Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine

 , ,
, ,  , and
, and 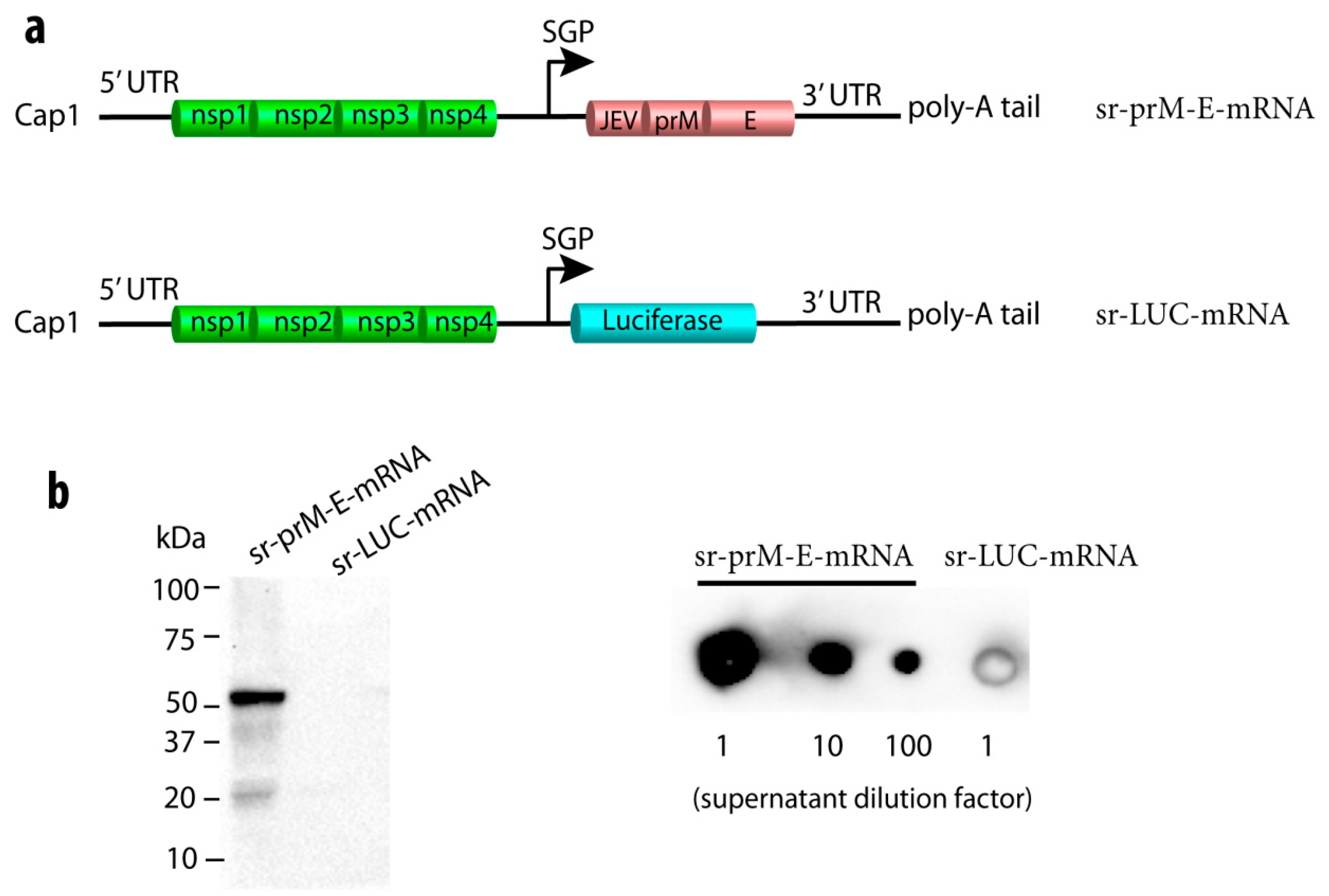{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells and Viruses
2.3. Synthesis of Self-Replicating mRNA Encoding ZIKV PrM-E or Luciferase
2.4. In Vitro Analysis of ZIKV E Protein Expression
2.5. Preparation of Formalin-Inactivated ZIKV Vaccine
2.6. Vaccination Experiments
2.7. Innate Immune Response and Expression
2.8. Zika Virus-Specific Antibody Titers
2.9. Zika Virus-Specific Cellular Immune Response
2.10. Zika Virus Challenge
2.11. ZIKV Virus Neutralization Tests
2.12. Determination of Zika Virus RNA in Blood
2.13. Statistical Analyses
3. Results
3.1. Construction of a Self-Replicating mRNA Expressing the ZIKV Pre-Membrane-Envelope Polyprotein
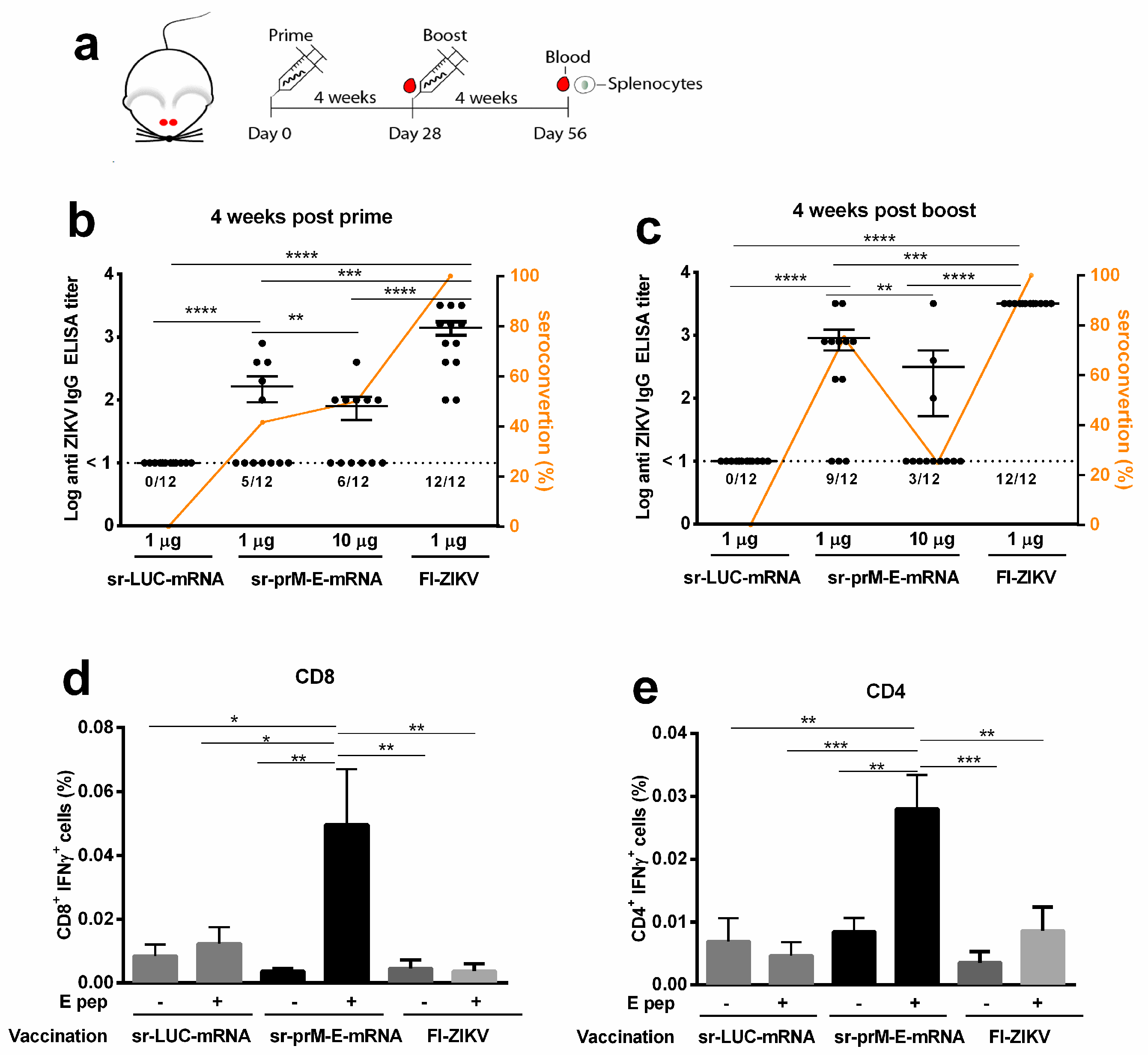
3.2. Immunogenicity of the Sr-PrM-E-mRNA ZIKV Vaccine in BALB/c Mice
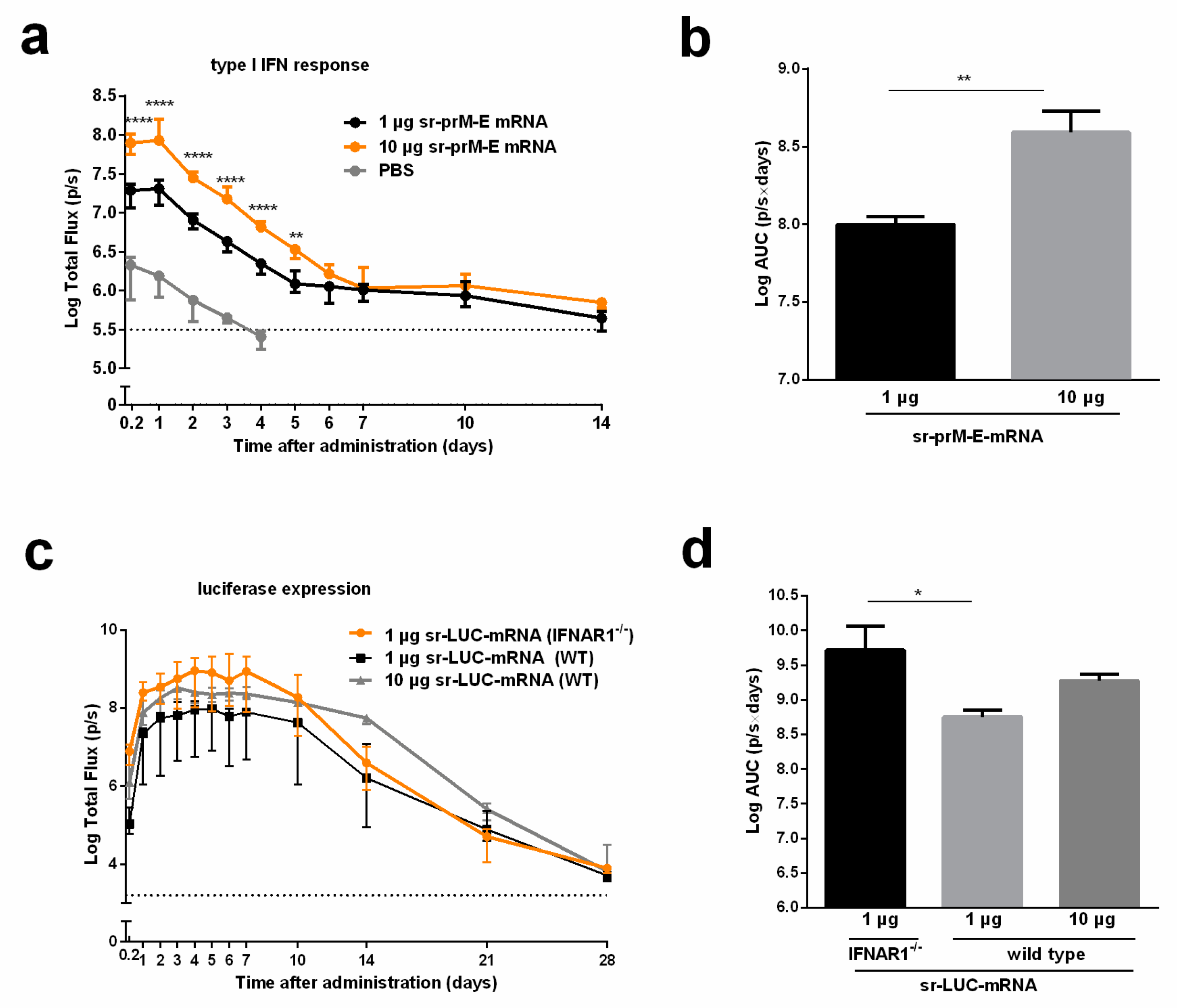
3.3. Immunogenicity of the Sr-PrM-E-mRNA ZIKV Vaccine in IFNAR1-/- and WT C57BL/6 Mice
3.4. The Interferon Response After Sr-PrM-E-mRNA ZIKV Vaccination Negatively Affects Antibody Titers
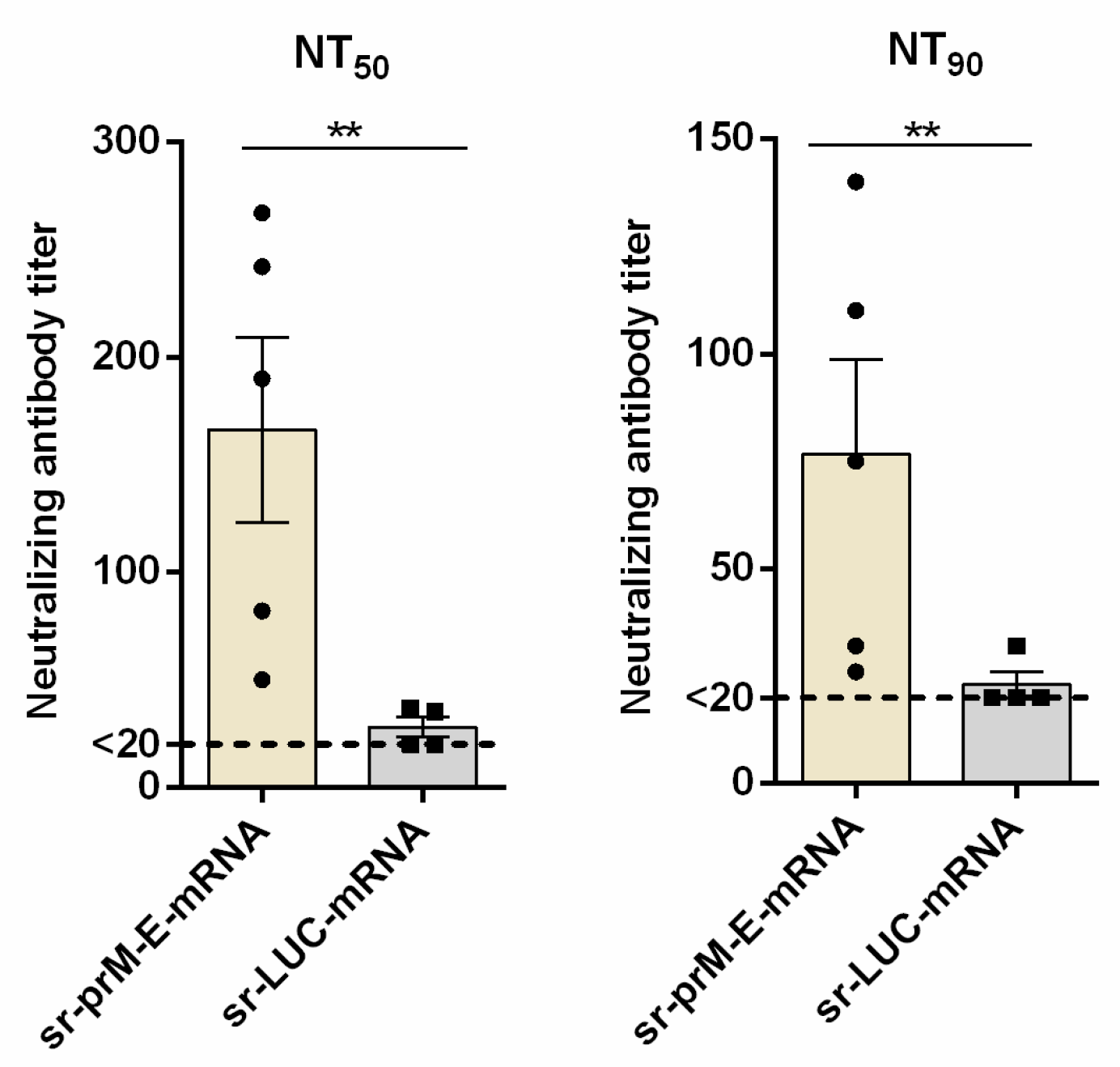
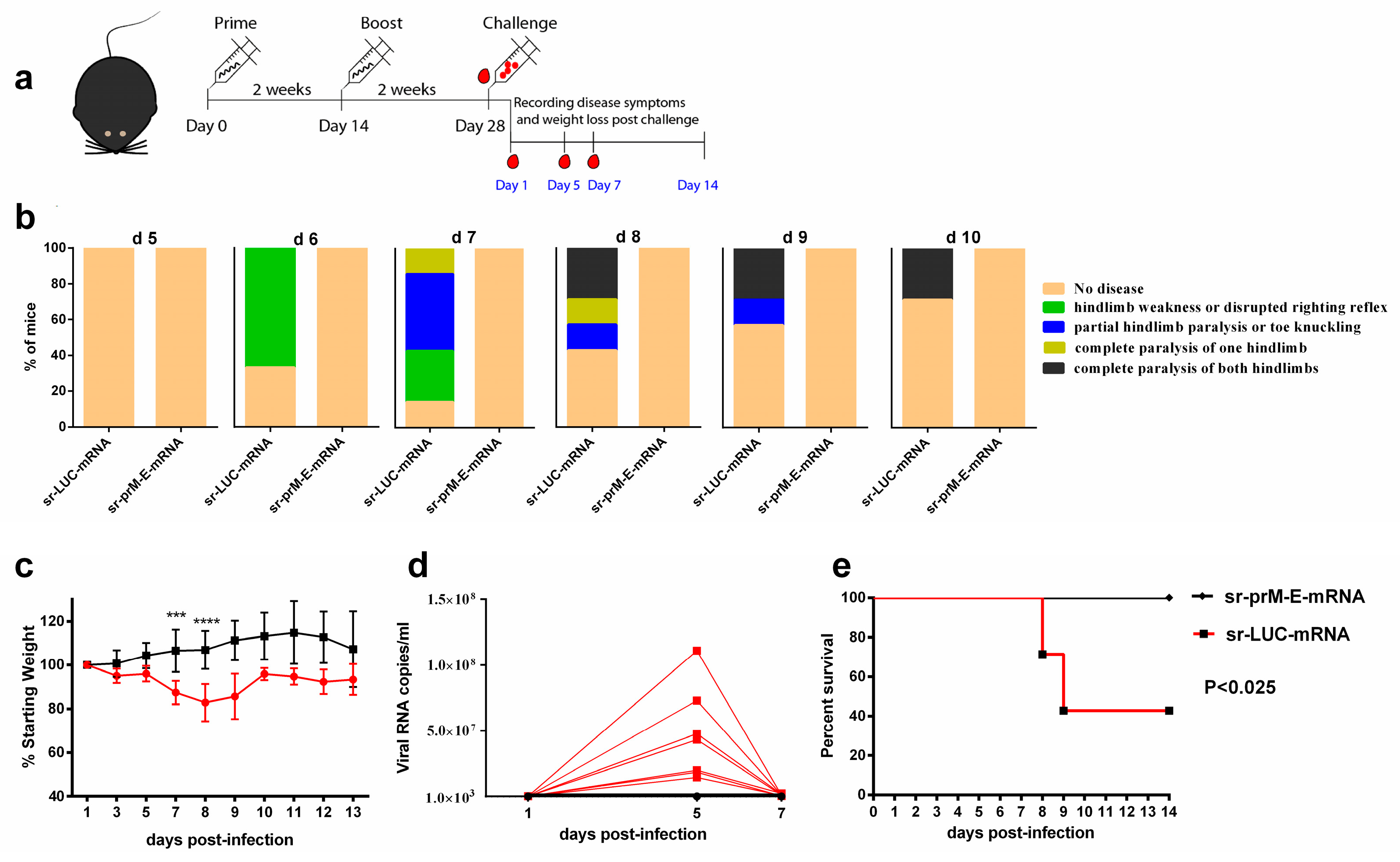
3.5. Protective Efficacy of the Sr-PrM-E-mRNA ZIKV Vaccine in IFNAR1-/- Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johansson, M.A.; Mier-y-Teran-Romero, L.; Reefhuis, J.; Gilboa, S.M.; Hills, S.L. Zika and the Risk of Microcephaly. N. Engl. J. Med. 2016, 375, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Nambala, P.; Su, W.C. Role of Zika Virus prM Protein in Viral Pathogenicity and Use in Vaccine Development. Front. Microbiol. 