The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Measurement of TEER
2.3. Viruses and Infections
2.4. RNA Isolation and qRT-PCR
2.5. Plaque Assays
2.6. MicroRNA Analysis
2.7. High Content Microscopy
2.8. Statistical Analysis
3. Results
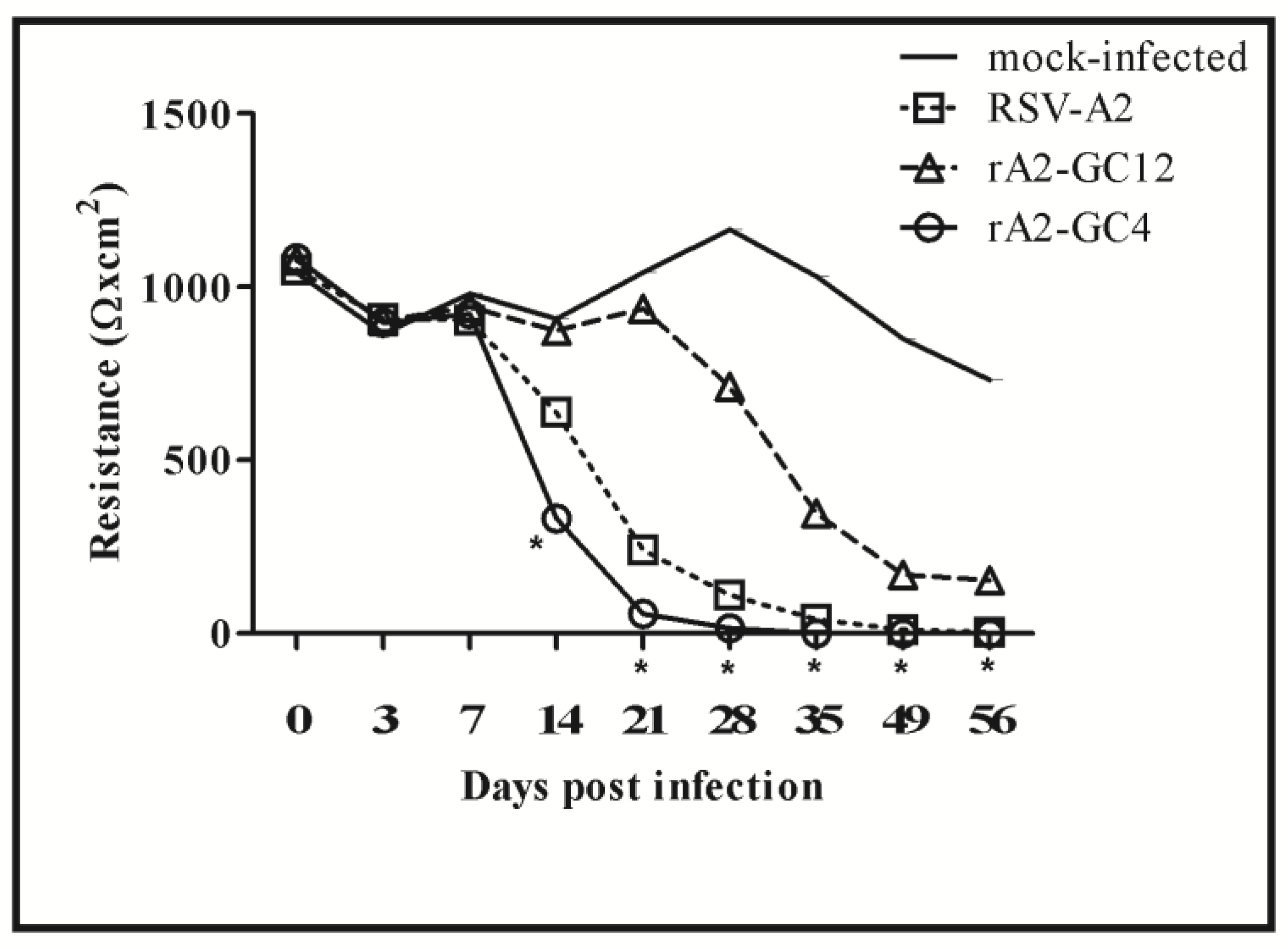
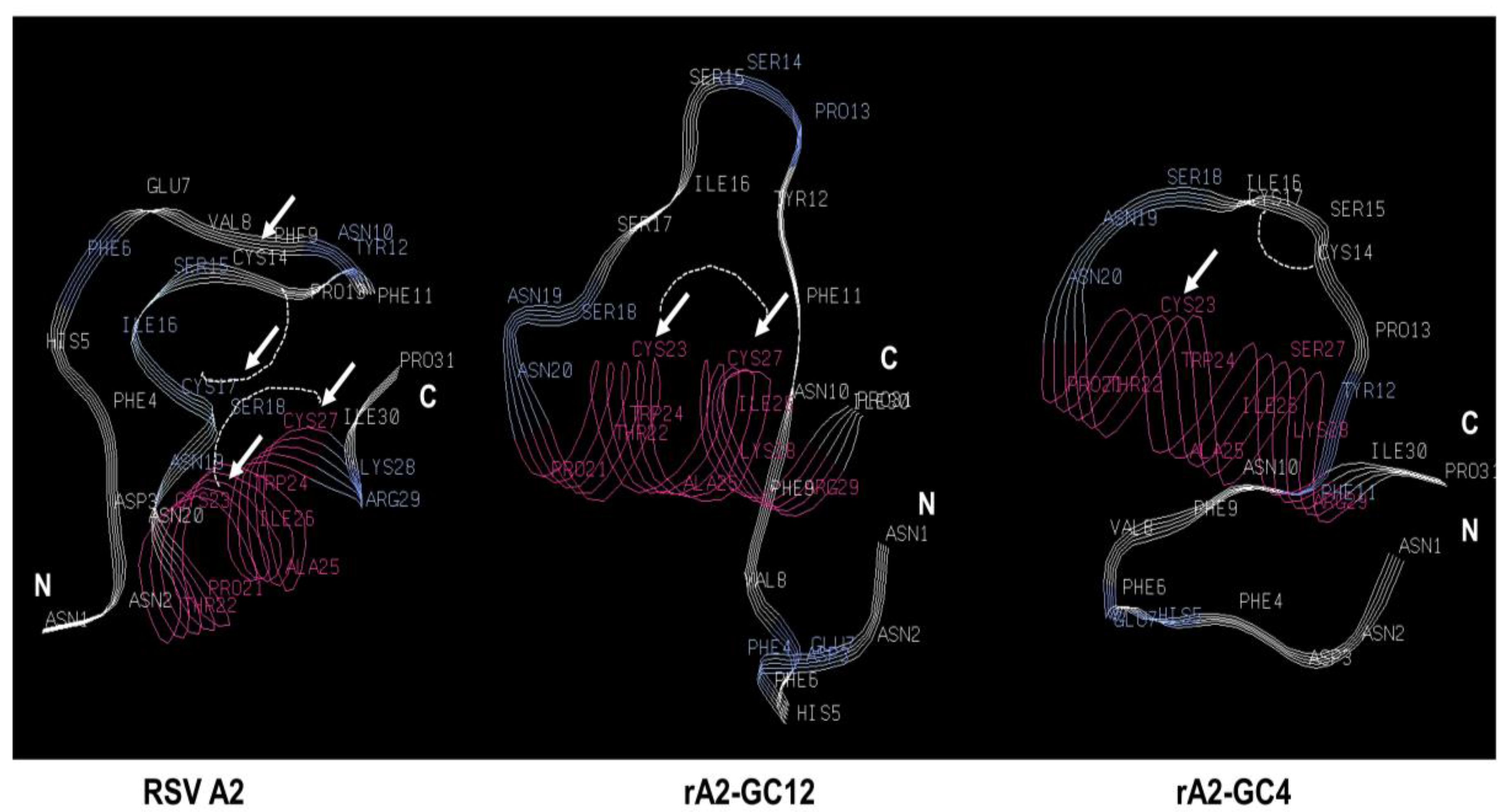
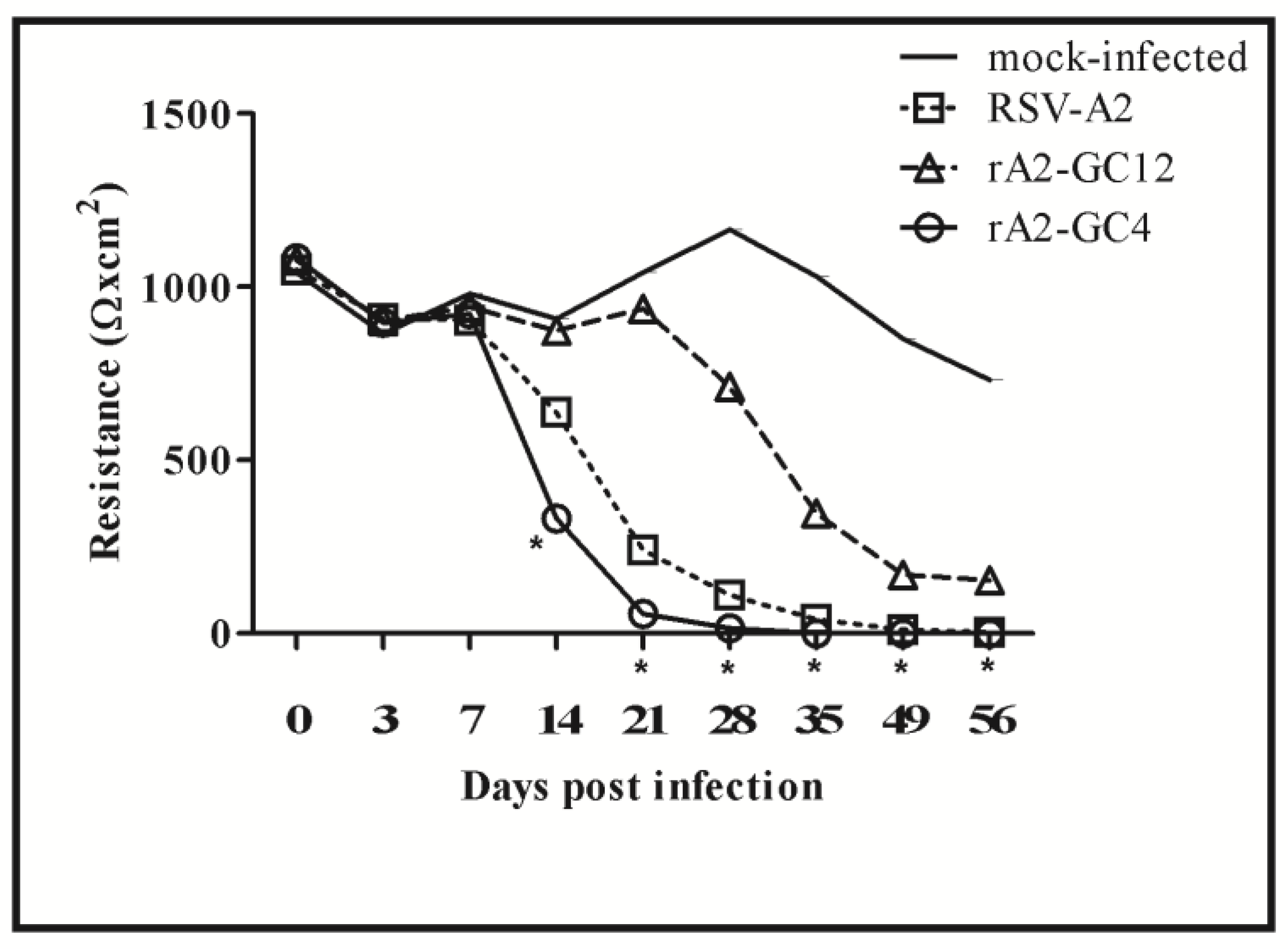
3.1. RSV G Protein CCR Modifies Polarization of Calu-3 Cells
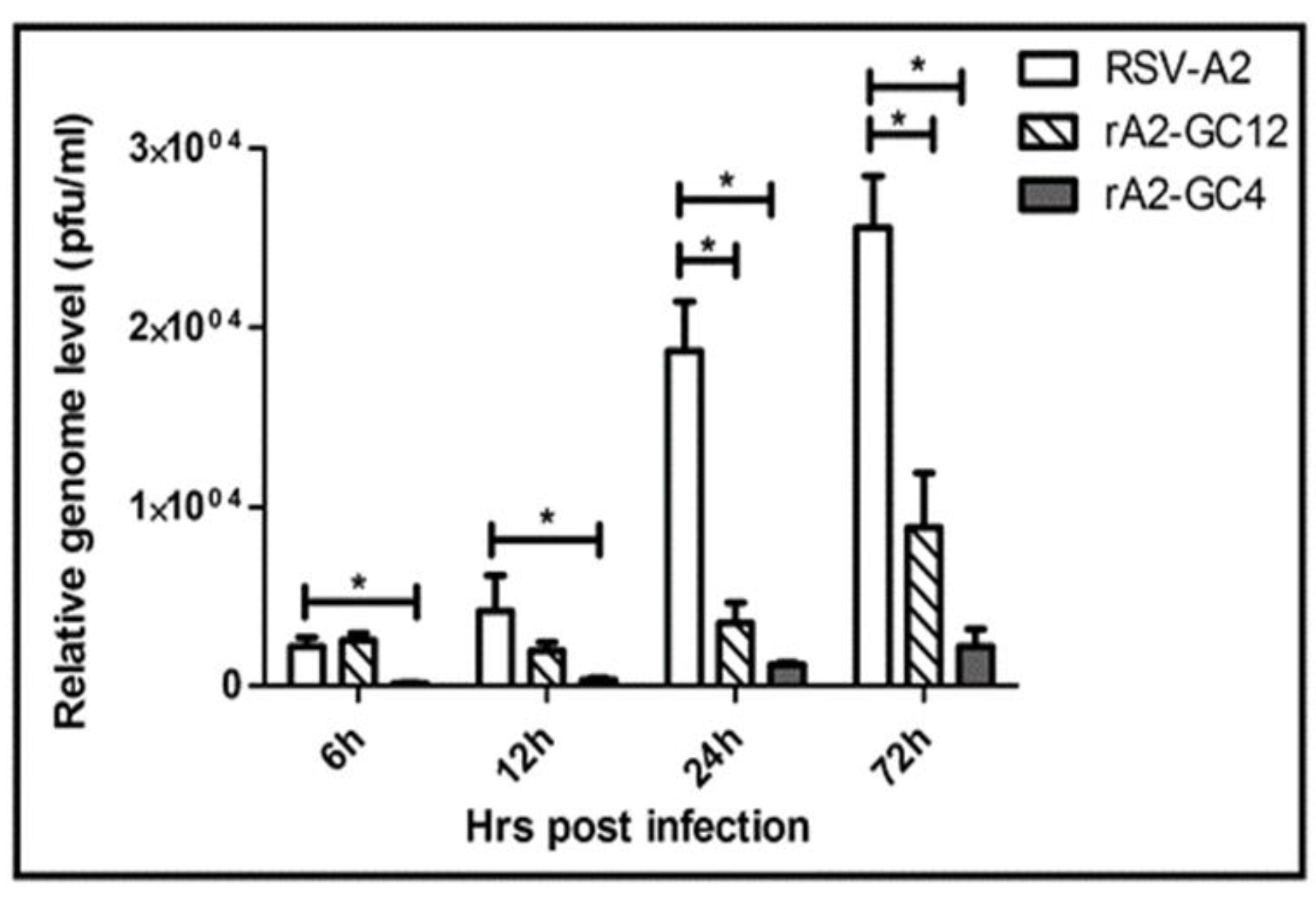
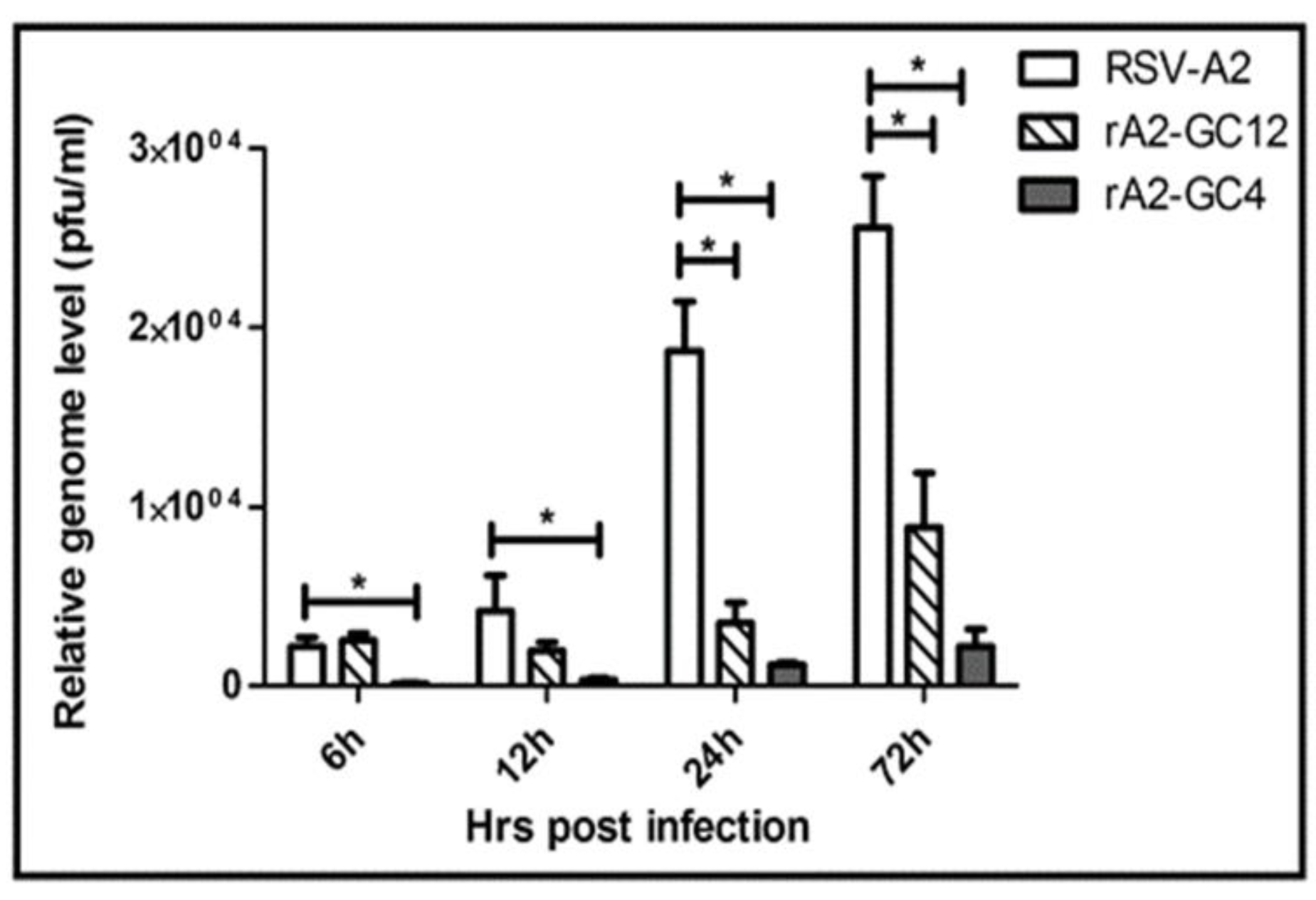
3.2. RSV G Protein CCR Modulates RSV Replication
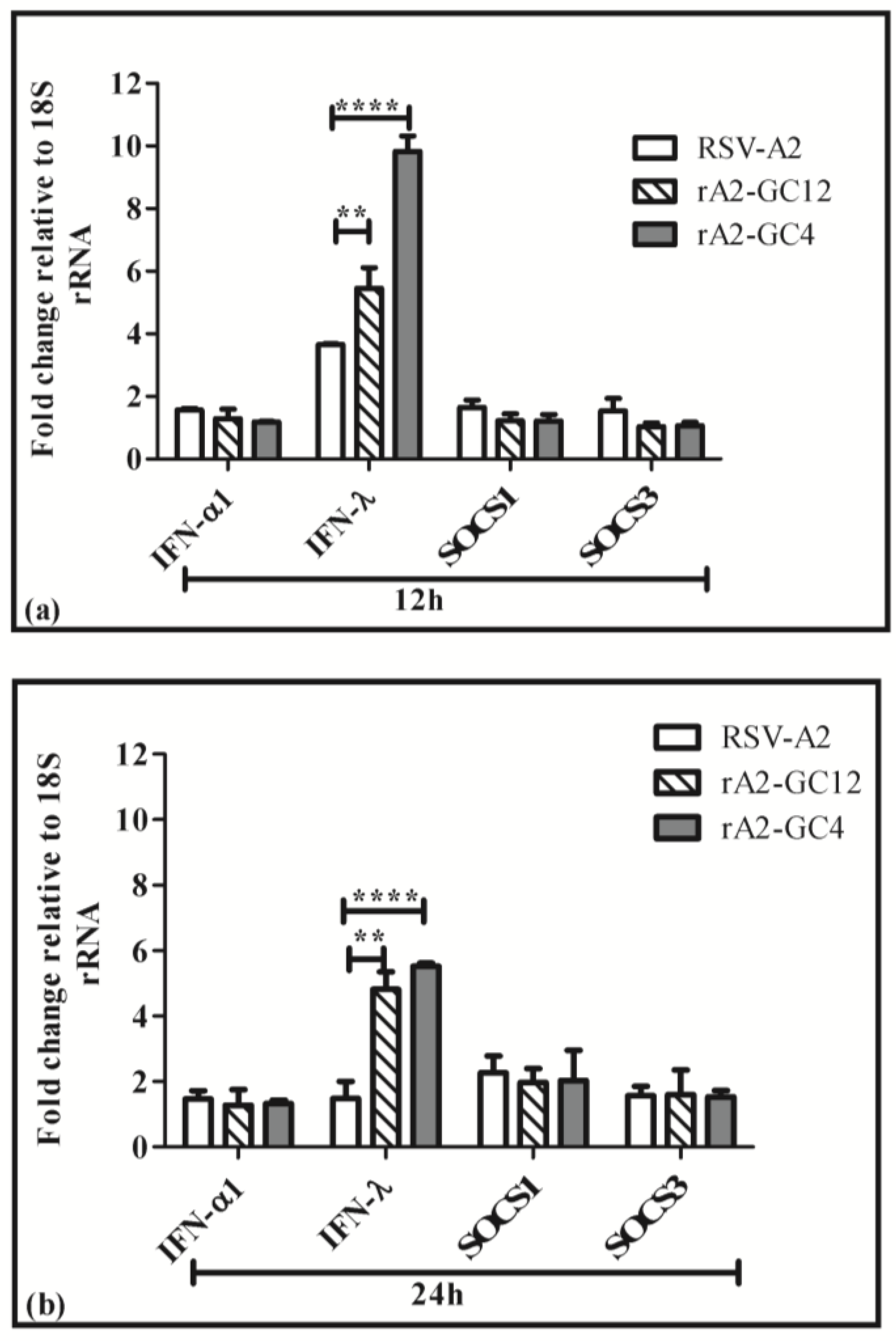
3.3. G Protein CCR Influences the Cell Cytokine and Chemokine Response to RSV Infection
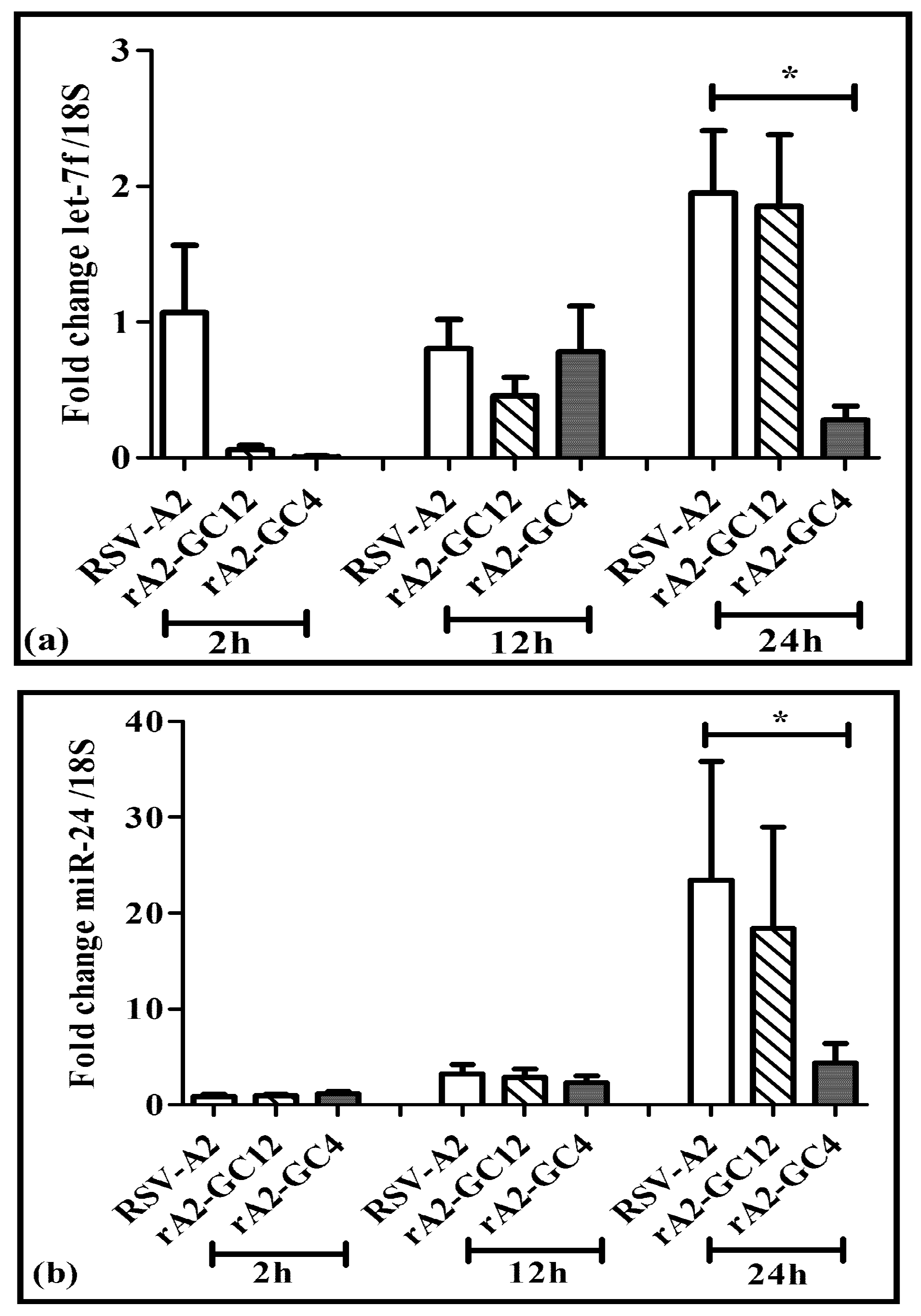
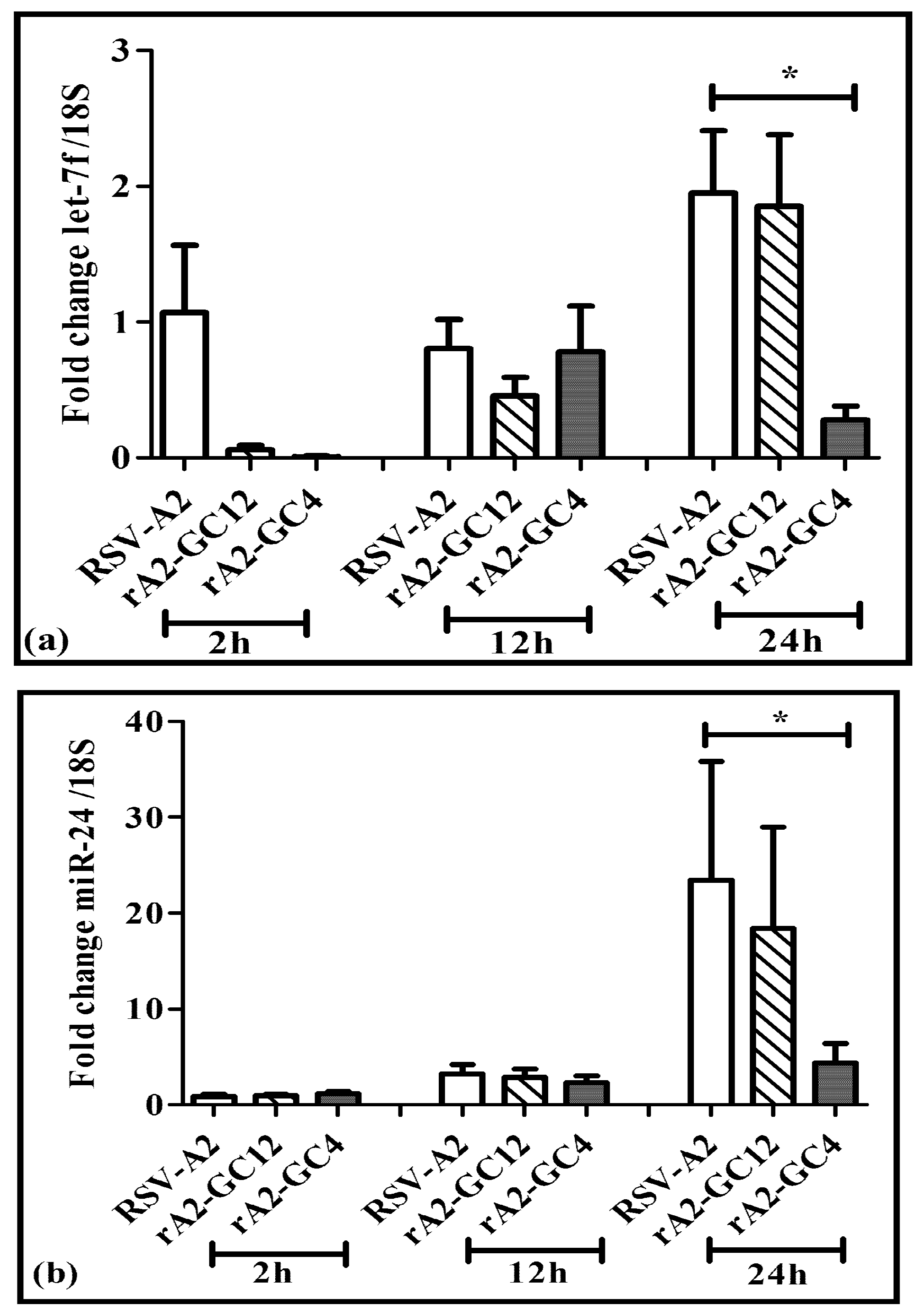
3.4. Perturbing the CX3C Motif Deregulates Host miRNA Responses
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shay, D.K.; Holman, R.C.; Newman, R.D.; Liu, L.L.; Stout, J.W.; Anderson, L.J. Bronchiolitis-associated hospitalizations among us children, 1980–1996. JAMA 1999, 282, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Stockman, L.J.; Curns, A.T.; Anderson, L.J.; Fischer-Langley, G. Respiratory syncytial virus-associated hospitalizations among infants and young children in the united states, 1997–2006. Pediatr. Infect. Dis. J. 2012, 31, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Thompson, W.W.; Viboud, C.G.; Ringholz, C.M.; Cheng, P.Y.; Steiner, C.; Abedi, G.R.; Anderson, L.J.; Brammer, L.; Shay, D.K. Hospitalizations associated with influenza and respiratory syncytial virus in the united states, 1993–2008. Clin. Infect. Dis. 2012, 54, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Respiratory syncytial virus activity—United States, July 2007–December 2008. MMWR Morb. Mortal. Wkly. Rep. 2008, 57, 1355–1358. