MVA-Vectored Pentameric Complex (PC) and gB Vaccines Improve Pregnancy Outcome after Guinea Pig CMV Challenge, but Only gB Vaccine Reduces Vertical Transmission

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Guinea Pigs
2.2. Cells and Virus
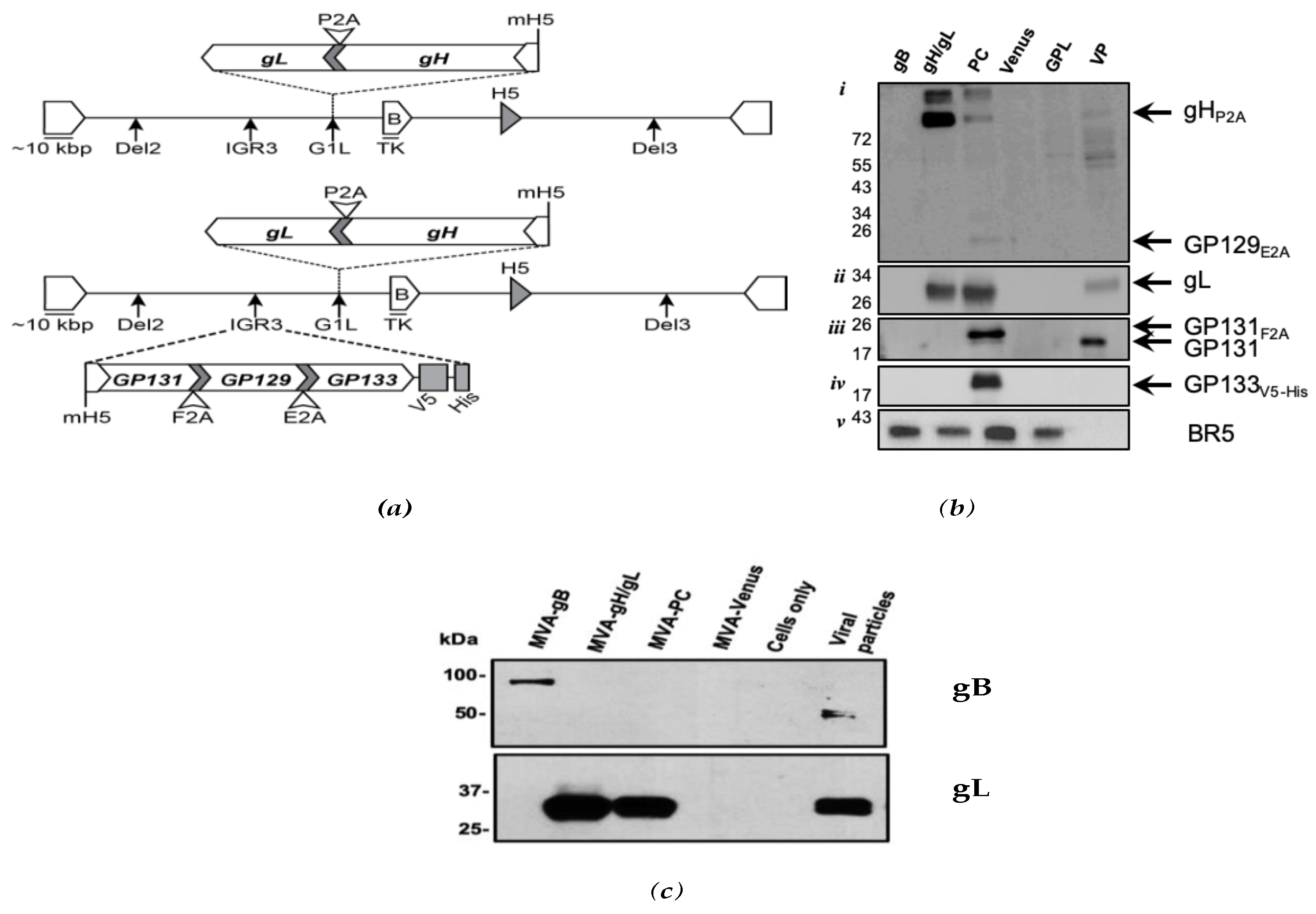
2.3. Generation of Vaccine Constructs and Western Blot Analyses
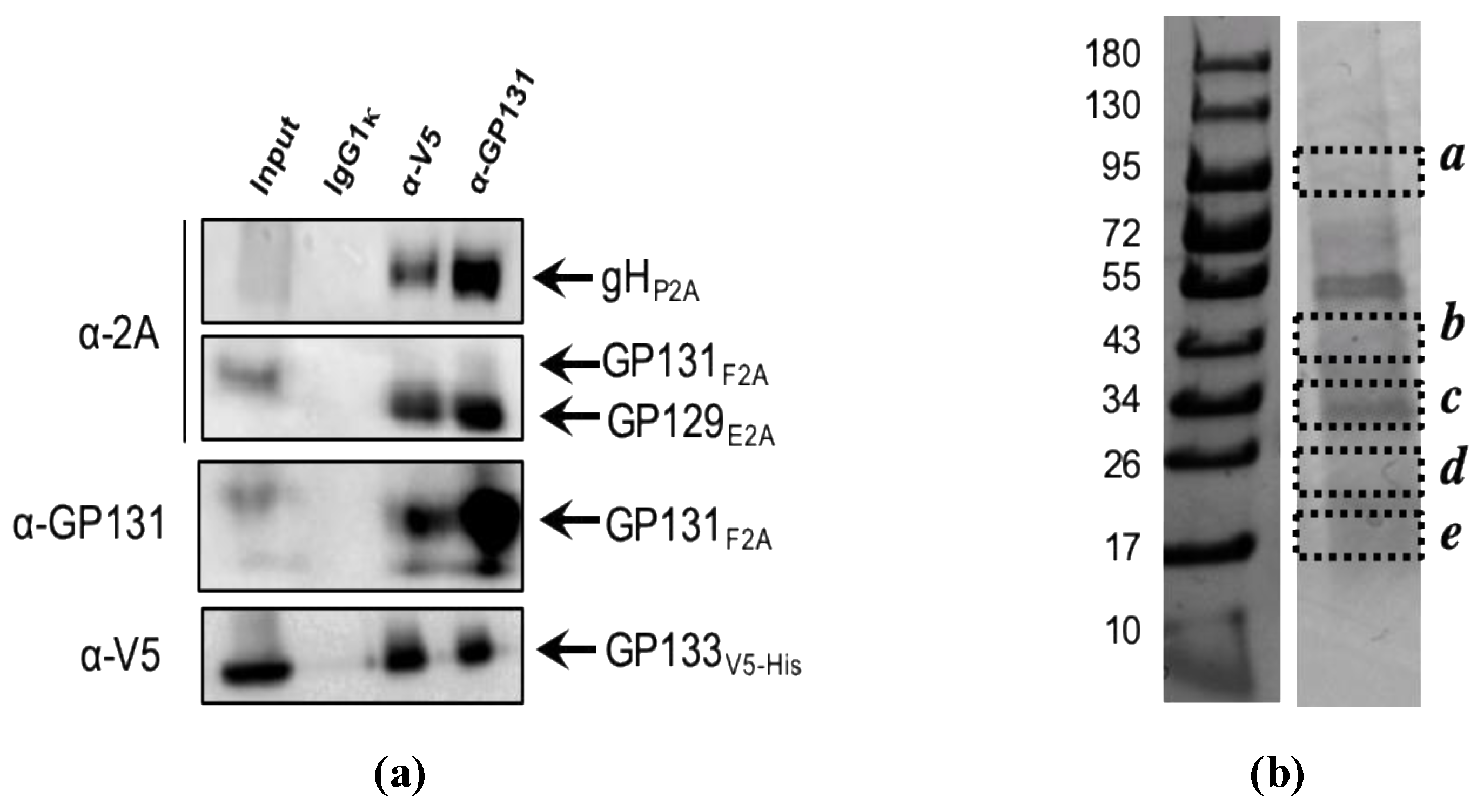
2.4. Mass Spectrometry Analyses
