Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine


Abstract
1. Introduction
2. Basics of T Cell Responses during Infection and Vaccination
3. Helping Put the Spotlight on Memory CD4+ T Cell Responses
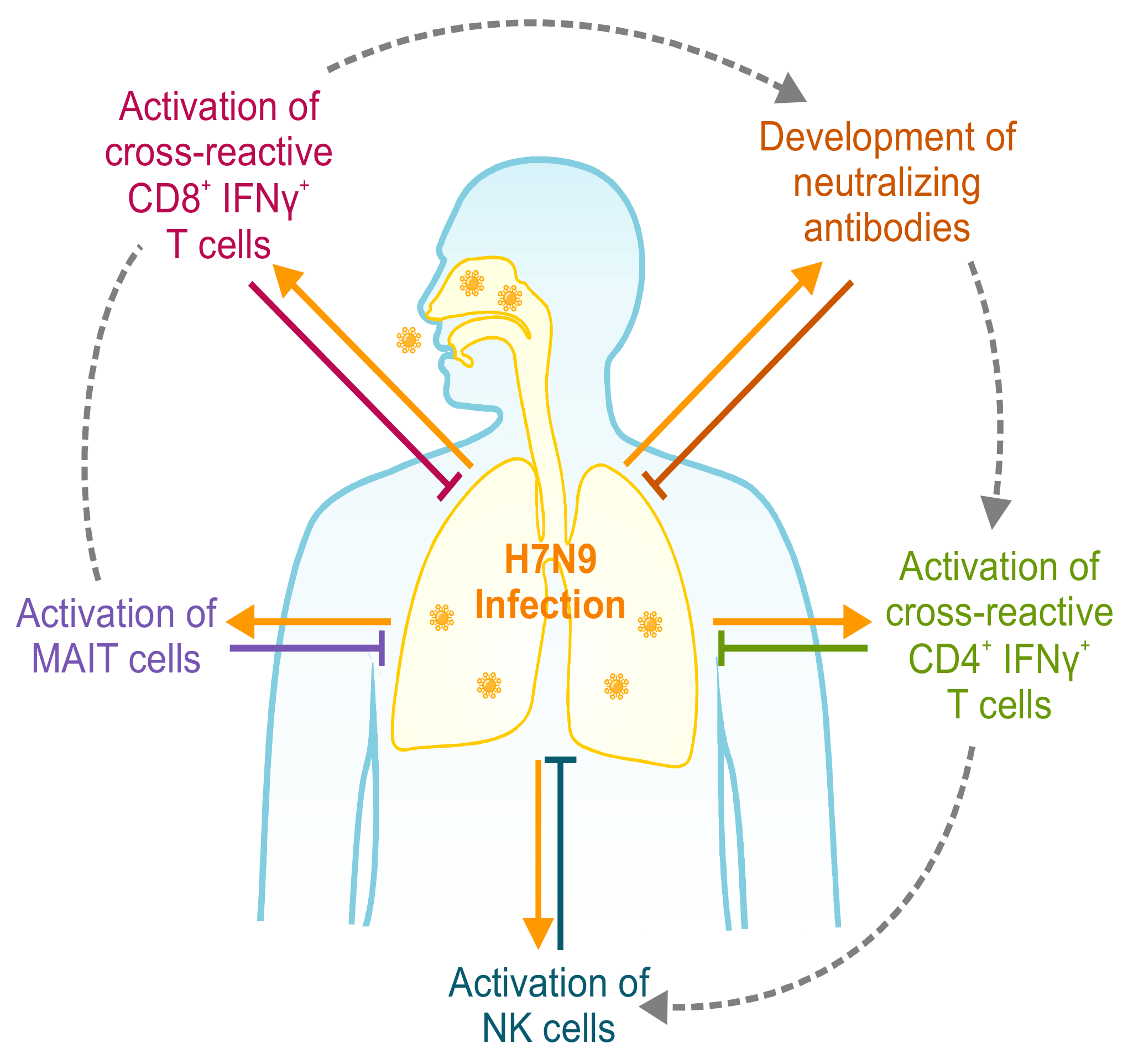
4. T Cell-Mediated Protection in Human Studies
5. Protect Globally—Think Locally! Trm for Influenza Infection
6. Universal Coverage: Epitopes, HLA, and Ethnicity
7. Universal Vaccine May Still Need to Thwart Viral Escape
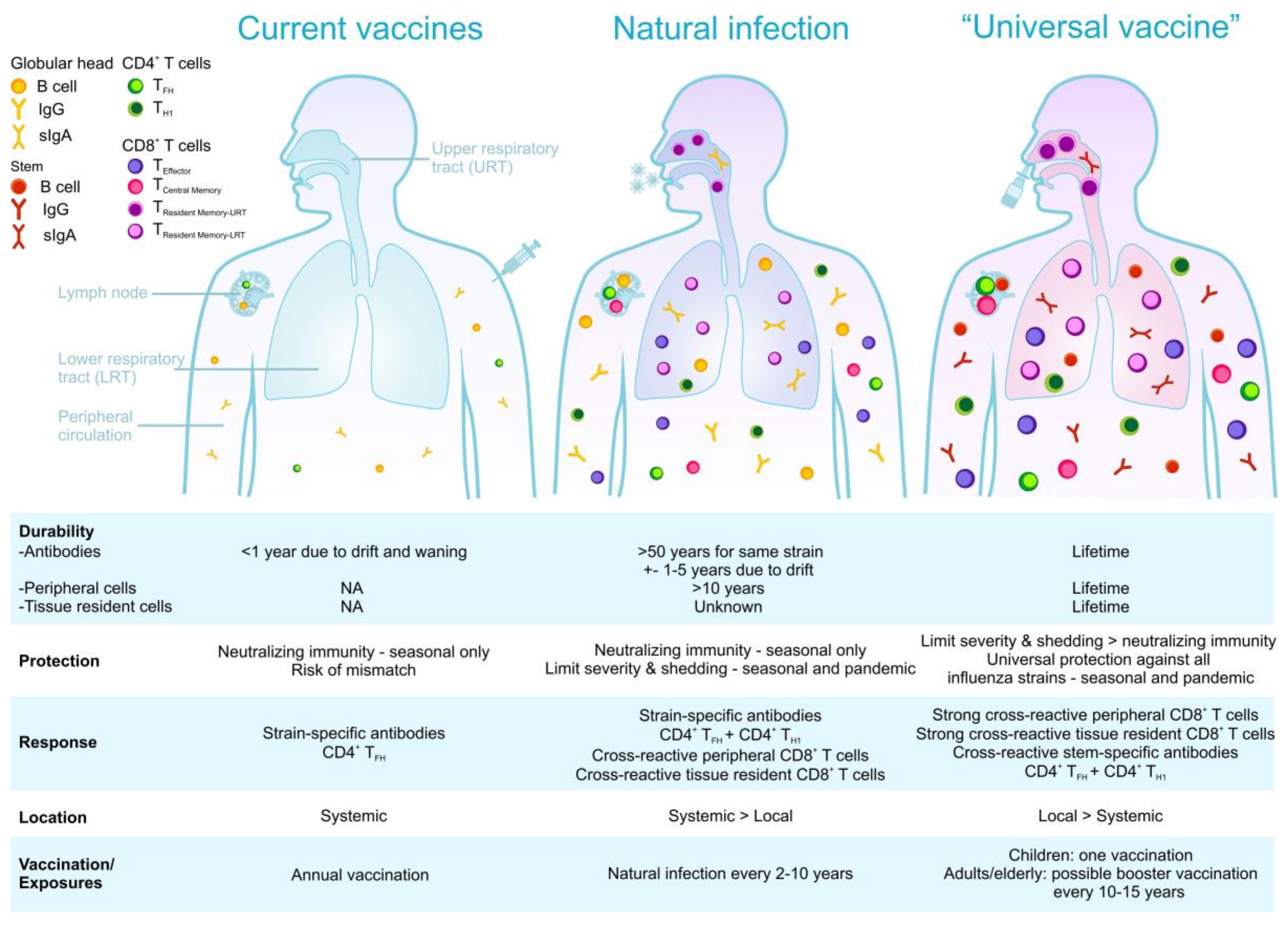
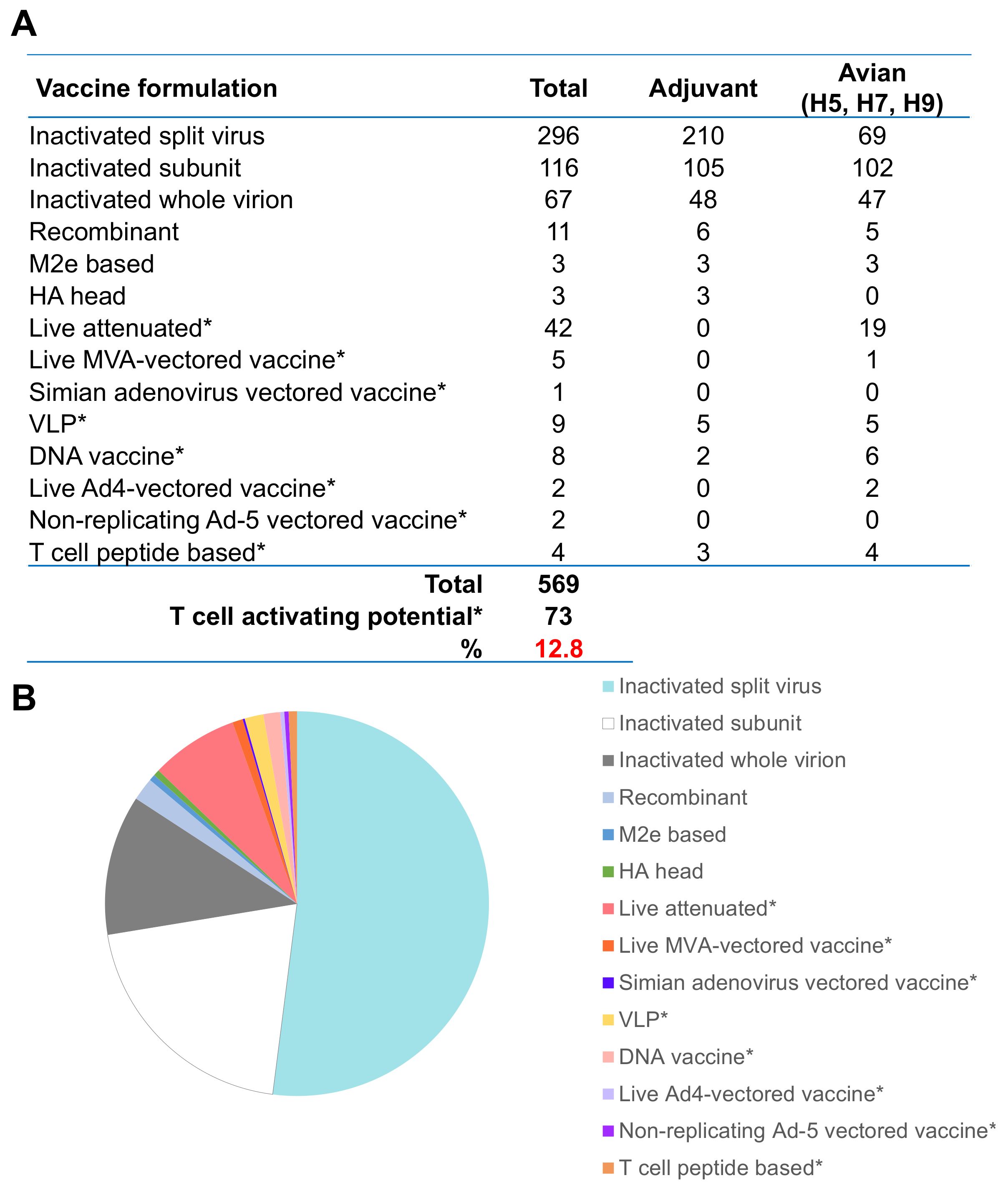
8. Current Developments in Influenza Vaccine Strategies and Future Perspectives towards a Universal Influenza Vaccine
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palache, A.; Abelin, A.; Hollingsworth, R.; Cracknell, W.; Jacobs, C.; Tsai, T.; Barbosa, P.; IFPMA Influenza Vaccine Supply (IFPMA IVS) task force. Survey of distribution of seasonal influenza vaccine doses in 201 countries (2004–2015): The 2003 World Health Assembly resolution on seasonal influenza vaccination coverage and the 2009 influenza pandemic have had very little impact on improving influenza control and pandemic preparedness. Vaccine 2017, 35, 4681–4686. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention; Appiah, G.D.; Blanton, L.; D’Mello, T.; Kniss, K.; Smith, S.; Mustaquim, D.; Steffens, C.; Dhara, R.; Cohen, J.; et al. Influenza activity—United States, 2014–15 season and composition of the 2015–16 influenza vaccine. MMWR 2015, 64, 583–590. [Google Scholar] [PubMed]
- Xie, H.; Wan, X.F.; Ye, Z.; Plant, E.P.; Zhao, Y.; Xu, Y.; Li, X.; Finch, C.; Zhao, N.; Kawano, T.; et al. H3N2 Mismatch of 2014–15 Northern Hemisphere Influenza Vaccines and Head-to-head Comparison between Human and Ferret Antisera derived Antigenic Maps. Sci. Rep. 2015, 5, 15279. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, C.S.; Levin, M.J. The rationale for quadrivalent influenza vaccines. Hum. Vaccines Immunother. 2012, 8, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Poon, L.L.; Zhu, H.C.; Ma, S.K.; Li, O.T.; Cheung, C.L.; Smith, G.J.; Peiris, J.S.; Guan, Y. Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 2010, 328, 1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Zost, S.J.; Thompson, A.J.; Oyen, D.; Nycholat, C.M.; McBride, R.; Paulson, J.C.; Hensley, S.E.; Wilson, I.A. A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine. PLoS Pathog. 2017, 13, e1006682. [Google Scholar] [CrossRef] [PubMed]
- Pebody, R.; McMenamin, J.; Nohynek, H. Live attenuated influenza vaccine (LAIV): Recent effectiveness results from the USA and implications for LAIV programmes elsewhere. Arch. Dis. Child. 2018, 103, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Caspard, H.; Gaglani, M.; Clipper, L.; Belongia, E.A.; McLean, H.Q.; Griffin, M.R.; Talbot, H.K.; Poehling, K.A.; Peters, T.R.; Veney, N.; et al. Effectiveness of live attenuated influenza vaccine and inactivated influenza vaccine in children 2–17 years of age in 2013–2014 in the United States. Vaccine 2016, 34, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Singanayagam, A.; Zambon, M.; Lalvani, A.; Barclay, W. Urgent challenges in implementing live attenuated influenza vaccine. Lancet Infect. Dis. 2018, 18, e25–e32. [Google Scholar] [CrossRef]
- Belongia, E.A.; Kieke, B.A.; Donahue, J.G.; Greenlee, R.T.; Balish, A.; Foust, A.; Lindstrom, S.; Shay, D.K.; Marshfield Influenza Study Group. Effectiveness of inactivated influenza vaccines varied substantially with antigenic match from the 2004–2005 season to the 2006–2007 season. J. Infect. Dis. 2009, 199, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Kaplan, B.; Zanin, M.; Debeauchamp, J.; Kercher, L.; Crumpton, J.C.; Seiler, P.; Sun, Y.; Tang, L.; Krauss, S.; et al. Impact of Adjuvants on the Immunogenicity and Efficacy of Split-Virion H7N9 Vaccine in Ferrets. J. Infect. Dis. 2015, 212, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Moise, L.; Tassone, R.; Gutierrez, A.H.; Terry, F.E.; Sangare, K.; Ardito, M.T.; Martin, W.D.; De Groot, A.S. H7N9 T-cell epitopes that mimic human sequences are less immunogenic and may induce Treg-mediated tolerance. Hum. Vaccines Immunother. 2015, 11, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Rockman, S.; Brown, L. Pre-pandemic and pandemic influenza vaccines. Hum. Vaccines 2010, 6, 792–801. [Google Scholar] [CrossRef]
- Bodewes, R.; Kreijtz, J.H.; Baas, C.; Geelhoed-Mieras, M.M.; de Mutsert, G.; van Amerongen, G.; van den Brand, J.M.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Vaccination against human influenza A/H3N2 virus prevents the induction of heterosubtypic immunity against lethal infection with avian influenza A/H5N1 virus. PLoS ONE 2009, 4, e5538. [Google Scholar] [CrossRef] [PubMed]
- Croft, N.P.; Purcell, A.W.; Tscharke, D.C. Quantifying epitope presentation using mass spectrometry. Mol. Immunol. 2015, 68, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Bui, H.H.; Sidney, J.; Zhang, Q.; Glenn, J.; Oseroff, C.; Mbawuike, I.N.; Alexander, J.; Newman, M.J.; Grey, H.; et al. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J. Virol. 2008, 82, 12241–12251. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Ha do, L.A.; Simmons, C.; de Jong, M.D.; Chau, N.V.; Schumacher, R.; Peng, Y.C.; McMichael, A.J.; Farrar, J.J.; Smith, G.L.; et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J. Clin. Investig. 2008, 118, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Parra, S.; Grant, E.; Loh, L.; Nguyen, T.H.; Campbell, K.A.; Tong, S.Y.; Miller, A.; Doherty, P.C.; Vijaykrishna, D.; Rossjohn, J.; et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc. Natl. Acad. Sci. USA 2014, 111, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Kreijtz, J.H.; de Mutsert, G.; Geelhoed-Mieras, M.M.; Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Osterhaus, A.D.; Fouchier, R.A.; Rimmelzwaan, G.F. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. J. Virol. 2014, 88, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.; de Mutsert, G.; van Baalen, C.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J. Virol. 2008, 82, 5161–5166. [Google Scholar] [CrossRef] [PubMed]
- Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Kreijtz, J.H.; de Mutsert, G.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Human T-cells directed to seasonal influenza A virus cross-react with 2009 pandemic influenza A (H1N1) and swine-origin triple-reassortant H3N2 influenza viruses. J. Gen. Virol. 2013, 94, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Hillaire, M.L.; van Trierum, S.E.; Kreijtz, J.H.; Bodewes, R.; Geelhoed-Mieras, M.M.; Nieuwkoop, N.J.; Fouchier, R.A.; Kuiken, T.; Osterhaus, A.D.; Rimmelzwaan, G.F. Cross-protective immunity against influenza pH1N1 2009 viruses induced by seasonal influenza A (H3N2) virus is mediated by virus-specific T-cells. J. Gen. Virol. 2011, 92, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.; Bodewes, R.; van Amerongen, G.; Kuiken, T.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine 2007, 25, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.; Bodewes, R.; van den Brand, J.M.; de Mutsert, G.; Baas, C.; van Amerongen, G.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Infection of mice with a human influenza A/H3N2 virus induces protective immunity against lethal infection with influenza A/H5N1 virus. Vaccine 2009, 27, 4983–4989. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R.; Kreijtz, J.H.; Geelhoed-Mieras, M.M.; van Amerongen, G.; Verburgh, R.J.; van Trierum, S.E.; Kuiken, T.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Vaccination against seasonal influenza A/H3N2 virus reduces the induction of heterosubtypic immunity against influenza A/H5N1 virus infection in ferrets. J. Virol. 2011, 85, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.P.; Doherty, P.C.; Branum, K.C.; Riberdy, J.M. Profound protection against respiratory challenge with a lethal H7N7 influenza A virus by increasing the magnitude of CD8(+) T-cell memory. J. Virol. 2000, 74, 11690–11696. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Bloom, J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. Elife 2014, 3, e03300. [Google Scholar] [CrossRef] [PubMed]
- Machkovech, H.M.; Bedford, T.; Suchard, M.A.; Bloom, J.D. Positive Selection in CD8+ T-Cell Epitopes of Influenza Virus Nucleoprotein Revealed by a Comparative Analysis of Human and Swine Viral Lineages. J. Virol. 2015, 89, 11275–11283. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, S.A.; Li, O.T.; Mak, P.W.; Mok, C.K.; Nicholls, J.M.; Guan, Y.; Waldmann, T.A.; Peiris, J.S.; Perera, L.P.; Poon, L.L. IL-15 adjuvanted multivalent vaccinia-based universal influenza Vaccine requires CD4+ T cells for heterosubtypic protection. Proc. Natl. Acad. Sci. USA 2014, 111, 5676–5681. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wan, Y.; Qiu, C.; Quinones-Parra, S.; Zhu, Z.; Loh, L.; Tian, D.; Ren, Y.; Hu, Y.; Zhang, X.; et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nat. Commun. 2015, 6, 6833. [Google Scholar] [CrossRef] [PubMed]
- Terajima, M.; Babon, J.A.; Co, M.D.; Ennis, F.A. Cross-reactive human B cell and T cell epitopes between influenza A and B viruses. Virol. J. 2013, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Nguyen, T.H.; Barclay, W.S.; Kedzierska, K. Knowns and unknowns of influenza B viruses. Future Microbiol. 2016, 11, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef] [PubMed]
- DiPiazza, A.; Nogales, A.; Poulton, N.; Wilson, P.C.; Martinez-Sobrido, L.; Sant, A.J. Pandemic 2009 H1N1 Influenza Venus reporter virus reveals broad diversity of MHC class II-positive antigen-bearing cells following infection in vivo. Sci. Rep. 2017, 7, 10857. [Google Scholar] [CrossRef] [PubMed]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999, 286, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Rimbert, M.; Echasserieau, K.; Saulquin, X.; Neveu, B.; Dechanet, J.; Cerundolo, V.; Bonneville, M. Selection of T cell clones expressing high-affinity public TCRs within Human cytomegalovirus-specific CD8 T cell responses. J. Immunol. 2005, 175, 6123–6132. [Google Scholar] [CrossRef] [PubMed]
- La Gruta, N.L.; Rothwell, W.T.; Cukalac, T.; Swan, N.G.; Valkenburg, S.A.; Kedzierska, K.; Thomas, P.G.; Doherty, P.C.; Turner, S.J. Primary CTL response magnitude in mice is determined by the extent of naive T cell recruitment and subsequent clonal expansion. J. Clin. Investig. 2010, 120, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Brehm, M.A.; Welsh, R.M.; Selin, L.K. Dynamics of memory T cell proliferation under conditions of heterologous immunity and bystander stimulation. J. Immunol. 2002, 169, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Mizukoshi, E.; Davis, A.R.; Bennink, J.R.; Rehermann, B. Cross-reactivity between hepatitis C virus and Influenza A virus determinant-specific cytotoxic T cells. J. Virol. 2001, 75, 11392–11400. [Google Scholar] [CrossRef] [PubMed]
- Nilges, K.; Hohn, H.; Pilch, H.; Neukirch, C.; Freitag, K.; Talbot, P.J.; Maeurer, M.J. Human papillomavirus type 16 E7 peptide-directed CD8+ T cells from patients with cervical cancer are cross-reactive with the coronavirus NS2 protein. J. Virol. 2003, 77, 5464–5474. [Google Scholar] [CrossRef] [PubMed]
- Clute, S.C.; Watkin, L.B.; Cornberg, M.; Naumov, Y.N.; Sullivan, J.L.; Luzuriaga, K.; Welsh, R.M.; Selin, L.K. Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J. Clin. Investig. 2005, 115, 3602–3612. [Google Scholar] [CrossRef] [PubMed]