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). Respiratory syncytial virus activity—United States, July 2011–January 2013. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 141–144. [Google Scholar]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Chin, J.; Magoffin, R.L.; Shearer, L.A.; Schieble, J.H.; Lennette, E.H. Field evaluation of a respiratory syncytial virus vaccine and a trivalent parainfluenza virus vaccine in a pediatric population. Am. J. Epidemiol. 1969, 89, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Weibel, R.E.; Stokes, J., Jr.; Leagus, M.B.; Mascoli, C.C.; Hilleman, M.R. Respiratory virus vaccines. V. Field evaluation for efficacy of heptavalent vaccine. Am. Rev. Respir. Dis. 1966, 94, 362–379. [Google Scholar] [PubMed]
- Lindell, D.M.; Morris, S.B.; White, M.P.; Kallal, L.E.; Lundy, P.K.; Hamouda, T.; Baker, J.R., Jr.; Lukacs, N.W. A novel inactivated intranasal respiratory syncytial virus vaccine promotes viral clearance without th2 associated vaccine-enhanced disease. PLoS ONE 2011, 6, e21823. [Google Scholar] [CrossRef] [PubMed]
- Connors, M.; Collins, P.L.; Firestone, C.Y.; Murphy, B.R. Respiratory syncytial virus (rsv) f, g, m2 (22k), and n proteins each induce resistance to rsv challenge, but resistance induced by m2 and n proteins is relatively short-lived. J. Virol. 1991, 65, 1634–1637. [Google Scholar] [PubMed]
- Stott, E.J.; Taylor, G.; Ball, L.A.; Anderson, K.; Young, K.K.; King, A.M.; Wertz, G.W. Immune and histopathological responses in animals vaccinated with recombinant vaccinia viruses that express individual genes of human respiratory syncytial virus. J. Virol. 1987, 61, 3855–3861. [Google Scholar] [PubMed]
- Anderson, L.J.; Dormitzer, P.R.; Nokes, D.J.; Rappuoli, R.; Roca, A.; Graham, B.S. Strategic priorities for respiratory syncytial virus (rsv) vaccine development. Vaccine 2013, 31 (Suppl. 2), B209–B215. [Google Scholar] [CrossRef] [PubMed]