2018, 9, 1797. [Google Scholar] [CrossRef] [PubMed]
- Besnard, M.; Lastere, S.; Teissier, A.; Cao-Lormeau, V.; Musso, D. Evidence of perinatal transmission of Zika virus, French Polynesia, December 2013 and February 2014. Eurosurveillance 2014, 19, 20751. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome—Case report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Fournier, E.; Leparc-Goffart, I.; Larre, P.; Cubizolle, S.; Sookhareea, C.; Lastere, S.; Ghawche, F. Increase in cases of Guillain-Barre syndrome during a Chikungunya outbreak, French Polynesia, 2014 to 2015. Eurosurveillance 2015, 20, 30079. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika Virus and Birth Defects—Reviewing the Evidence for Causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Hoen, B.; Schaub, B.; Funk, A.L.; Ardillon, V.; Boullard, M.; Cabie, A.; Callier, C.; Carles, G.; Cassadou, S.; Cesaire, R.; et al. Pregnancy Outcomes after ZIKV Infection in French Territories in the Americas. N. Engl. J. Med. 2018, 378, 985–994. [Google Scholar] [CrossRef]
- Lourenco, J.; Maia de Lima, M.; Faria, N.R.; Walker, A.; Kraemer, M.U.; Villabona-Arenas, C.J.; Lambert, B.; Marques de Cerqueira, E.; Pybus, O.G.; Alcantara, L.C.; et al. Epidemiological and ecological determinants of Zika virus transmission in an urban setting. Elife 2017, 6, e29820. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef]