2.5. Immunization Schedule and Immune Assays
2.6. Real-Time qPCR Analysis
2.7. Statistical Analyses
3. Results
3.1. Expression of GPCMV PC Subunits in MVA
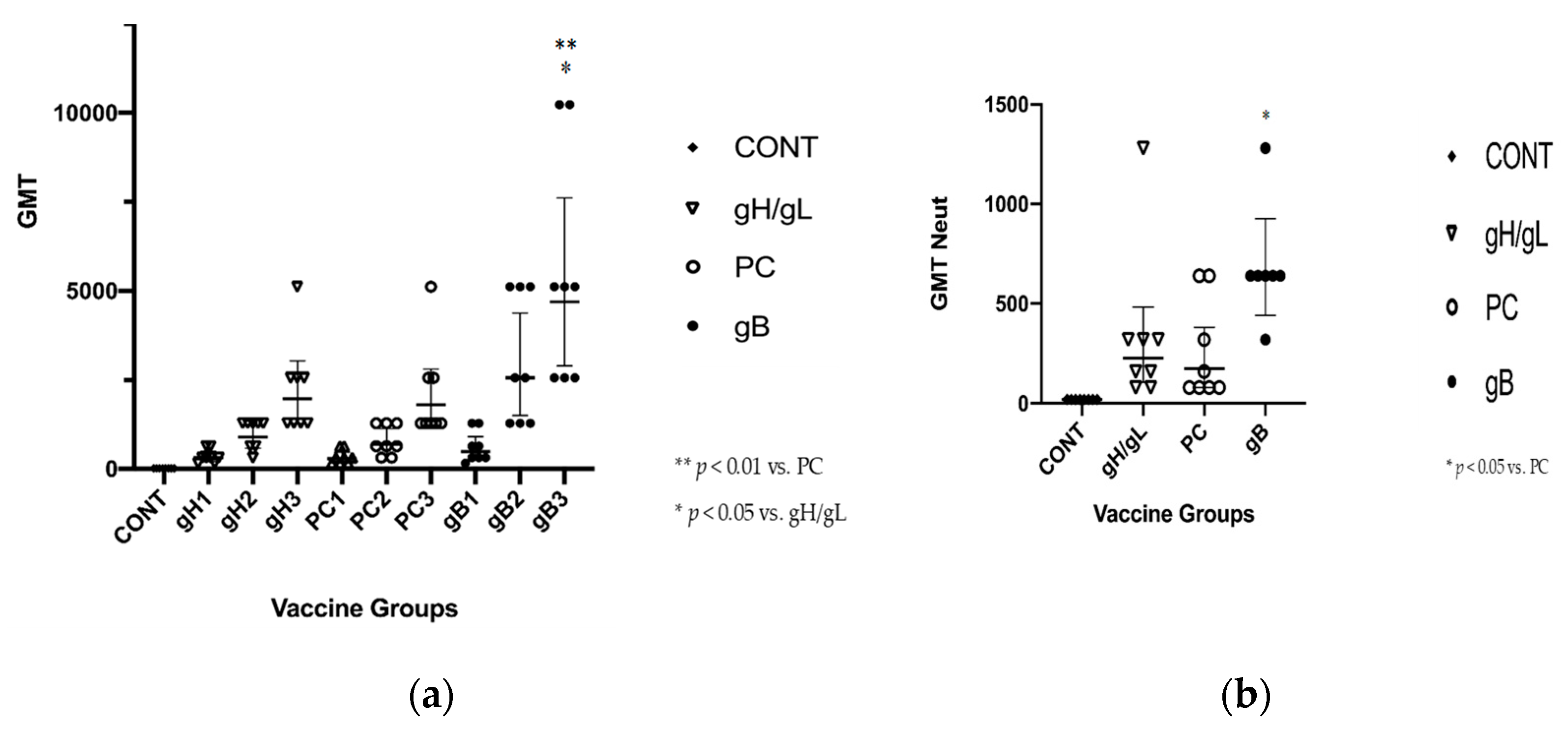
3.2. gB Vaccine Elicits Higher ELISA and Neutralization Responses than gH/gL or PC
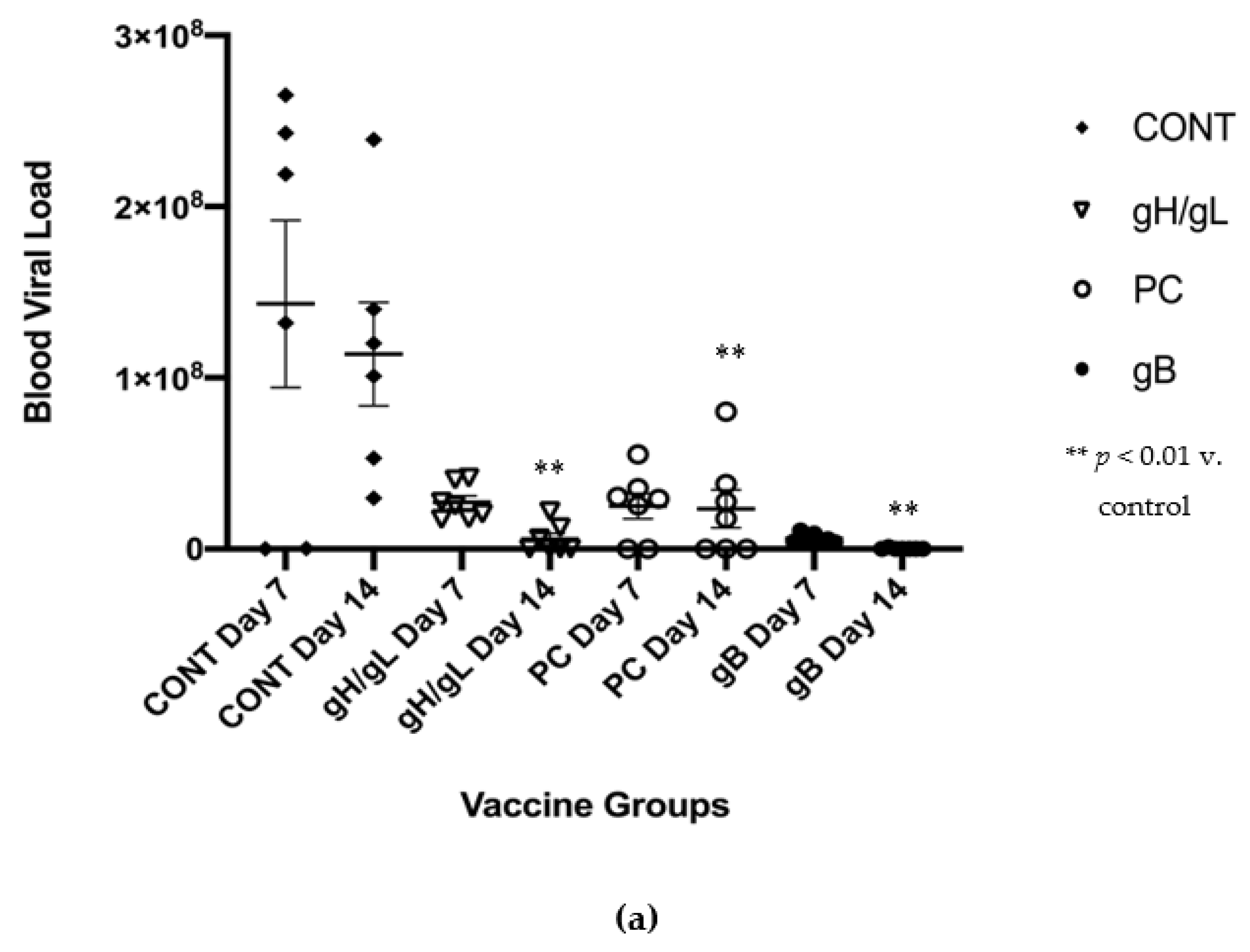
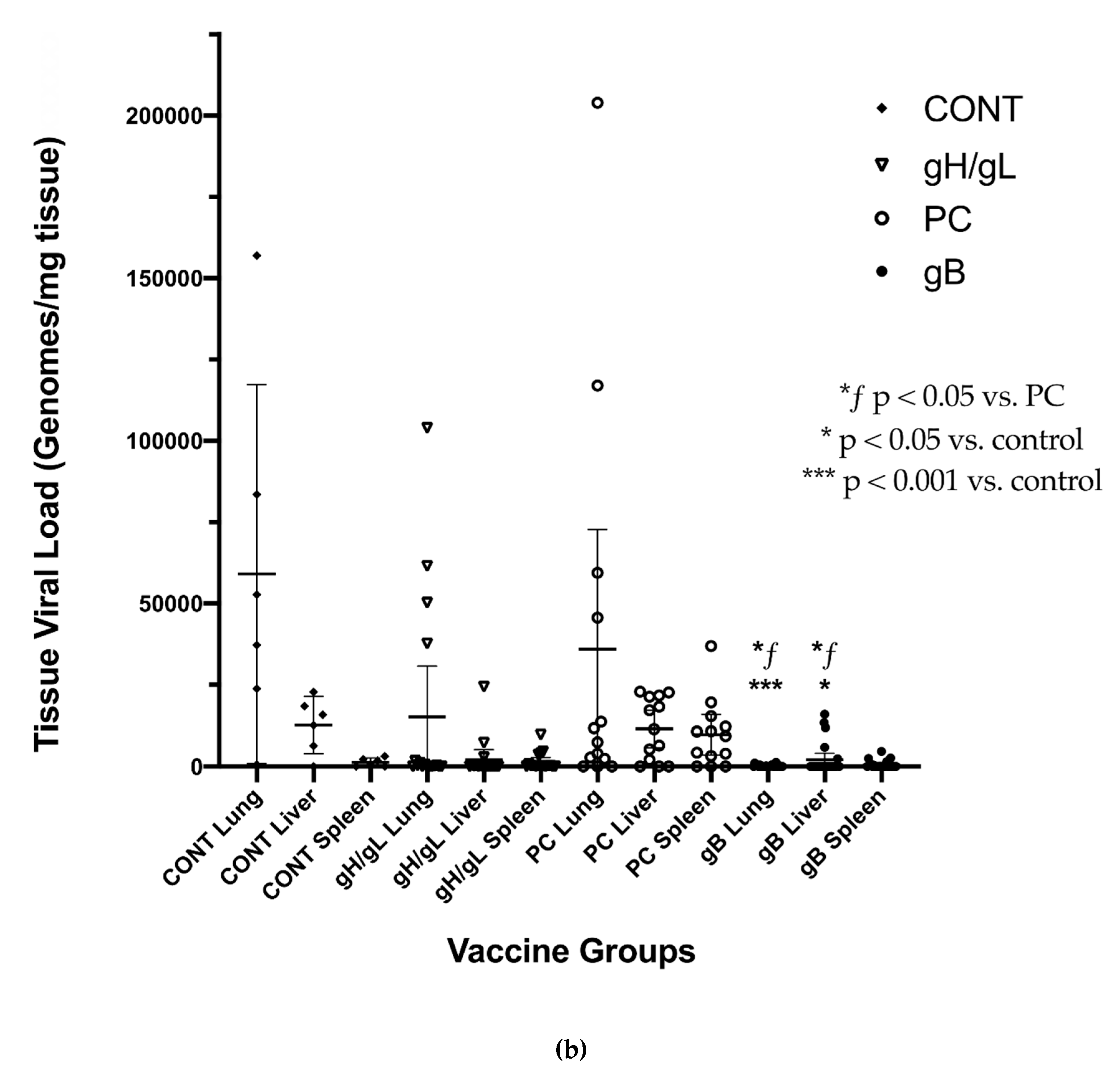
3.3. Impact of Preconception Vaccine on Maternal Viremia After GPCMV Challenge