- Castilow, E.M.; Varga, S.M. Overcoming t cell-mediated immunopathology to achieve safe rsv vaccination. Future Virol. 2008, 3, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Nokes, J.D.; Cane, P.A. New strategies for control of respiratory syncytial virus infection. Curr. Opin. Infect. Dis. 2008, 21, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.R.; Varga, S.M. Pulmonary immunity and immunopathology: Lessons from respiratory syncytial virus. Expert Rev. Vaccines 2008, 7, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Power, U.F. Respiratory syncytial virus (rsv) vaccines--two steps back for one leap forward. J. Clin. Virol. 2008, 41, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, D.A.; Baradaran, K.; McIntosh, K.; Patterson, J.L. Appearance of a soluble form of the g protein of respiratory syncytial virus in fluids of infected cells. J. Gen. Virol. 1987, 68 Pt 6, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.R.; Lichtenstein, D.; Ball, L.A.; Wertz, G.W. The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein g are synthesized from alternative initiation codons. J. Virol. 1994, 68, 4538–4546. [Google Scholar] [PubMed]
- Gorman, J.J.; Ferguson, B.L.; Speelman, D.; Mills, J. Determination of the disulfide bond arrangement of human respiratory syncytial virus attachment (g) protein by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Protein Sci. 1997, 6, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Tripp, R.A.; Jones, L.P.; Haynes, L.M.; Zheng, H.; Murphy, P.M.; Anderson, L.J. Cx3c chemokine mimicry by respiratory syncytial virus g glycoprotein. Nat. Immunol. 2001, 2, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M.E. Respiratory syncytial virus uses cx3cr1 as a receptor on primary human airway epithelial cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef] [PubMed]