- Ferguson, N.M.; Cucunuba, Z.M.; Dorigatti, I.; Nedjati-Gilani, G.L.; Donnelly, C.A.; Basanez, M.G.; Nouvellet, P.; Lessler, J. Epidemiology. Countering the Zika epidemic in Latin America. Science 2016, 353, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Madrinan, M.J.; Turell, M. History of Mosquitoborne Diseases in the United States and Implications for New Pathogens. Emerg. Infect. Dis. 2018, 24, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.S. ZIKA—How fast does this virus mutate? Zool. Res. 2016, 37, 110–115. [Google Scholar] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Larocca, R.A.; Abbink, P.; Peron, J.P.; Zanotto, P.M.; Iampietro, M.J.; Badamchi-Zadeh, A.; Boyd, M.; Ng’ang’a, D.; Kirilova, M.; Nityanandam, R.; et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Luo, H.; Xie, X.; Medeiros, D.B.A.; Wakamiya, M.; Tesh, R.B.; Barrett, A.D.; Wang, T.; et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 2017, 23, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Boigard, H.; Alimova, A.; Martin, G.R.; Katz, A.; Gottlieb, P.; Galarza, J.M. Zika virus-like particle (VLP) based vaccine. PLoS Negl. Trop. Dis. 2017, 11, e0005608. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Larocca, R.A.; De La Barrera, R.A.; Bricault, C.A.; Moseley, E.T.; Boyd, M.; Kirilova, M.; Li, Z.; Ng’ang’a, D.; Nanayakkara, O.; et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 2016, 353, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Stephenson, K.E.; Barouch, D.H. Zika virus vaccines. Nat. Rev. Microbiol. 2018, 16, 594–600. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Houser, K.V.; Yamshchikov, G.V.; Bellamy, A.R.; May, J.; Enama, M.E.; Sarwar, U.; Larkin, B.; Bailer, R.T.; Koup, R.; Paskel, M.; et al. DNA vaccine priming for seasonal influenza vaccine in children and adolescents 6 to 17 years of age: A phase 1 randomized clinical trial. PLoS ONE 2018, 13, e0206837. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C. DNA vaccines against Zika virus speed into clinical trials. Nat. Rev. Drug. Discov. 2016, 15, 521–522. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Bellamy, A.R.; Belshe, R.; Bernstein, D.I.; Edupuganti, S.; Patel, S.M.; Renehan, P.; Zajdowicz, T.; Schwartz, R.; Koup, R.; et al. DNA priming for seasonal influenza vaccine: A phase 1b double-blind randomized clinical trial. PLoS ONE 2015, 10, e0125914. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A.; Ulmer, J.B. Human clinical trials of plasmid DNA vaccines. Adv. Genet. 2005, 55, 25–40. [Google Scholar] [PubMed]
- Doukas, J.; Morrow, J.; Bellinger, D.; Hilgert, T.; Martin, T.; Jones, D.; Mahajan, R.; Rusalov, D.; Sullivan, S.; Rolland, A. Nonclinical biodistribution, integration, and toxicology evaluations of an H5N1 pandemic influenza plasmid DNA vaccine formulated with Vaxfectin(R). Vaccine 2011, 29, 5443–5452. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug. Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catani, J.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jeklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef]
- Aramaki, Y.; Takano, S.; Tsuchiya, S. Induction of apoptosis in macrophages by cationic liposomes. FEBS Lett. 1999, 460, 472–476. [Google Scholar] [CrossRef]
- Dokka, S.; Toledo, D.; Shi, X.; Castranova, V.; Rojanasakul, Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm. Res. 2000, 17, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Bae, C.S.; Ahn, T. Cargo-Free Nanoparticles Containing Cationic Lipids Induce Reactive Oxygen Species and Cell Death in HepG2 Cells. Biol. Pharm. Bull. 2016, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines 2013, 1, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Lienenklaus, S.; Cornitescu, M.; Zietara, N.; Lyszkiewicz, M.; Gekara, N.; Jablonska, J.; Edenhofer, F.; Rajewsky, K.; Bruder, D.; Hafner, M.; et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 2009, 183, 3229–3236. [Google Scholar] [CrossRef]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef]
- REED, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints12. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Leyman, B.; Huysmans, H.; Mc Cafferty, S.; Combes, F.; Cox, E.; Devriendt, B.; Sanders, N.N. Comparison of the Expression Kinetics and Immunostimulatory Activity of Replicating mRNA, Nonreplicating mRNA, and pDNA after Intradermal Electroporation in Pigs. Mol. Pharm. 