- Clute, S.C.; Naumov, Y.N.; Watkin, L.B.; Aslan, N.; Sullivan, J.L.; Thorley-Lawson, D.A.; Luzuriaga, K.; Welsh, R.M.; Puzone, R.; Celada, F.; et al. Broad cross-reactive TCR repertoires recognizing dissimilar Epstein-Barr and influenza A virus epitopes. J. Immunol. 2010, 185, 6753–6764. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, A.C.; Kurane, I.; Ennis, F.A.; Rothman, A.L. Analysis of murine CD8(+) T-cell clones specific for the Dengue virus NS3 protein: Flavivirus cross-reactivity and influence of infecting serotype. J. Virol. 1999, 73, 398–403. [Google Scholar] [PubMed]
- Valkenburg, S.A.; Josephs, T.M.; Clemens, E.B.; Grant, E.J.; Nguyen, T.H.; Wang, G.C.; Price, D.A.; Miller, A.; Tong, S.Y.; Thomas, P.G.; et al. Molecular basis for universal HLA-A*0201-restricted CD8+ T-cell immunity against influenza viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 4440–4445. [Google Scholar] [CrossRef] [PubMed]
- Cukalac, T.; Chadderton, J.; Zeng, W.; Cullen, J.G.; Kan, W.T.; Doherty, P.C.; Jackson, D.C.; Turner, S.J.; La Gruta, N.L. The influenza virus-specific CTL immunodominance hierarchy in mice is determined by the relative frequency of high-avidity T cells. J. Immunol. 2014, 192, 4061–4068. [Google Scholar] [CrossRef] [PubMed]
- Tscharke, D.C.; Croft, N.P.; Doherty, P.C.; La Gruta, N.L. Sizing up the key determinants of the CD8(+) T cell response. Nat. Rev. Immunol. 2015, 15, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Deliyannis, G.; Jackson, D.C.; Ede, N.J.; Zeng, W.; Hourdakis, I.; Sakabetis, E.; Brown, L.E. Induction of long-term memory CD8(+) T cells for recall of viral clearing responses against influenza virus. J. Virol. 2002, 76, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.M.; Li, C.K.; Chui, C.S.; Huang, A.K.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Wagar, L.E.; Rosella, L.; Crowcroft, N.; Lowcock, B.; Drohomyrecky, P.C.; Foisy, J.; Gubbay, J.; Rebbapragada, A.; Winter, A.L.; Achonu, C.; et al. Humoral and cell-mediated immunity to pandemic H1N1 influenza in a Canadian cohort one year post-pandemic: Implications for vaccination. PLoS ONE 2011, 6, e28063. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, E.; Lewis, M.W.; Hansen, S.G.; Strelow, L.I.; Nelson, J.A.; Sexton, G.J.; Hanifin, J.M.; Slifka, M.K. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 2003, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Hillaire, M.L.; Geelhoed-Mieras, M.M.; Osterhaus, A.D.; Fouchier, R.A.; Rimmelzwaan, G.F. Human influenza A virus-specific CD8+ T cell response is long-lived. J. Infect. Dis. 2015, 212, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.J.; Strutt, T.; Reome, J.; Hollenbaugh, J.A.; Roberts, A.D.; Woodland, D.L.; Swain, S.L.; Dutton, R.W. Priming with cold-adapted influenza A does not prevent infection but elicits long-lived protection against supralethal challenge with heterosubtypic virus. J. Immunol. 2007, 178, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Arevalo, M.T.; Chen, Y.; Chen, S.; Zeng, M. T-cell-mediated cross-strain protective immunity elicited by prime-boost vaccination with a live attenuated influenza Vaccine. Int. J. Infect. Dis. 2014, 27, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Slutter, B.; Pewe, L.L.; Lauer, P.; Harty, J.T. Cutting edge: Rapid boosting of cross-reactive memory CD8 T cells broadens the protective capacity of the Flumist Vaccine. J. Immunol. 2013, 190, 3854–3858. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, S.; Vertruyen, A.; Aristegui, J.; Esposito, S.; McKeith, D.D.; Klemola, T.; Biolek, J.; Kuhr, J.; Bujnowski, T.; Desgrandchamps, D.; et al. Superior relative efficacy of live attenuated influenza Vaccine compared with inactivated influenza Vaccine in young children with recurrent respiratory tract infections. Pediatr. Infect. Dis. J. 2006, 25, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Edwards, K.M.; Vesikari, T.; Black, S.V.; Walker, R.E.; Hultquist, M.; Kemble, G.; Connor, E.M.; Group C-TCES. Live attenuated versus inactivated influenza Vaccine in infants and young children. N. Engl. J. Med. 2007, 356, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Brokstad, K.A.; Ogra, P. Influenza virus: Immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza Vaccines. Scand. J. Immunol. 2004, 59, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hoft, D.F.; Babusis, E.; Worku, S.; Spencer, C.T.; Lottenbach, K.; Truscott, S.M.; Abate, G.; Sakala, I.G.; Edwards, K.M.; Creech, C.B.; et al. Live and inactivated influenza Vaccines induce similar humoral responses, but only live Vaccines induce diverse T-cell responses in young children. J. Infect. Dis. 2011, 204, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Mohn, K.G.I.; Zhou, F.; Brokstad, K.A.; Sridhar, S.; Cox, R.J. Boosting of Cross-Reactive and Protection-Associated T Cells in Children After Live Attenuated Influenza Vaccination. J. Infect. Dis. 2017, 215, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Martinez-Sobrido, L.; Eko, F.O.; Palese, P.; Garcia-Sastre, A.; Lyn, D.; Okenu, D.; Bandea, C.; Ananaba, G.A.; Black, C.M.; et al. Live-attenuated influenza viruses as delivery vectors for Chlamydia Vaccines. Immunology 2007, 122, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Begom, S.; Bermingham, A.; Ziegler, T.; Roberts, K.L.; Barclay, W.S.; Openshaw, P.; Lalvani, A. Predominance of heterosubtypic IFN-gamma-only-secreting effector memory T cells in pandemic H1N1 naive adults. Eur. J. Immunol. 2012, 42, 2913–2924. [Google Scholar] [CrossRef] [PubMed]
- He, X.S.; Holmes, T.H.; Zhang, C.; Mahmood, K.; Kemble, G.W.; Lewis, D.B.; Dekker, C.L.; Greenberg, H.B.; Arvin, A.M. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza Vaccines. J. Virol. 2006, 80, 11756–11766. [Google Scholar] [CrossRef] [PubMed]
- Mbawuike, I.N.; Piedra, P.A.; Cate, T.R.; Couch, R.B. Cytotoxic T lymphocyte responses of infants after natural infection or immunization with live cold-recombinant or inactivated influenza A virus Vaccine. J. Med. Virol. 1996, 50, 105–111. [Google Scholar] [CrossRef]
