Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections

Abstract
1. Introduction
1.1. A Pressing Need for Vaccines Against Parasitic Diseases
1.2. Why Does Commercial Vaccine Development Steer Clear of Parasitic Infectious Diseases?
1.2.1. Lack of Knowledge of the Biological Complexity of Parasites
1.2.2. Parasitic Infections Mainly Impact Poor People in Regions of Low Economic Power
1.2.3. Most Parasites Cause Chronic Disease and Disabilities but Do Not Kill the Host
1.2.4. Limitations of the Traditional Vaccine Platforms
1.3. Enlisting mRNA Vaccine Technology to Control Parasitic Diseases
2. Messenger RNA Vaccine Technology
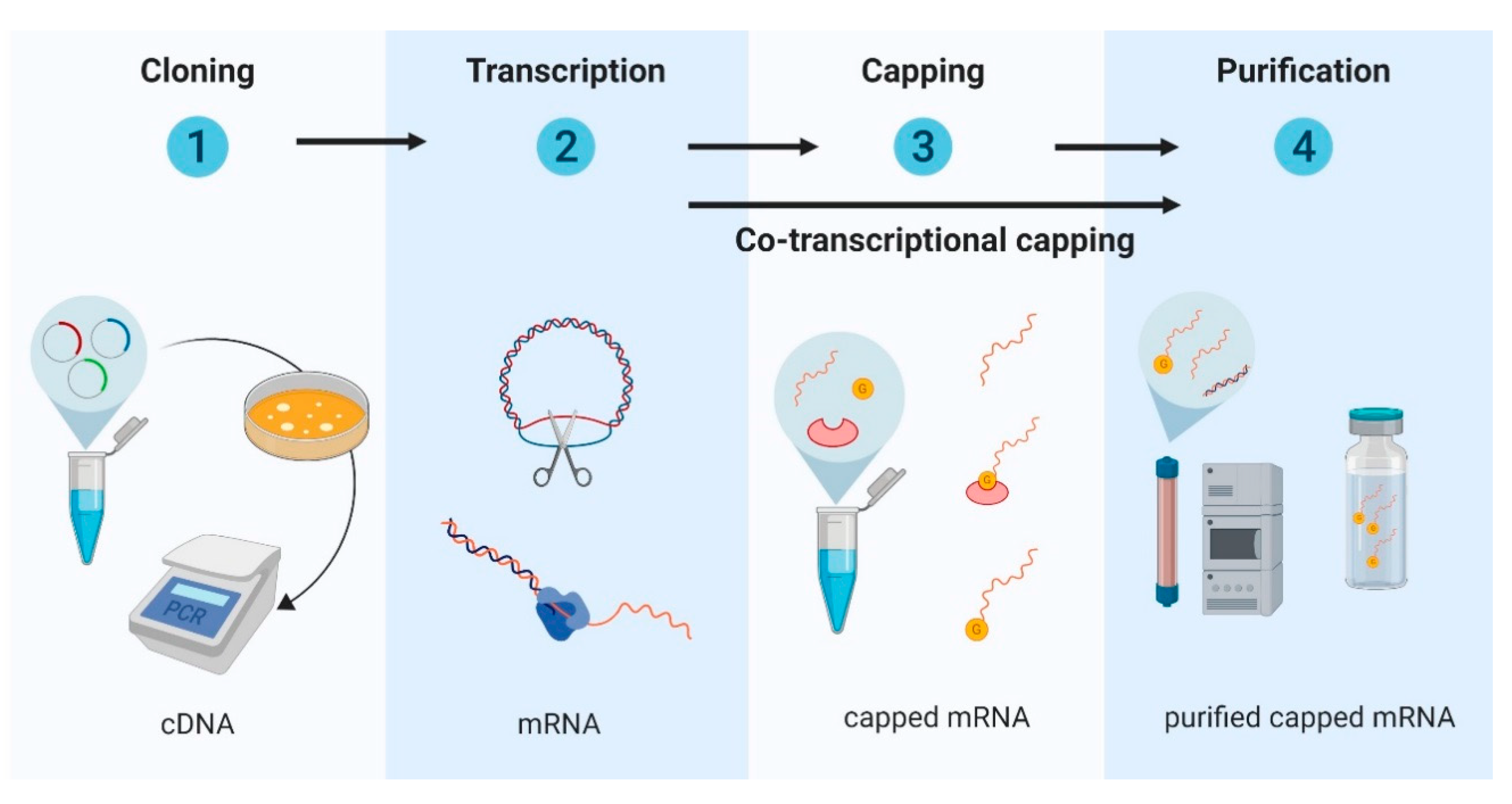
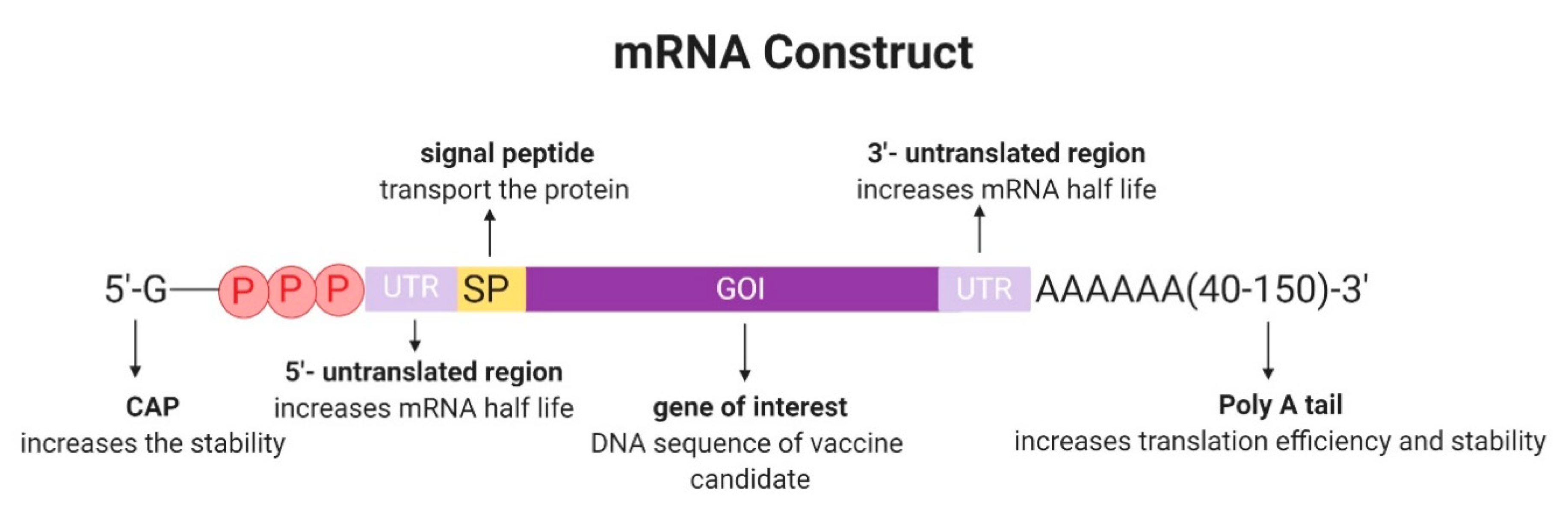
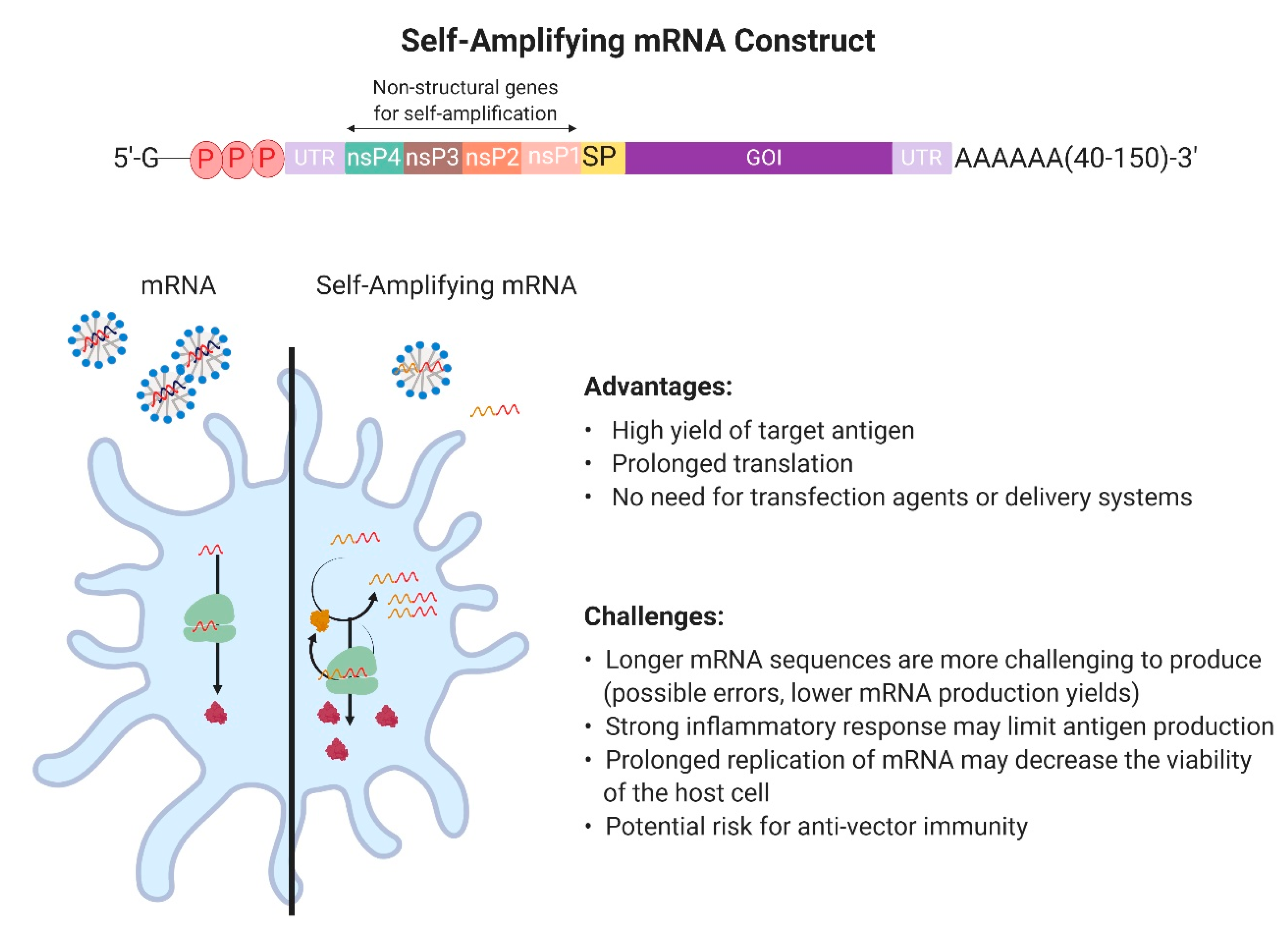
2.1. Design and Development of In Vitro Transcribed mRNA
2.2. Messenger RNA Delivery
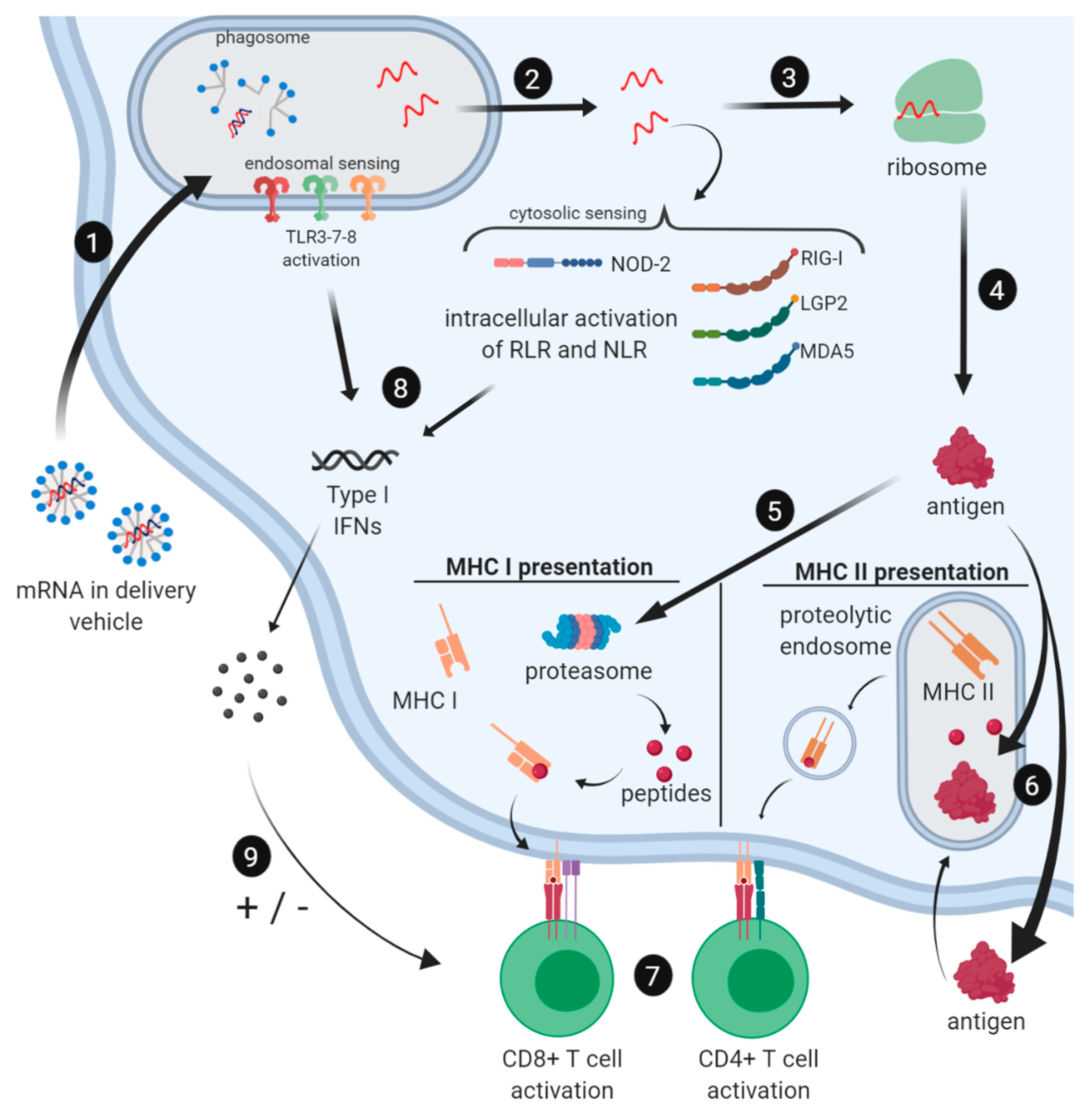
2.3. Immune Profile of mRNA Vaccines
2.3.1. Induction of the Innate Immune Response by mRNA Vaccines
2.3.2. Cellular and Humoral Immune Responses to mRNA Vaccines
2.4. Advantages and Limitations of the mRNA Platform for Vaccine Development Against Parasitic Infections
2.4.1. Production and Development
2.4.2. Multivalent mRNA Vaccines
2.4.3. Strong Cellular Immune Responses
2.4.4. Stability
2.4.5. Safety Profile
3. Messenger RNA Vaccines Against Parasitic Infections
3.1. Toxoplasma Gondii Infection
3.2. Malaria
3.3. Leishmania Donovani Infection
4. Concluding Remarks and Prospects of New mRNA Vaccines for Parasitic Diseases
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fenner, F. Global Eradication of Smallpox. Rev. Infect. Dis. 1982, 4, 916–930. [Google Scholar] [CrossRef]
- Norrby, E.; Uhnoo, I.; Brytting, M.; Zakikhany, K.; Lepp, T.; Olin, P. Polio close to eradication. Lakartidningen 2017, 114, 1–5. [Google Scholar]
- Achievements in Public Health, 1900–1999 Impact of Vaccines Universally Recommended for Children—United States, 1990–1998. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/00056803.htm#00003753.htm (accessed on 28 May 2019).