2018, 15, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Denies, S.; Cicchelero, L.; Van Audenhove, I.; Sanders, N.N. Combination of interleukin-12 gene therapy, metronomic cyclophosphamide and DNA cancer vaccination directs all arms of the immune system towards tumor eradication. J. Control. Release 2014, 187, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.J.; Hunt, A.R.; Davis, B. A single intramuscular injection of recombinant plasmid DNA induces protective immunity and prevents Japanese encephalitis in mice. J. Virol. 2000, 74, 4244–4252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morrison, T.E.; Diamond, M.S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J. Virol. 2017, 91, e00009–e00017. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, K.; Liljestrom, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, C. Cliniporator: Medical Electroporation of Tumors. In Handbook of Electroporation; Miklavcic, D., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–36. [Google Scholar]
- Giese, M. Delivery Technologies. In Introduction to Molecular Vaccinology; Giese, M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 233–258. [Google Scholar]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Cyster, J.G. Interfer’n with antibody responses. Sci. Immunol. 2016, 1, 1–3. [Google Scholar]
- Moseman, E.A.; Wu, T.; de la Torre, J.C.; Schwartzberg, P.L.; McGavern, D.B. Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Sci. Immunol. 2016, 1, 1–12. [Google Scholar]
- Sammicheli, S.; Kuka, M.; Di Lucia, P.; de Oya, N.J.; De Giovanni, M.; Fioravanti, J.; Cristofani, C.; Maganuco, C.G.; Fallet, B.; Ganzer, L.; et al. Inflammatory monocytes hinder antiviral B cell responses. Sci. Immunol. 2016, 1, 1–10. [Google Scholar]
- Fallet, B.; Narr, K.; Ertuna, Y.I.; Remy, M.; Sommerstein, R.; Cornille, K.; Kreutzfeldt, M.; Page, N.; Zimmer, G.; Geier, F.; et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol. 2016, 1, 1–10. [Google Scholar]
- Darville, T.; Andrews, C.W., Jr.; Sikes, J.D.; Fraley, P.L.; Braswell, L.; Rank, R.G. Mouse strain-dependent chemokine regulation of the genital tract T helper cell type 1 immune response. Infect. Immun. 2001, 69, 7419–7424. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.G.O.; Kitoko, J.Z.; Ferreira, F.M.; Suzart, V.G.; Papa, M.P.; Coelho, S.V.A.; Cavazzoni, C.B.; Paula-Neto, H.A.; Olsen, P.C.; Iwasaki, A.; et al. Critical role of CD4(+) T cells and IFNgamma signaling in antibody-mediated resistance to Zika virus infection. Nat. Commun. 2018, 9, 3136. [Google Scholar] [CrossRef] [PubMed]
- Regla-Nava, J.A.; Elong Ngono, A.; Viramontes, K.M.; Huynh, A.T.; Wang, Y.T.; Nguyen, A.T.; Salgado, R.; Mamidi, A.; Kim, K.; Diamond, M.S.; et al. Cross-reactive Dengue virus-specific CD8(+) T cells protect against Zika virus during pregnancy. Nat. Commun. 2018, 9, 3042. [Google Scholar] [CrossRef]