- Chirkova, T.; Lin, S.; Oomens, A.G.; Gaston, K.A.; Boyoglu-Barnum, S.; Meng, J.; Stobart, C.C.; Cotton, C.U.; Hartert, T.V.; Moore, M.L.; et al. Cx3cr1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J. Gen. Virol. 2015, 96, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Chirkova, T.; Boyoglu-Barnum, S.; Gaston, K.A.; Malik, F.M.; Trau, S.P.; Oomens, A.G.; Anderson, L.J. Respiratory syncytial virus g protein cx3c motif impairs human airway epithelial and immune cell responses. J. Virol. 2013, 87, 13466–13479. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.; Alvarez, R.; Jones, L.P.; Henderson, C.; Anderson, L.J.; Tripp, R.A. Respiratory syncytial virus g protein and g protein cx3c motif adversely affect cx3cr1+ t cell responses. J. Immunol. 2006, 176, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Tripp, R.A.; Dakhama, A.; Jones, L.P.; Barskey, A.; Gelfand, E.W.; Anderson, L.J. The g glycoprotein of respiratory syncytial virus depresses respiratory rates through the cx3c motif and substance p. J. Virol. 2003, 77, 6580–6584. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M.; Caidi, H.; Radu, G.U.; Miao, C.; Harcourt, J.L.; Tripp, R.A.; Anderson, L.J. Therapeutic monoclonal antibody treatment targeting respiratory syncytial virus (rsv) g protein mediates viral clearance and reduces the pathogenesis of rsv infection in balb/c mice. J. Infect. Dis. 2009, 200, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Miao, C.; Blanchard, E.G.; Caidi, H.; Radu, G.U.; Harcourt, J.L.; Haynes, L.M. Effect of chemokine receptor cx3cr1 deficiency in a murine model of respiratory syncytial virus infection. Comp. Med. 2012, 62, 14–20. [Google Scholar] [PubMed]