- Jourdan, P.M.; Lamberton, P.H.L.; Fenwick, A.; Addiss, D.G. Soil-transmitted helminth infections. Lancet 2018, 391, 252–265. [Google Scholar] [CrossRef]
- Filardy, A.A.; Guimarães-Pinto, K.; Nunes, M.P.; Zukeram, K.; Fliess, L.; Pereira, L.; Oliveira Nascimento, D.; Conde, L.; Morrot, A. Human Kinetoplastid Protozoan Infections: Where Are We Going Next? Front. Immunol. 2018, 9, 1943. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Kyu, H.H.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef]
- Hotez, P.J.; Fenwick, A.; Savioli, L.; Molyneux, D.H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 2009, 373, 1570–1575. [Google Scholar] [CrossRef]
- Hotez, P.J.; Damania, A.; Naghavi, M. Blue Marble Health and the Global Burden of Disease Study 2013. PLoS Negl. Trop. Dis. 2016, 10, e0004744. [Google Scholar] [CrossRef]
- Global Health Data Exchange, I. GBD Results Tool GHDx. Available online: http://ghdx.healthdata.org/gbd-results-tool?params=gbd-api-2017-permalink/4c9d166de41065b7a285b59edcf47405 (accessed on 3 May 2019).
- Hotez, P.J.; Fenwick, A.; Molyneux, D.H. Collateral Benefits of Preventive Chemotherapy—Expanding the War on Neglected Tropical Diseases. N. Engl. J. Med. 2019, 380, 2389–2391. [Google Scholar] [CrossRef]
- Stylianou, A.; Hadjichrysanthou, C.; Truscott, J.E.; Anderson, R.M. Developing a mathematical model for the evaluation of the potential impact of a partially efficacious vaccine on the transmission dynamics of Schistosoma mansoni in human communities. Parasit Vectors 2017, 10, 294. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Hotez, P.J.; Hertenstein, D.L.; Diemert, D.J.; Zapf, K.M.; Bottazzi, M.E.; Bethony, J.M.; Brown, S.T.; Lee, B.Y. Modeling the Economic and Epidemiologic Impact of Hookworm Vaccine and Mass Drug Administration (MDA) in Brazil, a High Transmission Setting. Vaccine 2016, 34, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Bottazzi, M.E.; Hotez, P.J. “Running the Gauntlet”: Formidable challenges in advancing neglected tropical diseases vaccines from development through licensure, and a “Call to Action”. Hum. Vaccines Immunother. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Anonymous Mosquirix H-W-2300. Available online: https://www.ema.europa.eu/en/mosquirix-h-w-2300 (accessed on 18 August 2019).
- Morrison, W.I.; Tomley, F. Development of vaccines for parasitic diseases of animals: Challenges and opportunities. Parasite Immunol. 2016, 38, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Abath, F.G.C.; Montenegro, S.M.L.; Gomes, Y.M. Vaccines against human parasitic diseases: An overview. Acta Trop. 1998, 71, 237–254. [Google Scholar] [CrossRef]
- Hotez, P.J. The global fight to develop antipoverty vaccines in the anti-vaccine era. Hum. Vaccines Immunother. 2018, 14, 2128–2131. [Google Scholar] [CrossRef]
- Perry, G.H. Parasites and human evolution. Evol. Anthropol. 2014, 23, 218–228. [Google Scholar] [CrossRef]
- Rogier, C. Challenge of developing anti-parasite vaccines in the tropics. Med. Trop. (Mars) 2007, 67, 328–334. [Google Scholar]
- Nadim, A.; Javadian, E.; Tahvildar-Bidruni, G.; Ghorbani, M. Effectiveness of leishmanization in the control of cutaneous leishmaniasis. Bulletin Société de Pathologie Exotique Filiales 1983, 76, 377–383. [Google Scholar]
- Sacks, D.L. Vaccines against tropical parasitic diseases: A persisting answer to a persisting problem. Nat. Immunol. 2014, 15, 403–405. [Google Scholar] [CrossRef]
- Ogutu, B.R.; Apollo, O.J.; McKinney, D.; Okoth, W.; Siangla, J.; Dubovsky, F.; Tucker, K.; Waitumbi, J.N.; Diggs, C.; Wittes, J.; et al. Blood Stage Malaria Vaccine Eliciting High Antigen-Specific Antibody Concentrations Confers No Protection to Young Children in Western Kenya. PLoS ONE 2009, 4, e4708. [Google Scholar] [CrossRef]