- Bonduelle, O.; Yahia, N.; Siberil, S.; Benhabiles, N.; Carrat, F.; Krivine, A.; Rozenberg, F.; Dimitrov, J.; Kaveri, S.V.; Curjol, A.; et al. Longitudinal and integrative biomodeling of effector and memory immune compartments after inactivated influenza vaccination. J. Immunol. 2013, 191, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.; Hazenfeld, S.; Brady, R.C.; Subbramanian, R.A. Comparison of antibody and T-cell responses elicited by licensed inactivated- and live-attenuated influenza Vaccines against H3N2 hemagglutinin. Hum. Immunol. 2011, 72, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Mohn, K.G.; Bredholt, G.; Brokstad, K.A.; Pathirana, R.D.; Aarstad, H.J.; Tondel, C.; Cox, R.J. Longevity of B-cell and T-cell responses after live attenuated influenza vaccination in children. J. Infect. Dis. 2015, 211, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, N.; Ye, Y.; Fenstermacher, K.J.; Liu, H.; Lane, A.P.; Pekosz, A. The M2 protein of live, attenuated influenza Vaccine encodes a mutation that reduces replication in human nasal epithelial cells. Vaccine 2017, 35, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Soloff, A.C.; Lu, X.; Montecalvo, A.; Nguyen, D.C.; Matsuoka, Y.; Robbins, P.D.; Swayne, D.E.; Donis, R.O.; Katz, J.M.; et al. Protection of mice and poultry from lethal H5N1 avian influenza virus through adenovirus-based immunization. J. Virol. 2006, 80, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A Vaccine, MVA-NP+M1. Clin. Infect. Dis. 2011, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, E.J.; Tan, A.C.; Short, K.R.; Brown, L.E.; Chua, B.Y.; Jackson, D.C. Reducing the impact of influenza-associated secondary pneumococcal infections. Immunol. Cell Biol. 2016, 94, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.J.; Lee, Y.T.; Kim, K.H.; Jung, Y.J.; Lee, Y.; Denning, T.L.; Kang, S.M. Effects of MF59 Adjuvant on Induction of Isotype-Switched IgG Antibodies and Protection after Immunization with T-Dependent Influenza Virus Vaccine in the Absence of CD4+ T Cells. J. Virol. 2016, 90, 6976–6988. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.L.; Valkenburg, S.A.; Wong, C.K.; Li, O.T.; Nicholls, J.M.; Rabadan, R.; Peiris, J.S.; Poon, L.L. Generation of Live Attenuated Influenza Virus by Using Codon Usage Bias. J. Virol. 2015, 89, 10762–10773. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C.; Pena, L.; Angel, M.; Solorzano, A.; Albrecht, R.; Perez, D.R.; Garcia-Sastre, A.; Palese, P. Live attenuated influenza viruses containing NS1 truncations as Vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 2009, 83, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Baz, M.; Boonnak, K.; Paskel, M.; Santos, C.; Powell, T.; Townsend, A.; Subbarao, K. Nonreplicating influenza A virus Vaccines confer broad protection against lethal challenge. MBio 2015, 6, e01487-15. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Decman, V.; Ali, M.A.; Abt, M.C.; Wolf, A.I.; Monticelli, L.A.; Mozdzanowska, K.; Angelosanto, J.M.; Artis, D.; Erikson, J.; et al. Cooperativity between CD8+ T cells, non-neutralizing antibodies, and alveolar macrophages is important for heterosubtypic influenza virus immunity. PLoS Pathog. 2013, 9, e1003207. [Google Scholar] [CrossRef] [PubMed]
- Forrest, B.D.; Pride, M.W.; Dunning, A.J.; Capeding, M.R.; Chotpitayasunondh, T.; Tam, J.S.; Rappaport, R.; Eldridge, J.H.; Gruber, W.C. Correlation of cellular immune responses with protection against culture-confirmed influenza virus in young children. Clin. Vaccine Immunol. 2008, 15, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Loh, L.; Wang, Z.; Sant, S.; Koutsakos, M.; Jegaskanda, S.; Corbett, A.J.; Liu, L.; Fairlie, D.P.; Crowe, J.; Rossjohn, J.; et al. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc. Natl. Acad. Sci. USA 2016, 113, 10133–10138. [Google Scholar] [CrossRef] [PubMed]
- Spitaels, J.; Roose, K.; Saelens, X. Influenza and Memory T Cells: How to Awake the Force. Vaccines 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- DiPiazza, A.; Richards, K.A.; Knowlden, Z.A.; Nayak, J.L.; Sant, A.J. The Role of CD4 T Cell Memory in Generating Protective Immunity to Novel and Potentially Pandemic Strains of Influenza. Front. Immunol. 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Riberdy, J.M.; Christensen, J.P.; Branum, K.; Doherty, P.C. Diminished primary and secondary influenza virus-specific CD8(+) T-cell responses in CD4-depleted Ig(-/-) mice. J. Virol. 2000, 74, 9762–9765. [Google Scholar] [CrossRef] [PubMed]
- Belz, G.T.; Wodarz, D.; Diaz, G.; Nowak, M.A.; Doherty, P.C. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J. Virol. 2002, 76, 12388–12393. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Lanzavecchia, A. Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 2013, 31, 705–742. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Wheatley, A.K.; Loh, L.; Clemens, E.B.; Sant, S.; Nussing, S.; Fox, A.; Chung, A.W.; Laurie, K.L.; Hurt, A.C.; et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Chan, C.; Qiu, X.; Shannon, I.; White, C.L.; Sant, A.J.; Nayak, J.L. Selective pre-priming of HA-specific CD4 T cells restores immunological reactivity to HA on heterosubtypic influenza infection. PLoS ONE 2017, 12, e0176407. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.A.; Nayak, J.; Chaves, F.A.; DiPiazza, A.; Knowlden, Z.A.; Alam, S.; Treanor, J.J.; Sant, A.J. Seasonal Influenza Can Poise Hosts for CD4 T-Cell Immunity to H7N9 Avian Influenza. J. Infect. Dis. 2015, 212, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Nayak, J.L.; Richards, K.A.; Yang, H.; Treanor, J.J.; Sant, A.J. Effect of influenza A(H5N1) Vaccine prepandemic priming on CD4+ T-cell responses. J. Infect. Dis. 2015, 211, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Gostic, K.M.; Ambrose, M.; Worobey, M.; Lloyd-Smith, J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 2016, 354, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Dilzer, A.M.; Meents, D.L.; Swain, S.L. CD4 T cell-mediated protection from lethal influenza: Perforin and antibody-mediated mechanisms give a one-two punch. J. Immunol. 