- Zellweger, R.M.; Eddy, W.E.; Tang, W.W.; Miller, R.; Shresta, S. CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J. Immunol. 2014, 193, 4117–4124. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Z.; Portela Catani, J.P.; Mc Cafferty, S.; Couck, L.; Van Den Broeck, W.; Gorlé, N.; Vandenbroucke, R.E.; Devriendt, B.; Ulbert, S.; Cnops, L.; et al. Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine. Vaccines 2019, 7, 96. https://doi.org/10.3390/vaccines7030096
Zhong Z, Portela Catani JP, Mc Cafferty S, Couck L, Van Den Broeck W, Gorlé N, Vandenbroucke RE, Devriendt B, Ulbert S, Cnops L, et al. Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine. Vaccines. 2019; 7(3):96. https://doi.org/10.3390/vaccines7030096
Chicago/Turabian StyleZhong, Zifu, João Paulo Portela Catani, Séan Mc Cafferty, Liesbeth Couck, Wim Van Den Broeck, Nina Gorlé, Roosmarijn E. Vandenbroucke, Bert Devriendt, Sebastian Ulbert, Lieselotte Cnops, and et al. 2019. "Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine" Vaccines 7, no. 3: 96. https://doi.org/10.3390/vaccines7030096
APA StyleZhong, Z., Portela Catani, J. P., Mc Cafferty, S., Couck, L., Van Den Broeck, W., Gorlé, N., Vandenbroucke, R. E., Devriendt, B., Ulbert, S., Cnops, L., Michels, J., Ariën, K. K., & Sanders, N. N. (2019). Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine. Vaccines, 7(3), 96. https://doi.org/10.3390/vaccines7030096