- Wykes, M.N. Why haven’t we made an efficacious vaccine for malaria? EMBO Rep. 2013, 14, 661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poveda, C.; Biter, A.B.; Bottazzi, M.E.; Strych, U. Establishing preferred product characterization for the evaluation of RNA vaccine antigens. Vaccines 2019, in press. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.; De Koker, S.; Saelens, X.; Vanham, G.; Grooten, J. Challenges and advances towards the rational design of mRNA vaccines. Trends Mol. Med. 2013, 19, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E.; Dahlberg, J.E.; Lund, E. Reverse 5’ caps in RNAs made in vitro by phage RNA polymerases. RNA 1995, 1, 957–967. [Google Scholar] [PubMed]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl (3’-deoxy)GpppG. RNA 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.K.M.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Self-Replicating RNA Viruses for RNA Therapeutics. Molecules 2018, 23, 3310. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-amplifying mRNA vaccines. Adv. Genet. 2015, 89, 179–233. [Google Scholar] [PubMed]
- Duthie, M.S.; Van Hoeven, N.; MacMillen, Z.; Picone, A.; Mohamath, R.; Erasmus, J.; Hsu, F.-C.; Stinchcomb, D.T.; Reed, S.G. Heterologous Immunization with Defined RNA and Subunit Vaccines Enhances T Cell Responses That Protect Against Leishmania donovani. Front. Immunol 2018, 9, 2420. [Google Scholar] [CrossRef] [PubMed]
- Johanning, F.W.; Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.A.; Pike, M.J.; Curiel, D.T. A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res. 1995, 23, 1495–1501. [Google Scholar] [CrossRef]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljeström, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.-M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Sun, X. Recent advances in mRNA vaccine delivery. Nano Res. 2018, 11, 5338–5354. [Google Scholar] [CrossRef]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Park, S.-J.; Yim, Y.; Kim, J.; Choi, C.; Won, C.; Min, D.-H. Recent Advances in RNA Therapeutics and RNA Delivery Systems Based on Nanoparticles. Adv. Ther. 2018, 1, 1800065. [Google Scholar] [CrossRef]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs from Ebola Virus Disease. J Infect. Dis. 2018, 217, 451–455. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8+ T Cell Immunity to mRNA Vaccines. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Liu, M.A. Immunologic basis of vaccine vectors. Immunity 2010, 33, 504–515. [Google Scholar] [CrossRef]
- Lee, B.L.; Barton, G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014, 24, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.R.; Bruns, A.M.; Horvath, C.M. MDA5 and LGP2: Accomplices and Antagonists of Antiviral Signal Transduction. J. Virol. 2014, 88, 8194–8200. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F. Modulation of T-cell function by type I interferon. Immunol. Cell Biol. 2012, 90, 492–497. [Google Scholar] [CrossRef]
- Agarwal, P.; Raghavan, A.; Nandiwada, S.L.; Curtsinger, J.M.; Bohjanen, P.R.; Mueller, D.L.; Mescher, M.F. Gene Regulation and Chromatin Remodeling by IL-12 and Type I IFN in Programming for CD8 T Cell Effector Function and Memory. J. Immunol. 2009, 183, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef]
- Wiesel, M.; Crouse, J.; Bedenikovic, G.; Sutherland, A.; Joller, N.; Oxenius, A. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol. 2012, 42, 320–329. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibori, T.; Su, L.; Arduini, R.M.; Baker, D.P.; David, M. Cutting Edge: Role of STAT1, STAT3, and STAT5 in IFN-αβ Responses in T Lymphocytes. J. Immunol. 2005, 174, 609–613. [Google Scholar] [CrossRef]
- Terawaki, S.; Chikuma, S.; Shibayama, S.; Hayashi, T.; Yoshida, T.; Okazaki, T.; Honjo, T. IFN-α Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity. J. Immunol 2011, 186, 2772–2779. [Google Scholar] [CrossRef]