2006, 177, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Cowling, B.J.; Chan, K.H.; Fang, V.J.; Lau, L.L.H.; So, H.C.; Fung, R.O.P.; Ma, E.S.K.; Kwong, A.S.K.; Chan, C.W.; Tsui, W.W.S.; et al. Comparative epidemiology of pandemic and seasonal influenza A in households. N. Engl. J. Med. 2010, 362, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Epstein, S.L. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: An experiment of nature. J. Infect. Dis. 2006, 193, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Slepushkin, A.N. The effect of a previous attack of A1 influenza on susceptibility to A2 virus during the 1957 outbreak. Bull. World Health Organ. 1959, 20, 297–301. [Google Scholar] [PubMed]
- Oshansky, C.M.; Gartland, A.J.; Wong, S.S.; Jeevan, T.; Wang, D.; Roddam, P.L.; Caniza, M.A.; Hertz, T.; Devincenzo, J.P.; Webby, R.J.; et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 2014, 189, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.C.; Wang, L.; Goonetilleke, N.; Fragaszy, E.B.; Bermingham, A.; Copas, A.; Dukes, O.; Millett, E.R.; Nazareth, I.; Nguyen-Van-Tam, J.S.; et al. Natural T Cell-mediated Protection against Seasonal and Pandemic Influenza. Results of the Flu Watch Cohort Study. Am. J. Respir. Crit. Care Med. 2015, 191, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Le, N.M.; Horby, P.; van Doorn, H.R.; Nguyen, V.T.; Nguyen, H.H.; Nguyen, T.C.; Vu, D.P.; Nguyen, M.H.; Diep, N.T.; et al. Severe pandemic H1N1 2009 infection is associated with transient NK and T deficiency and aberrant CD8 responses. PLoS ONE 2012, 7, e31535. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Oshansky, C.M.; Guo, X.J.; Ralston, J.; Wood, T.; Reynolds, G.; Seeds, R.; Newbern, C.; Waite, B.; Widdowson, M.A.; et al. Severe influenza is characterized by prolonged immune activation: Results from the SHIVERS cohort study. J. Infect. Dis. 2017, 217, 245–256. [Google Scholar]
- Zhao, Y.; Zhang, Y.H.; Denney, L.; Young, D.; Powell, T.J.; Peng, Y.C.; Li, N.; Yan, H.P.; Wang, D.Y.; Shu, Y.L.; et al. High levels of virus-specific CD4+ T cells predict severe pandemic influenza A virus infection. Am. J. Respir. Crit. Care Med. 2012, 186, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A. Cytotoxic T-cell immunity to influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Cui, G.; Wei, Y.; Chen, J.; Zuo, J.; Cao, H.; Chen, Y.; Yao, H.; Tian, Z.; Li, L. Severe H7N9 infection is associated with decreased antigen-presenting capacity of CD14+ cells. PLoS ONE 2014, 9, e92823. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Lynfield, R.; Dwyer, D.E.; Losso, M.H.; Cozzi-Lepri, A.; Wentworth, D.; Lane, H.C.; Dewar, R.; Rupert, A.; Metcalf, J.A.; et al. The association between serum biomarkers and disease outcome in influenza A(H1N1)pdm09 virus infection: Results of two international observational cohort studies. PLoS ONE 2013, 8, e57121. [Google Scholar] [CrossRef] [PubMed]
- Fiore-Gartland, A.; Panoskaltsis-Mortari, A.; Agan, A.A.; Mistry, A.J.; Thomas, P.G.; Matthay, M.A.; Investigators, P.P.; Hertz, T.; Randolph, A.G. Cytokine Profiles of Severe Influenza Virus-Related Complications in Children. Front. Immunol. 2017, 8, 1423. [Google Scholar] [CrossRef] [PubMed]
- Slutter, B.; Pewe, L.L.; Kaech, S.M.; Harty, J.T. Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity 2013, 39, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Thomas, P.G. Balancing Immune Protection and Immune Pathology by CD8(+) T-Cell Responses to Influenza Infection. Front. Immunol. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Barcena, M.; Koster, A.J.; Kasper, J.; Kirkpatrick, C.J.; Scott, D.P.; de Vries, R.D.; Herold, S.; Rimmelzwaan, G.F.; Kuiken, T.; et al. Human CD8+ T Cells Damage Noninfected Epithelial Cells during Influenza Virus Infection In Vitro. Am. J. Respir. Cell Mol. Biol. 2017, 57, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Mohn, K.G.; Cox, R.J.; Tunheim, G.; Berdal, J.E.; Hauge, A.G.; Jul-Larsen, A.; Norwegian Pandemic, G.; Peters, B.; Oftung, F.; Jonassen, C.M.; et al. Immune Responses in Acute and Convalescent Patients with Mild, Moderate and Severe Disease during the 2009 Influenza Pandemic in Norway. PLoS ONE 2015, 10, e0143281. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Chan, P.K.; Wong, C.K.; Wong, K.T.; Choi, K.W.; Joynt, G.M.; Lam, P.; Chan, M.C.; Wong, B.C.; Lui, G.C.; et al. Viral clearance and inflammatory response patterns in adults hospitalized for pandemic 2009 influenza A(H1N1) virus pneumonia. Antivir. Ther. 2011, 16, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Mok, C.K.; Liu, X.Q.; Li, X.B.; He, J.F.; Guan, W.D.; Xu, Y.H.; Pan, W.Q.; Chen, L.Y.; Lin, Y.P.; et al. Clinical, virological and immunological features from patients infected with re-emergent avian-origin human H7N9 influenza disease of varying severity in Guangdong province. PLoS ONE 2015, 10, e0117846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef] [PubMed]
- de Bree, G.J.; Daniels, H.; Schilfgaarde, M.; Jansen, H.M.; Out, T.A.; van Lier, R.A.; Jonkers, R.E. Characterization of CD4+ memory T cell responses directed against common respiratory pathogens in peripheral blood and lung. J. Infect. Dis. 2007, 195, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Duan, S.; DeBeauchamp, J.; Zanin, M.; Kercher, L.; Sonnberg, S.; Fabrizio, T.; Jeevan, T.; Crumpton, J.C.; Oshansky, C.; et al. The immune correlates of protection for an avian influenza H5N1 Vaccine in the ferret model using oil-in-water adjuvants. Sci. Rep. 2017, 7, 44727. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed]
