| GO:0060337 Type I interferon signaling pathway | MX2, SAMHD1, XAF1, GBP2, IFIT1, IFITM1, IFITM2, HLA-C | 0.15 |
| GO:0002237 Response to molecule of bacterial origin | CXCL2, CXCL8, CD24, MALT1 | 0.20 |
| GO:0045766 Positive regulation of angiogenesis | CXCL8, GATA2, GATA6, WNT5A, CCBE1, C3, HIF1A, ITGB2, TGFBR2, TWIST1 | 0.22 |
| GO: 0040037 Negative regulation of fibroblast growth, factor receptor signaling pathway | WNT4, WNT5A, SPRY1, SULF2 | 0.22 |
| Term | Gene Symbol | Benjamini (p-Value) |
|---|---|---|
| hsa04060 Cytokine-cytokine receptor interaction | CCL17, CCL26, CXCL2, CXCL5, CXCL8, CD70, TNFRSF19, TNFRSF1B, TNFRSF21, IFNGR1, IL1RAP, IL7R, LIFR, TGFBR2, TNFSF10, TNFSF13B, TNFSF14, TNFSF9 | 0.015 |
| hsa05133 Pertussis | CXCL5, CXCL8, CASP1, C1R, C1S, C3, C4BPA, ITGB2, TLR4 | 0.041 |
| hsa05134 Legionellosis | CXCL2, CXCL8, CASP1, C3, EEF1A2, ITGB2, TLR4 | 0.11 |
| hsa00760 Nicotinate and nicotinamide metabolism | NT5C3A, CD38, AOX1, NNMT, NNT | 0.21 |
| hsa04610 Complement and coagulation cascades | C1R, C1S, C3, C4BPA, CFB, CFI, PLAT | 0.22 |
| hsa05150 Staphylococcus aureus infection | C1R, C1S, C3, CFB, CFI, ITGB2 | 0.27 |
| hsa05152 Tuberculosis | CD74, MALT1, CTSS, CIITA, C3, EEA1, ITGB2, IFNGR1, TLR4, VDR | 0.55 |
| hsa04145 Phagosome | CTSS, C1R, C3, EEA1, ITGB2, HLA-C, TLR4, TUBA1A, TUBB3 | 0.51 |
| hsa04612 Antigen processing and presentation | CD74, IFI30, B2M, CTSS, CIITA, HLA-C | 0.55 |
| hsa05146 Amoebiasis | CXCL8, COL4A5, COL4A6, COL5A1, FN1, ITGB2, TLR4 | 0.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Vicente, M.; González-Sanz, R.; Cuesta, I.; Monzón, S.; Resino, S.; Martínez, I. Downregulation of A20 Expression Increases the Immune Response and Apoptosis and Reduces Virus Production in Cells Infected by the Human Respiratory Syncytial Virus. Vaccines 2020, 8, 100. https://doi.org/10.3390/vaccines8010100
Martín-Vicente M, González-Sanz R, Cuesta I, Monzón S, Resino S, Martínez I. Downregulation of A20 Expression Increases the Immune Response and Apoptosis and Reduces Virus Production in Cells Infected by the Human Respiratory Syncytial Virus. Vaccines. 2020; 8(1):100. https://doi.org/10.3390/vaccines8010100
Chicago/Turabian StyleMartín-Vicente, María, Rubén González-Sanz, Isabel Cuesta, Sara Monzón, Salvador Resino, and Isidoro Martínez. 2020. "Downregulation of A20 Expression Increases the Immune Response and Apoptosis and Reduces Virus Production in Cells Infected by the Human Respiratory Syncytial Virus" Vaccines 8, no. 1: 100. https://doi.org/10.3390/vaccines8010100
APA StyleMartín-Vicente, M., González-Sanz, R., Cuesta, I., Monzón, S., Resino, S., & Martínez, I. (2020). Downregulation of A20 Expression Increases the Immune Response and Apoptosis and Reduces Virus Production in Cells Infected by the Human Respiratory Syncytial Virus. Vaccines, 8(1), 100. https://doi.org/10.3390/vaccines8010100