- Beissert, T.; Koste, L.; Perkovic, M.; Walzer, K.C.; Erbar, S.; Selmi, A.; Diken, M.; Kreiter, S.; Türeci, Ö.; Sahin, U. Improvement of In Vivo Expression of Genes Delivered by Self-Amplifying RNA Using Vaccinia Virus Immune Evasion Proteins. Hum. Gene Ther. 2017, 28, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Sarén, T.; Ramachandran, M.; Martikainen, M.; Yu, D. Insertion of the Type-I IFN Decoy Receptor B18R in a miRNA-Tagged Semliki Forest Virus Improves Oncolytic Capacity but Results in Neurotoxicity. Mol. Ther. Oncol. 2017, 7, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, N.; Dowdy, S.F. Enhanced generation of iPSCs from older adult human cells by a synthetic five-factor self-replicative RNA. PLoS ONE 2017, 12, e0182018. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, e52. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.; Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. B-cell activation by armed helper T cells. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J.; et al. Induction of Robust B Cell Responses after Influenza mRNA Vaccination Is Accompanied by Circulating Hemagglutinin-Specific ICOS+ PD-1+ CXCR3+ T Follicular Helper Cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.G.; Hu, Y.; Khan, A.M.; Zhou, J.; Salmon, J.; Chikhlikar, P.R.; Jung, K.-O.; Marques, E.T.A.; August, J.T. Dendritic Cell Mediated Delivery of Plasmid DNA Encoding LAMP/HIV-1 Gag Fusion Immunogen Enhances T Cell Epitope Responses in HLA DR4 Transgenic Mice. PLoS ONE 2010, 5, e8574. [Google Scholar] [CrossRef]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- WHO. Review of Vaccine Price Data; Submitted by WHO European Region; Member States through the WHO/UNICEF; Joint Reporting Form for 2013; WHO: Geneva, Switzerland, 2013; p. 60. [Google Scholar]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.-L.; Neu, A.; Sprangers, R. A general method for rapid and cost-efficient large-scale production of 5′ capped RNA. RNA 2016, 22, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; Gregorio, E.D.; Geall, A.J.; Brazzoli, M.; Bertholet, S. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLoS ONE 2016, 11, e0161193. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Golombek, S.; Steinle, H.; Hann, L.; Velic, A.; Macek, B.; Krajewski, S.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Efficient reduction of synthetic mRNA induced immune activation by simultaneous delivery of B18R encoding mRNA. J. Biol. Eng. 2019, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Stitz, L.; Vogel, A.; Schnee, M.; Voss, D.; Rauch, S.; Mutzke, T.; Ketterer, T.; Kramps, T.; Petsch, B. A thermostable messenger RNA based vaccine against rabies. PLoS Negl. Trop. Dis. 2017, 11, e0006108. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Drane, D.; Gowans, E.J. Long-term storage of DNA-free RNA for use in vaccine studies. BioTechniques 2007, 43, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Kirschman, J.L.; Bhosle, S.; Vanover, D.; Blanchard, E.L.; Loomis, K.H.; Zurla, C.; Murray, K.; Lam, B.C.; Santangelo, P.J. Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA–protein correlations at the level of single cells. Nucleic Acids Res. 2017, 45, e113. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Latest development on RNA-based drugs and vaccines. Future Sci. OA 2018, 4, FSO300. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, Y.; Zhao, X.; Lee, R.J.; Teng, L.; Zhou, C. Enhancing the Therapeutic Delivery of Oligonucleotides by Chemical Modification and Nanoparticle Encapsulation. Molecules 2017, 22, 1724. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Reesor, E.K.; Xu, Y.; Zope, H.R.; Zetter, B.R.; Shi, J. Biomaterials for mRNA delivery. Biomater. Sci. 2015, 3, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Hinz, T.; Kallen, K.; Britten, C.M.; Flamion, B.; Granzer, U.; Hoos, A.; Huber, C.; Khleif, S.; Kreiter, S.; Rammensee, H.-G.; et al. The European Regulatory Environment of RNA-Based Vaccines. Methods Mol. Biol. 2017, 1499, 203–222. [Google Scholar] [PubMed]
- Luo, F.; Zheng, L.; Hu, Y.; Liu, S.; Wang, Y.; Xiong, Z.; Hu, X.; Tan, F. Induction of Protective Immunity against Toxoplasma gondii in Mice by Nucleoside Triphosphate Hydrolase-II (NTPase-II) Self-amplifying RNA Vaccine Encapsulated in Lipid Nanoparticle (LNP). Front. Microbiol. 2017, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Baeza Garcia, A.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B.; et al. Neutralization of the Plasmodium- encoded MIF ortholog confers protective immunity against malaria infection. Nat. Commun. 2018, 9, 2714. [Google Scholar] [CrossRef]
- mRNA Vaccines and Therapeutics Market Forecast 2019–2029. Visiongain: London, UK, 2019.