- De Bree, G.J.; van Leeuwen, E.M.; Out, T.A.; Jansen, H.M.; Jonkers, R.E.; van Lier, R.A. Selective accumulation of differentiated CD8+ T cells specific for respiratory viruses in the human lung. J. Exp. Med. 2005, 202, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Purwar, R.; Campbell, J.; Murphy, G.; Richards, W.G.; Clark, R.A.; Kupper, T.S. Resident memory T cells (T(RM)) are abundant in human lung: Diversity, function, and antigen specificity. PLoS ONE 2011, 6, e16245. [Google Scholar] [CrossRef] [PubMed]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyarto, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8(+) T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Turner, D.; Pham, Q.; Wherry, E.J.; Lefrancois, L.; Farber, D.L. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011, 187, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.L.; Bickham, K.L.; Thome, J.J.; Kim, C.Y.; D’Ovidio, F.; Wherry, E.J.; Farber, D.L. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2013, 7, 501. [Google Scholar] [CrossRef] [PubMed]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, Y.; Lee, Y.T.; Bouchard, K.R.; Benechet, A.; Khanna, K.; Cauley, L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014, 95, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Suarez-Ramirez, J.E.; Wu, T.; Redman, J.M.; Bouchard, K.; Hadley, G.A.; Cauley, L.S. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J. Virol. 2011, 85, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Piet, B.; de Bree, G.J.; Smids-Dierdorp, B.S.; van der Loos, C.M.; Remmerswaal, E.B.; von der Thusen, J.H.; van Haarst, J.M.; Eerenberg, J.P.; ten Brinke, A.; van der Bij, W.; et al. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Investig. 2011, 121, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Pizzolla, A.; Nguyen, T.H.; Sant, S.; Jaffar, J.; Loudovaris, T.; Mannering, S.I.; Thomas, P.G.; Westall, G.P.; Kedzierska, K.; Wakim, L.M. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J. Clin. Investig. 2018, 128, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Oja, A.E.; Piet, B.; Helbig, C.; Stark, R.; van der Zwan, D.; Blaauwgeers, H.; Remmerswaal, E.B.M.; Amsen, D.; Jonkers, R.E.; Moerland, P.D.; et al. Trigger-happy resident memory CD4(+) T cells inhabit the human lungs. Mucosal Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Smith, J.; Caminschi, I.; Lahoud, M.H.; Villadangos, J.A. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol. 2015, 8, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. Transforming Growth Factor-beta Signaling Controls the Formation and Maintenance of Gut-Resident Memory T Cells by Regulating Migration and Retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Takamura, S.; Yagi, H.; Hakata, Y.; Motozono, C.; McMaster, S.R.; Masumoto, T.; Fujisawa, M.; Chikaishi, T.; Komeda, J.; Itoh, J.; et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J. Exp. Med. 2016, 213, 3057–3073. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Gupta, N.; Mintern, J.D.; Villadangos, J.A. Enhanced survival of lung tissue-resident memory CD8(+) T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 2013, 14, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Bevan, M.J. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl. Acad. Sci. USA 2010, 107, 17872–17879. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. A Vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012, 491, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Gilchuk, P.; Hill, T.M.; Guy, C.; McMaster, S.R.; Boyd, K.L.; Rabacal, W.A.; Lu, P.; Shyr, Y.; Kohlmeier, J.E.; Sebzda, E.; et al. A Distinct Lung-Interstitium-Resident Memory CD8(+) T Cell Subset Confers Enhanced Protection to Lower Respiratory Tract Infection. Cell Rep. 2016, 16, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Deliyannis, G.; Kedzierska, K.; Lau, Y.F.; Zeng, W.; Turner, S.J.; Jackson, D.C.; Brown, L.E. Intranasal lipopeptide primes lung-resident memory CD8+ T cells for long-term pulmonary protection against influenza. Eur. J. Immunol. 2006, 36, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Gasper, D.J.; Neldner, B.; Plisch, E.H.; Rustom, H.; Carrow, E.; Imai, H.; Kawaoka, Y.; Suresh, M. Effective Respiratory CD8 T-Cell Immunity to Influenza Virus Induced by Intranasal Carbomer-Lecithin-Adjuvanted Non-replicating Vaccines. PLoS Pathog. 2016, 12, e1006064. [Google Scholar] [CrossRef] [PubMed]
- Pizzolla, A.; Nguyen, T.H.O.; Smith, J.M.; Brooks, A.G.; Kedzieska, K.; Heath, W.R.; Reading, P.C.; Wakim, L.M. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Slutter, B.; Van Braeckel-Budimir, N.; Abboud, G.; Varga, S.M.; Salek-Ardakani, S.; Harty, J.T. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362. [Google Scholar] [CrossRef] [PubMed]
- IPD-IMGT/HLA. IMGT/HLA Statistics. 2017. Available online: https://www.ebi.ac.uk/ipd/imgt/hla/stats.html (accessed on 22 December 2017).
- Day, E.B.; Charlton, K.L.; La Gruta, N.L.; Doherty, P.C.; Turner, S.J. Effect of MHC class I diversification on influenza epitope-specific CD8+ T cell precursor frequency and subsequent effector function. J. Immunol. 2011, 186, 6319–6328. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Reinhold, B.B.; Zhang, G.L.; Ivanov, A.R.; Karger, B.L.; Reinherz, E.L. Physical detection of influenza A epitopes identifies a stealth subset on human lung epithelium evading natural CD8 immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; Schafer, F.; Bardet, W.; Buchli, R.; Air, G.M.; Hildebrand, W.H. HLA class I molecules consistently present internal influenza epitopes. Proc. Natl. Acad. Sci. USA 2009, 106, 540–545. [Google Scholar] [CrossRef] [PubMed]
- NCBI. dbMHC Anthropology. Available online: https://www.ncbi.nlm.nih.gov/gv/mhc/ihwg.cgi?cmd=page&page=AnthroMain (accessed on 22 December 2017).