- Boyoglu-Barnum, S.; Todd, S.O.; Chirkova, T.; Barnum, T.R.; Gaston, K.A.; Haynes, L.M.; Tripp, R.A.; Moore, M.L.; Anderson, L.J. An anti-g protein monoclonal antibody treats rsv disease more effectively than an anti-f monoclonal antibody in balb/c mice. Virology 2015, 483, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.I.; Piepenhagen, P.A.; Kishko, M.; DiNapoli, J.M.; Groppo, R.P.; Zhang, L.; Almond, J.; Kleanthous, H.; Delagrave, S.; Parrington, M. Cx3cr1 is expressed in differentiated human ciliated airway cells and co-localizes with respiratory syncytial virus on cilia in a g protein-dependent manner. PLoS ONE 2015, 10, e0130517. [Google Scholar] [CrossRef] [PubMed]
- Boyoglu-Barnum, S.; Gaston, K.A.; Todd, S.O.; Boyoglu, C.; Chirkova, T.; Barnum, T.R.; Jorquera, P.; Haynes, L.M.; Tripp, R.A.; Moore, M.L.; et al. A respiratory syncytial virus (rsv) anti-g protein f(ab')2 monoclonal antibody suppresses mucous production and breathing effort in rsv ra2-line19f-infected balb/c mice. J. Virol. 2013, 87, 10955–10967. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.L.; Karron, R.A.; Tripp, R.A. Anti-g protein antibody responses to respiratory syncytial virus infection or vaccination are associated with inhibition of g protein cx3c-cx3cr1 binding and leukocyte chemotaxis. J. Infect. Dis. 2004, 190, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Choi, Y.; Haynes, L.M.; Harcourt, J.L.; Anderson, L.J.; Jones, L.P.; Tripp, R.A. Vaccination to induce antibodies blocking the cx3c-cx3cr1 interaction of respiratory syncytial virus g protein reduces pulmonary inflammation and virus replication in mice. J. Virol. 2010, 84, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Grantham, M.L.; Wu, W.H.; Lalime, E.N.; Lorenzo, M.E.; Klein, S.L.; Pekosz, A. Palmitoylation of the influenza a virus m2 protein is not required for virus replication in vitro but contributes to virus virulence. J. Virol. 