- Iavarone, C.; O’hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef]
- Beaumier, C.M.; Gillespie, P.M.; Strych, U.; Hayward, T.; Hotez, P.J.; Bottazzi, M.E. Status of vaccine research and development of vaccines for Chagas disease. Vaccine 2016, 34, 2996–3000. [Google Scholar] [CrossRef]
- Jones, K.; Versteeg, L.; Damania, A.; Keegan, B.; Kendricks, A.; Pollet, J.; Cruz-Chan, J.V.; Gusovsky, F.; Hotez, P.J.; Bottazzi, M.E. Vaccine-linked chemotherapy improves benznidazole efficacy for acute Chagas disease. Infect. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.A.; Versteeg, L.; Wang, Q.; Pollet, J.; Zhan, B.; Gusovsky, F.; Bottazzi, M.E.; Hotez, P.J.; Jones, K.M. A therapeutic vaccine prototype induces protective immunity and reduces cardiac fibrosis in a mouse model of chronic Trypanosoma cruzi infection. PLoS Negl. Tro. Dis. 2019, 13, e0007413. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Virgilio, F.; Pontes, C.; Dominguez, M.R.; Ersching, J.; Rodrigues, M.M.; Vasconcelos, J.R. CD8(+) T cell-mediated immunity during Trypanosoma cruzi infection: A path for vaccine development? Mediators Inflamm. 2014, 2014, 243786. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease/Parasite | Prevalence | DALY’s | Deaths |
|---|---|---|---|
| Ascariasis Ascaris lumbricoides | 447,008,998 | 860,833 | 3206 |
| Trichuriasis Trichuris trichiura | 289,617,741 | 212,664 | N/A |
| Hookworm disease Ancylostoma duodenale and Necator americanus | 229,217,130 | 845,010 | N/A |
| Schistosomiasis Schistosoma spp. | 142,788,542 | 1,431,447 | 8837 |
| Malaria Plasmodium spp. | 136,085,123 | 45,014,578 | 619,827 |
| Chagas disease Trypanosoma cruzi | 6,196,959 | 232,143 | 7853 |
| Leishmaniasis Leishmania sp. | 4,130,197 | 774,211 | 7527 |
| Sleeping Sickness Trypanosoma brucei | 4896 | 78,990 | 1364 |
| Vaccine Platform | Advantages | Disadvantages |
|---|---|---|
| Killed/Attenuated Parasites |
|
|
| Subunit/Recombinant Protein |
|
|
| Viral Vector |
|
|
| DNA |
|
|
| RNA |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Versteeg, L.; Almutairi, M.M.; Hotez, P.J.; Pollet, J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines 2019, 7, 122. https://doi.org/10.3390/vaccines7040122
Versteeg L, Almutairi MM, Hotez PJ, Pollet J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines. 2019; 7(4):122. https://doi.org/10.3390/vaccines7040122
Chicago/Turabian StyleVersteeg, Leroy, Mashal M. Almutairi, Peter J. Hotez, and Jeroen Pollet. 2019. "Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections" Vaccines 7, no. 4: 122. https://doi.org/10.3390/vaccines7040122
APA StyleVersteeg, L., Almutairi, M. M., Hotez, P. J., & Pollet, J. (2019). Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines, 7(4), 122. https://doi.org/10.3390/vaccines7040122