- Hertz, T.; Oshansky, C.M.; Roddam, P.L.; DeVincenzo, J.P.; Caniza, M.A.; Jojic, N.; Mallal, S.; Phillips, E.; James, I.; Halloran, M.E.; et al. HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection. Proc. Natl. Acad. Sci. USA 2013, 110, 13492–13497. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; Takeshita, L.Y.; Santos, E.J.; Kempson, F.; Maia, M.H.; da Silva, A.L.; Teles e Silva, A.L.; Ghattaoraya, G.S.; Alfirevic, A.; Jones, A.R.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef] [PubMed]
- Clemens, E.B.; Grant, E.J.; Wang, Z.; Gras, S.; Tipping, P.; Rossjohn, J.; Miller, A.; Tong, S.Y.; Kedzierska, K. Towards identification of immune and genetic correlates of severe influenza disease in Indigenous Australians. Immunol. Cell Biol. 2016, 94, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Munkhbat, B.; Hagihara, M.; Tsuritani, I.; Abe, H.; Tsuji, K. HLA-A*24-B*07-DRB1*01 haplotype implicated with genetic disposition of peak bone mass in healthy young Japanese women. Hum. Immunol. 1998, 59, 243–249. [Google Scholar] [CrossRef]
- Qu, H.Q.; Polychronakos, C. The effect of the MHC locus on autoantibodies in type 1 diabetes. J. Med. Genet. 2009, 46, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus—Present and future perspectives. Nat. Rev. Endocrinol. 2011, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; O’Brien, S.J. The influence of HLA genotype on AIDS. Annu. Rev. Med. 2003, 54, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Leslie, A.J.; Honeyborne, I.; Ramduth, D.; Thobakgale, C.; Chetty, S.; Rathnavalu, P.; Moore, C.; Pfafferott, K.J.; Hilton, L.; et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 2004, 432, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.; Walker, B.D. HIV and HLA class I: An evolving relationship. Immunity 2012, 37, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Deaths related to 2009 pandemic influenza A (H1N1) among American Indian/Alaska Natives—12 States, 2009. MMWR 2009, 58, 1341–1344. [Google Scholar]
- Flint, S.M.; Davis, J.S.; Su, J.Y.; Oliver-Landry, E.P.; Rogers, B.A.; Goldstein, A.; Thomas, J.H.; Parameswaran, U.; Bigham, C.; Freeman, K.; et al. Disproportionate impact of pandemic (H1N1) 2009 influenza on Indigenous people in the Top End of Australia’s Northern Territory. Med. J. Aust. 2010, 192, 617–622. [Google Scholar] [PubMed]
- Investigators, A.I.; Webb, S.A.; Pettila, V.; Seppelt, I.; Bellomo, R.; Bailey, M.; Cooper, D.J.; Cretikos, M.; Davies, A.R.; Finfer, S.; et al. Critical care services and 2009 H1N1 influenza in Australia and New Zealand. N. Engl. J. Med. 2009, 361, 1925–1934. [Google Scholar]
- Kumar, A.; Zarychanski, R.; Pinto, R.; Cook, D.J.; Marshall, J.; Lacroix, J.; Stelfox, T.; Bagshaw, S.; Choong, K.; Lamontagne, F.; et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009, 302, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- La Ruche, G.; Tarantola, A.; Barboza, P.; Vaillant, L.; Gueguen, J.; Gastellu-Etchegorry, M.; Epidemic Intelligence Team at In VS. The 2009 pandemic H1N1 influenza and indigenous populations of the Americas and the Pacific. EuroSurveillance 2009, 14, 19366. [Google Scholar] [CrossRef] [PubMed]
- Darrah, P.A.; Patel, D.T.; De Luca, P.M.; Lindsay, R.W.; Davey, D.F.; Flynn, B.J.; Hoff, S.T.; Andersen, P.; Reed, S.G.; Morris, S.L.; et al. Multifunctional TH1 cells define a correlate of Vaccine-mediated protection against Leishmania major. Nat. Med. 2007, 13, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Daucher, M.; Price, D.A.; Brenchley, J.M.; Lamoreaux, L.; Metcalf, J.A.; Rehm, C.; Nies-Kraske, E.; Urban, E.; Yoder, C.; Rock, D.; et al. Virological outcome after structured interruption of antiretroviral therapy for human immunodeficiency virus infection is associated with the functional profile of virus-specific CD8+ T cells. J. Virol. 2008, 82, 4102–4114. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Cellerai, C.; Bellutti Enders, F.; Kostler, J.; Codarri, L.; Tapia, G.; Boyman, O.; Castro, E.; Gaudieri, S.; James, I.; et al. Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc. Natl. Acad. Sci. USA 2007, 104, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.C.; de Mutsert, G.; Graus, Y.M.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. The magnitude and specificity of influenza A virus-specific cytotoxic T-lymphocyte responses in humans is related to HLA-A and -B phenotype. J. Virol. 2002, 76, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Burrows, S.R.; Silins, S.L.; Moss, D.J.; Khanna, R.; Misko, I.S.; Argaet, V.P. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J. Exp. Med. 1995, 182, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Apple, R.; Clare-Salzler, M.; Trembleau, S.; Mathis, D.; Adorini, L.; Sercarz, E. Determinant capture as a possible mechanism of protection afforded by major histocompatibility complex class II molecules in autoimmune disease. J. Exp. Med. 1993, 178, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.C.; McKenzie, I.F. Quantitative variation in H-2-antigen expression. I. Estimation of H-2K and H-2D expression in different strains of mice. Immunogenetics 1980, 11, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Rowland-Jones, S.L.; Powis, S.H.; Sutton, J.; Mockridge, I.; Gotch, F.M.; Murray, N.; Hill, A.B.; Rosenberg, W.M.; Trowsdale, J.; McMichael, A.J. An antigen processing polymorphism revealed by HLA-B8-restricted cytotoxic T lymphocytes which does not correlate with TAP gene polymorphism. Eur. J. Immunol. 1993, 23, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Tourdot, S.; Gould, K.G. Competition between MHC class I alleles for cell surface expression alters CTL responses to influenza A virus. J. Immunol. 2002, 169, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Tussey, L.G.; Rowland-Jones, S.; Zheng, T.S.; Androlewicz, M.J.; Cresswell, P.; Frelinger, J.A.; McMichael, A.J. Different MHC class I alleles compete for presentation of overlapping viral epitopes. Immunity 1995, 3, 65–77. [Google Scholar] [CrossRef]
- Valkenburg, S.A.; Gras, S.; Guillonneau, C.; Hatton, L.A.; Bird, N.A.; Twist, K.A.; Halim, H.; Jackson, D.C.; Purcell, A.W.; Turner, S.J.; et al. Preemptive priming readily overcomes structure-based mechanisms of virus escape. Proc. Natl. Acad. Sci. USA 2013, 110, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Kreijtz, J.H.; Geelhoed-Mieras, M.M.; Nieuwkoop, N.J.; Spronken, M.I.; van de Vijver, D.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Differential Recognition of Influenza A Viruses by M158-66 Epitope-Specific CD8+ T Cells Is Determined by Extraepitopic Amino Acid Residues. J. Virol. 2015, 90, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Kreijtz, J.H.; Rimmelzwaan, G.F. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef] [PubMed]
- Voeten, J.T.; Bestebroer, T.M.; Nieuwkoop, N.J.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J. Virol. 2000, 74, 6800–6807. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, S.A.; Quinones-Parra, S.; Gras, S.; Komadina, N.; McVernon, J.; Wang, Z.; Halim, H.; Iannello, P.; Cole, C.; Laurie, K.; et al. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat. Commun. 2013, 4, 2663. [Google Scholar] [CrossRef] [PubMed]
- Berkhoff, E.G.; Boon, A.C.; Nieuwkoop, N.J.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. A mutation in the HLA-B*2705-restricted NP383-391 epitope affects the human influenza A virus-specific cytotoxic T-lymphocyte response in vitro. J. Virol. 2004, 78, 5216–5222. [Google Scholar] [CrossRef] [PubMed]