2009, 83, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.C.; Parsons, K.; Barr, I.; Lowther, S.; Middleton, D.; Hansbro, P.M.; Wark, P.A. Critical role of constitutive type i interferon response in bronchial epithelial cell to influenza infection. PLoS ONE 2012, 7, e32947. [Google Scholar] [CrossRef] [PubMed]
- Saedisomeolia, A.; Wood, L.G.; Garg, M.L.; Gibson, P.G.; Wark, P.A. Lycopene enrichment of cultured airway epithelial cells decreases the inflammation induced by rhinovirus infection and lipopolysaccharide. J. Nutr. Biochem. 2009, 20, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Hill, T.; Li, K.; Peters, C.J.; Tseng, C.T. Severe acute respiratory syndrome (sars) coronavirus-induced lung epithelial cytokines exacerbate sars pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 2009, 83, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Hill, T.E.; Yoshikawa, N.; Popov, V.L.; Galindo, C.L.; Garner, H.R.; Peters, C.J.; Tseng, C.T. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS ONE 2010, 5, e8729. [Google Scholar] [CrossRef] [PubMed]
- Grainger, C.I.; Greenwell, L.L.; Lockley, D.J.; Martin, G.P.; Forbes, B. Culture of calu-3 cells at the air interface provides a representative model of the airway epithelial barrier. Pharm. Res. 2006, 23, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.L.; Caidi, H.; Anderson, L.J.; Haynes, L.M. Evaluation of the calu-3 cell line as a model of in vitro respiratory syncytial virus infection. J. Virol. Methods 2011, 174, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Couch, R.B.; Englund, J.A.; Whimbey, E. Respiratory viral infections in immunocompetent and immunocompromised persons. Am. J. Med. 1997, 102, 2–9, discussion 25–26. [Google Scholar] [CrossRef]