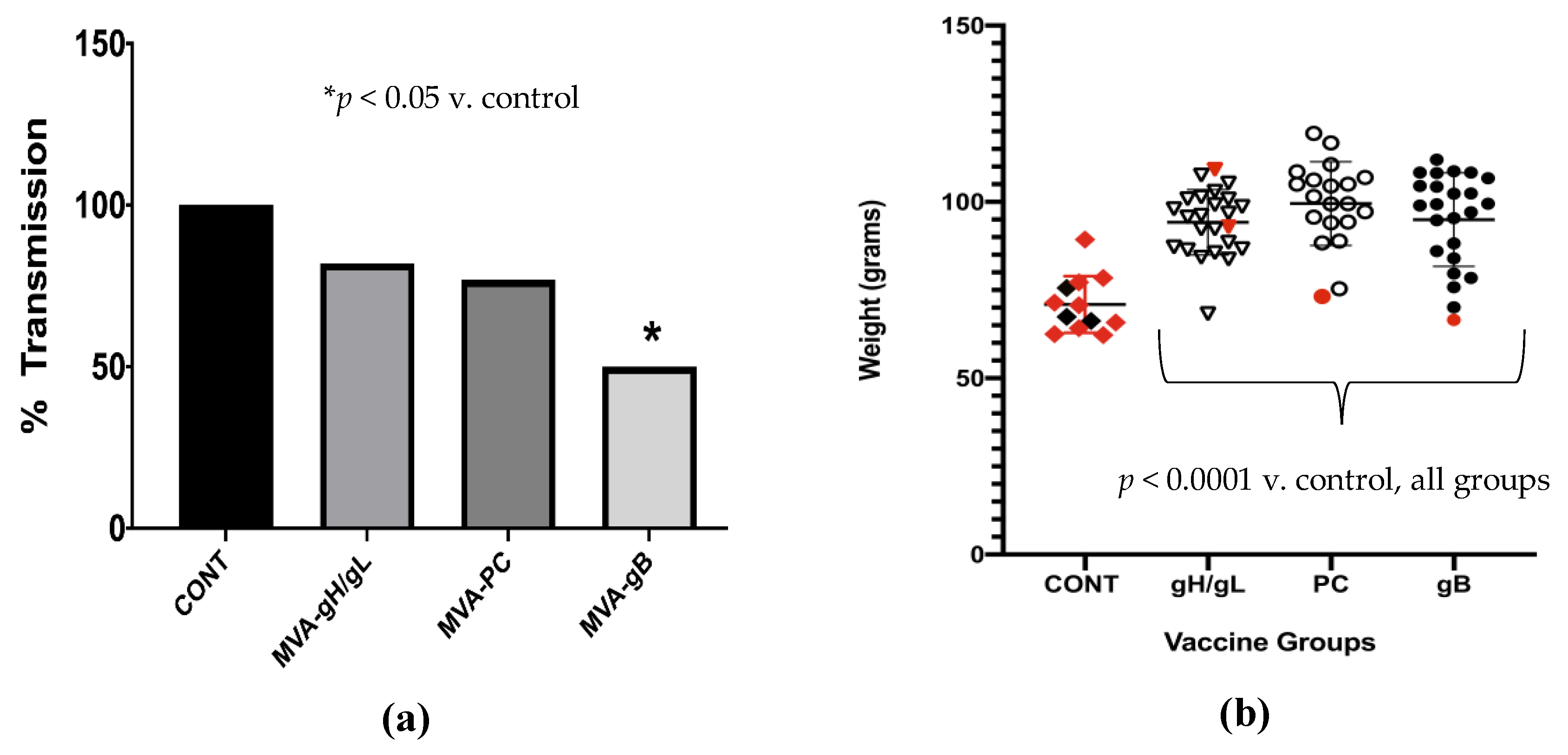
3.4. Impact of Preconception Vaccine on Pup Birth Weight, Mortality, and Congenital Infection
4. Discussion
5. Conclusions
- A vectored MVA vaccine, constructed by exploiting the 2A peptide system for optimization of multi-subunit herpesvirus vaccines, effectively expresses the GPCMV pentamer subunits.
- Vectored MVA vaccines, expressing the GPCMV homologs of gB, gH/gL and the PC, are immunogenic, and elicit ELISA and neutralizing antibody responses.
- ELISA responses to vaccination were significantly better in the MVA-gpgB vaccine group, compared to the MVA-gp75/gL and MVA-gpPC vaccine groups.
- Neutralization responses were significantly better upon completion of the three-dose vaccine series in the MVA-gpgB vaccine group, compared to the MVA-gpPC vaccine group.
- All vaccine strategies resulted in reduced maternal DNAemia at day 14, following a SG-GPCMV challenge during pregnancy, although reductions were greater for MVA-gpgB vaccine (698-fold), than the MVA-gp75/gL (19.5-fold) and MVA-gpPC (4.9-fold) groups, respectively.
- Preconception vaccination improved pup birth weight and reduced mortality in all groups, but only the gB vaccine resulted in significant reductions in congenital GPCMV transmission.