- Berkhoff, E.G.; Geelhoed-Mieras, M.M.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Assessment of the extent of variation in influenza A virus cytotoxic T-lymphocyte epitopes by using virus-specific CD8+ T-cell clones. J. Gen. Virol. 2007, 88, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Price, G.E.; Ou, R.; Jiang, H.; Huang, L.; Moskophidis, D. Viral escape by selection of cytotoxic T cell-resistant variants in influenza A virus pneumonia. J. Exp. Med. 2000, 191, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Rimmelzwaan, G.F.; Boon, A.C.; Voeten, J.T.; Berkhoff, E.G.; Fouchier, R.A.; Osterhaus, A.D. Sequence variation in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. Virus Res. 2004, 103, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, S.A.; Gras, S.; Guillonneau, C.; La Gruta, N.L.; Thomas, P.G.; Purcell, A.W.; Rossjohn, J.; Doherty, P.C.; Turner, S.J.; Kedzierska, K. Protective efficacy of cross-reactive CD8+ T cells recognising mutant viral epitopes depends on peptide-MHC-I structural interactions and T cell activation threshold. PLoS Pathog. 2010, 6, e1001039. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.; Kedzierski, L.; Valkenburg, S.A.; Laurie, K.; Liu, Y.C.; Denholm, J.T.; Richards, M.J.; Rimmelzwaan, G.F.; Kelso, A.; Doherty, P.C.; et al. Cross-reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12599–12604. [Google Scholar] [CrossRef] [PubMed]
- Berkhoff, E.G.; Geelhoed-Mieras, M.M.; Verschuren, E.J.; van Baalen, C.A.; Gruters, R.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. The loss of immunodominant epitopes affects interferon-gamma production and lytic activity of the human influenza virus-specific cytotoxic T lymphocyte response in vitro. Clin. Exp. Immunol. 2007, 148, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Berkhoff, E.G.; de Wit, E.; Geelhoed-Mieras, M.M.; Boon, A.C.; Symons, J.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Functional constraints of influenza A virus epitopes limit escape from cytotoxic T lymphocytes. J. Virol. 2005, 79, 11239–11246. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; McCoy, W.; Schafer, F.; Bardet, W.; Buchli, R.; Fremont, D.H.; Hildebrand, W.H. T-cell tolerance for variability in an HLA class I-presented influenza A virus epitope. J. Virol. 2009, 83, 9206–9214. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, I.; Larionova, N.; Rudenko, L. Live Attenuated Reassortant Vaccines Based on A/Leningrad/134/17/57 Master Donor Virus Against H5 Avian Influenza. Open Microbiol. J. 2017, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Leitman, E.M.; Thobakgale, C.F.; Adland, E.; Ansari, M.A.; Raghwani, J.; Prendergast, A.J.; Tudor-Williams, G.; Kiepiela, P.; Hemelaar, J.; Brener, J.; et al. Role of HIV-specific CD8+ T cells in pediatric HIV cure strategies after widespread early viral escape. J. Exp. Med. 2017, 214, 3239–3261. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Oldstone, M.B.; Palese, P. Protective immunity and susceptibility to infectious diseases: Lessons from the 1918 influenza pandemic. Nat. Immunol. 2007, 8, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Recommendations on the Composition of Influenza Virus Vaccines. Available online: http://www.who.int/influenza/Vaccines/virus/recommendations/en/ (accessed on 20 November 2017).
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza Vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Giarola-Silva, S.; Coelho-Dos-Reis, J.G.A.; Mourao, M.M.; Campi-Azevedo, A.C.; Nakagaki Silva, E.E.; Luiza-Silva, M.; Martins, M.A.; Silveira-Cassette, A.C.O.; Batista, M.A.; Peruhype-Magalhaes, V.; et al. Distinct patterns of cellular immune response elicited by influenza non-adjuvanted and AS03-adjuvanted monovalent H1N1(pdm09) Vaccine. Antivir. Res. 2017, 144, 70–82. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Rappuoli, R.; De Gregorio, E.; Tsai, T.; Del Giudice, G. MF59 adjuvant: The best insurance against influenza strain diversity. Expert Rev Vaccines 2011, 10, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Couch, R.B.; Bayas, J.M.; Caso, C.; Mbawuike, I.N.; Lopez, C.N.; Claeys, C.; El Idrissi, M.; Herve, C.; Laupeze, B.; Oostvogels, L.; et al. Superior antigen-specific CD4+ T-cell response with AS03-adjuvantation of a trivalent influenza Vaccine in a randomised trial of adults aged 65 and older. BMC Infect. Dis. 2014, 14, 425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; McElhaney, J.E.; Walrond, L.; Cyr, T.D.; Merani, S.; Kollmann, T.R.; Halperin, S.A.; Scheifele, D.W. Cellular immune responses of older adults to four influenza Vaccines: Results of a randomized, controlled comparison. Hum. Vaccines Immunother. 2017, 13, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. Strategies to induce broadly protective antibody responses to viral glycoproteins. Expert Rev. Vaccines 2017, 16, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Suguitan, A.L., Jr.; Pinna, D.; Silacci, C.; Fernandez-Rodriguez, B.M.; Vanzetta, F.; Santos, C.; Luke, C.J.; Torres-Velez, F.J.; Temperton, N.J.; et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza Vaccine. J. Clin. Investig. 2010, 120, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.J.; McCaw, J.M.; Brown, L.; Jackson, D.; Kedzierska, K.; McVernon, J. Prior population immunity reduces the expected impact of CTL-inducing Vaccines for pandemic influenza control. PLoS ONE 2015, 10, e0120138. [Google Scholar] [CrossRef] [PubMed]
- Zacour, M.; Ward, B.J.; Brewer, A.; Tang, P.; Boivin, G.; Li, Y.; Warhuus, M.; McNeil, S.A.; LeBlanc, J.J.; Hatchette, T.F.; et al. Standardization of Hemagglutination Inhibition Assay for Influenza Serology Allows for High Reproducibility between Laboratories. Clin. Vaccines Immunol. 2016, 23, 236–342. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, S.; D’Alessio, F.; Houard, S.; Remarque, E.J.; Stockhofe, N.; Engelhardt, O.G. Workshop report: Immunoassay standardisation for “universal” influenza Vaccines. Influenza Other Respir. Viruses 2017, 11, 194–201. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Vaccine Requirement | Hurdles | Solutions |
|---|---|---|
| Induce protective T cell responses | What are the correlates of protection for T cells?
|
|
| Consensus on an acceptable level of protection for T cell vaccines |
| |
| Provide universal influenza immunity | Targeting heterologous T cell responses that induce broad protection against diverse influenza virus strains and subtypes. |
|
| Providing population-wide coverage across diverse HLA profiles and ethnicities. |
| |
| Circumventing virus escape of T cell immunity. |
| |
| Establish local immunity at the site of infection | Seeding durable, effective Trm memory populations in the lung and upper respiratory tract. |
|
| Synergize multiple immune mechanisms | Combining long-lasting broadly-reactive T and B cell immunity. |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemens, E.B.; Van de Sandt, C.; Wong, S.S.; Wakim, L.M.; Valkenburg, S.A. Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine. Vaccines 2018, 6, 18. https://doi.org/10.3390/vaccines6020018
Clemens EB, Van de Sandt C, Wong SS, Wakim LM, Valkenburg SA. Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine. Vaccines. 2018; 6(2):18. https://doi.org/10.3390/vaccines6020018
Chicago/Turabian StyleClemens, E. Bridie, Carolien Van de Sandt, Sook San Wong, Linda M. Wakim, and Sophie A. Valkenburg. 2018. "Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine" Vaccines 6, no. 2: 18. https://doi.org/10.3390/vaccines6020018
APA StyleClemens, E. B., Van de Sandt, C., Wong, S. S., Wakim, L. M., & Valkenburg, S. A. (2018). Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine. Vaccines, 6(2), 18. https://doi.org/10.3390/vaccines6020018