- Dakhama, A.; Vitalis, T.Z.; Hegele, R.G. Persistence of respiratory syncytial virus (rsv) infection and development of rsv-specific igg1 response in a guinea-pig model of acute bronchiolitis. Eur. Respir. J. 1997, 10, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.G.; Hayashi, S.; Bramley, A.M.; Hogg, J.C. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest 1994, 105, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Isaia, G.; Teodosiu, O.; Popescu, G.; Athanasiu, P.; Sternberg, I.; Dumitriu, Z. Persistence of viruses in the nasopharynx of apparently healthy children aged 0–5 years. Results of investigations performed in 1982–83. Virologie 1985, 36, 175–179. [Google Scholar] [PubMed]
- Mejias, A.; Chavez-Bueno, S.; Gomez, A.M.; Somers, C.; Estripeaut, D.; Torres, J.P.; Jafri, H.S.; Ramilo, O. Respiratory syncytial virus persistence: Evidence in the mouse model. Pediatr. Infect. Dis. J. 2008, 27, S60–S62. [Google Scholar] [CrossRef] [PubMed]
- Sikkel, M.B.; Quint, J.K.; Mallia, P.; Wedzicha, J.A.; Johnston, S.L. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatr. Infect. Dis. J. 2008, 27, S63–S70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peeples, M.E.; Boucher, R.C.; Collins, P.L.; Pickles, R.J. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 2002, 76, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.L.; Haynes, L.M. Establishing a liquid-covered culture of polarized human airway epithelial calu-3 cells to study host cell response to respiratory pathogens in vitro. J. Vis. Exp. JoVE 2013. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.N.; Whitehead, S.S.; Collins, P.L. Contribution of the respiratory syncytial virus g glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology 2001, 289, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.S.; Juhasz, K.; Firestone, C.Y.; Collins, P.L.; Murphy, B.R. Recombinant respiratory syncytial virus (rsv) bearing a set of mutations from cold-passaged rsv is attenuated in chimpanzees. J. Virol. 1998, 72, 4467–4471. [Google Scholar] [PubMed]