- Future investigations of glycoprotein vaccines based on MVA vectors are warranted in the GPCMV model.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheeran, M.C.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Isaac Mde, L.; Amaral, F.R.; Carvalheiro, C.G.; Aragon, D.C.; Manfredi, A.K.; Boppana, S.B.; Britt, W.J. Congenital cytomegalovirus infection as a cause of sensorineural hearing loss in a highly immune population. Pediatr. Infect. Dis. J. 2011, 30, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Riga, M.; Korres, G.; Chouridis, P.; Naxakis, S.; Danielides, V. Congenital cytomegalovirus infection inducing non-congenital sensorineural hearing loss during childhood; a systematic review. Int. J. Pediatr. Otorhinolaryngol. 2018, 115, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Itell, H.L.; Nelson, C.S.; Martinez, D.R.; Permar, S.R. Maternal immune correlates of protection against placental transmission of cytomegalovirus. Placenta 2017, 60 (Suppl. 1), S73–S79. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Searching for a serological correlate of protection for a CMV vaccine. J. Infect. Dis. 2018, 217, 1861–1864. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R.; Permar, S.R.; Plotkin, S.A. Progress toward development of a vaccine against congenital cytomegalovirus infection. Clin. Vaccine Immunol. 2017, 24, e00268-17. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.J.; La Rosa, C.; Chiuppesi, F.; Contreras, H.; Dadwal, S.; Wussow, F.; Bautista, S.; Nakamura, R.; Zaia, J.A. A fifty-year odyssey: Prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev. Vaccines 2018, 17, 889–911. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F.; Duliegè, A.M.; Boppana, S.; Sekulovich, R.; Percell, S.; Britt, W.; Burke, R.L. A subunit cytomegalovirus vaccine based on recombinant envelope glycoprotein B and a new adjuvant. J. Infect. Dis. 1999, 180, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F. Development and evidence for efficacy of CMV glycoprotein B vaccine with MF59 adjuvant. J. Clin. Virol. 2009, 46 (Suppl. 4), S73–S76. [Google Scholar] [CrossRef] [PubMed]
- Sabbaj, S.; Pass, R.F.; Goepfert, P.A.; Pichon, S. Glycoprotein B vaccine is capable of boosting both antibody and CD4 T-cell responses to cytomegalovirus in chronically infected women. J. Infect. Dis. 2011, 203, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Reap, E.A.; Katen, K.; Watson, A.; Smith, K.; Norberg, P.; Olmsted, R.A.; Hoeper, A.; Morris, J.; Negri, S.; et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 2009, 28, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; de la Rosa, E.C.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.S.; Herold, B.C.; Permar, S.R. A new era in cytomegalovirus vaccinology: Considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. NPJ Vaccines 2018, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Cao, Z.; Wang, S.; Flora, M.; Adler, S.P.; McVoy, M.A.; Snapper, C.M. Novel trimeric human cytomegalovirus glycoprotein B elicits a high-titer neutralizing antibody response. Vaccine 2018, 36, 5580–5590. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Ryckman, B.J.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 2008, 82, 11837–11850. [Google Scholar] [CrossRef] [PubMed]
- Fouts, A.E.; Comps-Agrar, L.; Stengel, K.F.; Ellerman, D.; Schoeffler, A.J.; Warming, S.; Eaton, D.L.; Feierbach, B. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc. Natl. Acad. Sci. USA 2014, 111, 8209–8214. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 2011, 85, 11638–11645. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Johnson, D.C. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2012, 2, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS ONE 2013, 8, e59863. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 2008, 82, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Knoche, A.J.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 2010, 84, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Monroe, J.; Linton, C.; Archer, J.; Beard, C.W.; Barnett, S.W.; Palladino, G.; Mason, P.W.; Carfi, A.; Lilja, A.E. Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 2014, 32, 3796–3804. [Google Scholar] [CrossRef] [PubMed]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, I.; Wen, Y.; Ciferri, C.; Schulze, A.; Fühner, V.; Leong, M.; Gerber, A.; Gerrein, R.; Nandi, A.; Lilja, A.E.; et al. Expression of the human cytomegalovirus pentamer complex for vaccine use in a CHO system. Biotechnol. Bioeng. 2015, 112, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Chiuppesi, F.; Martinez, J.; Campo, J.; Johnson, E.; Flechsig, C.; Newell, M.; Tran, E.; Ortiz, J.; La Rosa, C.; et al. Human cytomegalovirus vaccine based on the envelope gH/gL pentamer complex. PLoS Pathog. 2014, 10, e1004524. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Chin, A.L.; Liu, J.; Jardetzky, T.S.; Mudd, J.O.; Orloff, S.L.; Streblow, D.; Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Duarte, G.; et al. HCMV trimer- and pentamer-specific antibodies synergize for virus neutralization but do not correlate with congenital transmission. Proc. Natl. Acad. Sci. USA 2019, 116, 3728–3733. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.A.; Lockridge, K.M.; Salamat, S.; Tinling, S.P.; Yue, Y.; Zhou, S.S.; Gospe, S.M., Jr.; Britt, W.J.; Tarantal, A.F. Nonhuman primate models of intrauterine cytomegalovirus infection. ILAR J. 2006, 47, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Developing a vaccine against congenital cytomegalovirus (CMV) infection: What have we learned from animal models? Where should we go next? Future Virol. 2013, 8, 1161–1182. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Lilja, A.E.; Shenk, T. Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc. Natl. Acad. Sci. USA 2008, 105, 19950–19955. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Kaur, A.; Lilja, A.; Diamond, D.J.; Walter, M.R.; Barry, P.A. The susceptibility of primary cultured rhesus macaque kidney epithelial cells to rhesus cytomegalovirus strains. J. Gen. Virol. 2016, 97, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Yue, Y.; Martinez, J.; Deere, J.D.; Longmate, J.; Herrmann, A.; Barry, P.A.; Diamond, D.J. A vaccine based on the rhesus cytomegalovirus UL128 complex induces broadly neutralizing antibodies in rhesus macaques. J. Virol. 2013, 87, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nozawa, N.; Katano, H.; Fukui, Y.; Tsuda, M.; Tsutsui, Y.; Kurane, I.; Inoue, N. Characterization of the guinea pig cytomegalovirus genome locus that encodes homologs of human cytomegalovirus major immediate-early genes, UL128, and UL130. Virology 2009, 391, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, M.; Yan, D.; Fouts, A.; Xu, M.; Estevez, A.; Austin, C.D.; Bazan, F.; Feierbach, B. Characterization of the guinea pig CMV gH/gL/GP129/GP131/GP133 complex in infection and spread. Virology 2013, 441, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Gnanandarajah, J.S.; Gillis, P.A.; Hernandez-Alvarado, N.; Higgins, L.; Markowski, T.W.; Sung, H.; Lumley, S.; Schleiss, M.R. Identification by mass spectrometry and immune response analysis of guinea pig cytomegalovirus (GPCMV) pentameric complex proteins GP129, 131 and 133. Viruses 2014, 6, 727–751. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.; Choi, K.Y.; Root, M.; McGregor, A. A homolog pentameric complex dictates viral epithelial tropism, pathogenicity and congenital infection rate in guinea pig cytomegalovirus. PLoS Pathog. 2016, 12, e1005755. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, M.R.; Yan, D.; Vij, R.; Hongo, J.A.; Nakamura, G.; Vernes, J.M.; Meng, Y.G.; Lein, S.; Chan, P.; Ross, J.; et al. A neutralizing anti-gH/gL monoclonal antibody is protective in the guinea pig model of congenital CMV infection. PLoS Pathog. 2014, 10, e1004060. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.C.; Gillis, P.; Hernandez-Alvarado, N.; Fernández-Alarcón, C.; Schmit, M.; Zabeli, J.C.; Wussow, F.; Diamond, D.J.; Schleiss, M.R. Comparison of monovalent glycoprotein B with bivalent gB/pp65 (GP83) vaccine for congenital cytomegalovirus infection in a guinea pig model: Inclusion of GP83 reduces gB antibody response but both vaccine approaches provide equivalent protection against pup mortality. Vaccine 2015, 33, 4013–4018. [Google Scholar] [PubMed]
- Chiuppesi, F.; Nguyen, J.; Park, S.; Contreras, H.; Kha, M.; Meng, Z.; Kaltcheva, T.; Iniguez, A.; Martinez, J.; La Rosa, C.; et al. Multiantigenic modified vaccinia virus Ankara vaccine vectors to elicit potent humoral and cellular immune responses against human cytomegalovirus in mice. J. Virol. 2018, 92, e01012-18. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Chiuppesi, F.; Meng, Z.; Martinez, J.; Nguyen, J.; Barry, P.A.; Diamond, D.J. Exploiting 2A peptides to elicit potent neutralizing antibodies by a multi-subunit herpesvirus glycoprotein complex. J. Virol. Methods 2018, 251, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Manuel, E.R.; Wang, Z.; Li, Z.; La Rosa, C.; Zhou, W.; Diamond, D.J. Intergenic region 3 of modified vaccinia ankara is a functional site for insert gene expression and allows for potent antigen-specific immune responses. Virology 2010, 403, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Shors, S.T.; Murphy, B.R.; Moss, B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996, 14, 1451–1458. [Google Scholar] [CrossRef]
- Wang, Z.; Martinez, J.; Zhou, W.; La Rosa, C.; Srivastava, T.; Dasgupta, A.; Rawal, R.; Li, Z.; Britt, W.J.; Diamond, D. Modified H5 promoter improves stability of insert genes while maintaining immunogenicity during extended passage of genetically engineered MVA vaccines. Vaccine 2010, 28, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, M.; Sodeik, B.; Ericsson, M.; Wolffe, E.J.; Shida, H.; Hiller, G.; Griffiths, G. Assembly of vaccinia virus: The second wrapping cisterna is derived from the trans Golgi network. J. Virol. 1994, 68, 130–147. [Google Scholar] [PubMed]
- Protein details for Mesocricetus auratus ((assembly MesAur1.0). Available online: https://www.ncbi.nlm.nih.gov/genome/proteins/11998?genome_assembly_id=40397&gi=-1 (accessed on 4 November 2019).
- Caviid betaherpesvirus 2 (Guinea pig cytomegalovirus). Available online: https://www.ncbi.nlm.nih.gov/taxonomy/?term=caviid+betaherpesvirus+2 (accessed on 4 November 2019).
- The global proteome machine. Available online: https://www.thegpm.org/crap (accessed on 4 November 2019).
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.; Choi, K.Y.; McGregor, A. Cytomegalovirus UL128 homolog mutants that form a pentameric complex produce virus with impaired epithelial trophoblast cell tropism and altered pathogenicity in the guinea pig. Virology 2017, 509, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R.; Bierle, C.J.; Swanson, E.C.; McVoy, M.A.; Wang, J.B.; Al-Mahdi, Z.; Geballe, A.P. Vaccination with a live attenuated cytomegalovirus devoid of a protein kinase R inhibitory gene results in reduced maternal viremia and improved pregnancy outcome in a guinea pig congenital infection model. J. Virol. 2015, 89, 9727–9738. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.R.; Bialek, S.R.; Boppana, S.B.; Griffiths, P.D.; Laughlin, C.A.; Ljungman, P.; Mocarski, E.S.; Pass, R.F.; Read, J.S.; Schleiss, M.R.; et al. Priorities for CMV vaccine development. Vaccine 2013, 32, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J. Congenital human cytomegalovirus infection and the enigma of maternal immunity. J. Virol. 2017, 91, e02392-16. [Google Scholar] [CrossRef] [PubMed]
- Permar, S.R.; Schleiss, M.R.; Plotkin, S.A. Advancing our understanding of protective maternal immunity as a guide for development of vaccines to reduce congenital cytomegalovirus infections. J. Virol. 2018, 92, e00030-18. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Fukuchi, S.; Hashimoto, K.; Fukui, Y.; Tsuda, M.; Kataoka, M.; Katano, H.; Inoue, N. Guinea pig cytomegalovirus GP129/131/133, homologues of human cytomegalovirus UL128/130/131A, are necessary for infection of monocytes and macrophages. J. Gen. Virol. 2014, 95, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- McVoy, M.A.; Wang, J.B.; Dittmer, D.P.; Bierle, C.J.; Swanson, E.C.; Fernández-Alarcón, C.; Hernandez-Alvarado, N.; Zabeli, J.C.; Schleiss, M.R. Repair of a mutation disrupting the guinea pig cytomegalovirus pentameric complex acquired during fibroblast passage restores pathogenesis in immune-suppressed guinea pigs and in the context of congenital infection. J. Virol. 2016, 90, 7715–7727. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Inclusion of the viral pentamer complex in a vaccine design greatly improves protection against congenital cytomegalovirus in the guinea pig model. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Root, M.; McGregor, A. A novel non-replication-competent cytomegalovirus capsid mutant vaccine strategy is effective in reducing congenital infection. J. Virol. 2016, 90, 7902–7919. [Google Scholar] [CrossRef] [PubMed]
- Deere, J.D.; Chang, W.L.W.; Villalobos, A.; Schmidt, K.A.; Deshpande, A.; Castillo, L.D.; Fike, J.; Walter, M.R.; Barry, P.A.; Hartigan-O’Connor, D.J. Neutralization of rhesus cytomegalovirus IL-10 reduces horizontal transmission and alters long-term immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 13036–13041. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPCMV | HCMV | % Identity | % Similarity | MW (kDa) |
|---|---|---|---|---|
| GP129 | UL128 | 34 | 48 | 22.8 |
| GP131 | UL130 | 29 | 36 | 24.3 |
| GP133 | UL131 | 18 | 29 | 17.0/19.7 * |
| GP115 | gL | 30 | 49 | 31.6 |
| GP75 | gH | 42 | 64 | 84.0 |
| Origin | 2A | Peptide Sequence |
|---|---|---|
| Porcine teschovirus-11 | P2A | GSGATNFSLLKQAGDVEENPG*P |
| Thosea asigna virus | T2A | GSGEGRGSLLTCGDVEENPG*P |
| Foot and Mouth picornavirus | F2A | GSGVKQTLNFDLLKLAGDVESNPG*P |
| Equine rhinitis A virus | E2A | GSGQCTNYALLKLAGDVESNPG*P |
| ORF | MW (kDa) | ID | Unique Spectra | Unique Peptide Sequences | % Coverage | Max Xcorr | Probability |
|---|---|---|---|---|---|---|---|
| GP133V5-HIS | 19.67 | e | 11 | 9 | 43 | 4.61 | 100 |
| GP115 (gL) | 29.82 | c | 5 | 4 | 16 | 3.70 | 100 |
| GP75 (gH) | 83.85 | a | 10 | 10 | 17 | 4.90 | 100 |
| Vaccine Group | Number Vaccinated | Number Pregnant | Total Pups | Live Pups | Pup Mortality (%) | Pregnancy Duration Post-Challenge (Days) |
|---|---|---|---|---|---|---|
| Venus | 8 | 6 | 12 | 3 | 75 | 12.0 |
| gH/gL | 8 | 7 | 24 | 22 | 8.3 | 16.0 |
| PC | 8 | 7 | 20 | 19 | 5.0 | 11.6 |
| gB | 8 | 7 | 24 | 23 | 4.2 | 11.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, H.; Wussow, F.; Fernández-Alarcón, C.; Bierle, C.; Nguyen, J.; Diamond, D.J.; Schleiss, M.R. MVA-Vectored Pentameric Complex (PC) and gB Vaccines Improve Pregnancy Outcome after Guinea Pig CMV Challenge, but Only gB Vaccine Reduces Vertical Transmission. Vaccines 2019, 7, 182. https://doi.org/10.3390/vaccines7040182
Contreras H, Wussow F, Fernández-Alarcón C, Bierle C, Nguyen J, Diamond DJ, Schleiss MR. MVA-Vectored Pentameric Complex (PC) and gB Vaccines Improve Pregnancy Outcome after Guinea Pig CMV Challenge, but Only gB Vaccine Reduces Vertical Transmission. Vaccines. 2019; 7(4):182. https://doi.org/10.3390/vaccines7040182
Chicago/Turabian StyleContreras, Heidi, Felix Wussow, Claudia Fernández-Alarcón, Craig Bierle, Jenny Nguyen, Don J. Diamond, and Mark R. Schleiss. 2019. "MVA-Vectored Pentameric Complex (PC) and gB Vaccines Improve Pregnancy Outcome after Guinea Pig CMV Challenge, but Only gB Vaccine Reduces Vertical Transmission" Vaccines 7, no. 4: 182. https://doi.org/10.3390/vaccines7040182
APA StyleContreras, H., Wussow, F., Fernández-Alarcón, C., Bierle, C., Nguyen, J., Diamond, D. J., & Schleiss, M. R. (2019). MVA-Vectored Pentameric Complex (PC) and gB Vaccines Improve Pregnancy Outcome after Guinea Pig CMV Challenge, but Only gB Vaccine Reduces Vertical Transmission. Vaccines, 7(4), 182. https://doi.org/10.3390/vaccines7040182