- Kim, Y.K.; Choi, E.H.; Lee, H.J. Genetic variability of the fusion protein and circulation patterns of genotypes of the respiratory syncytial virus. J. Med. Virol. 2007, 79, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Caidi, H.; Harcourt, J.L.; Tripp, R.A.; Anderson, L.J.; Haynes, L.M. Combination therapy using monoclonal antibodies against respiratory syncytial virus (rsv) g glycoprotein protects from rsv disease in balb/c mice. PLoS ONE 2012, 7, e51485. [Google Scholar] [CrossRef] [PubMed]
- Sullender, W. Antigenic analysis of chimeric and truncated g proteins of respiratory syncytial virus. Virology 1995, 209, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G.; McCray, P.B., Jr.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, P.; Hegele, R.G. Rsv fusion: Time for a new model. Viruses 2013, 5, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained protein kinase d activation mediates respiratory syncytial virus-induced airway barrier disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tuffery, P. Pep-fold: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [PubMed]
- Oshansky, C.M.; Krunkosky, T.M.; Barber, J.; Jones, L.P.; Tripp, R.A. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type i interferon response to infection by a toll-like receptor pathway. Viral Immunol. 2009, 22, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Bakre, A.; Mitchell, P.; Coleman, J.K.; Jones, L.P.; Saavedra, G.; Teng, M.; Tompkins, S.M.; Tripp, R.A. Respiratory syncytial virus modifies micrornas regulating host genes that affect virus replication. J. Gen. Virol. 2012, 93, 2346–2356. [Google Scholar] [CrossRef] [PubMed]
- Melendi, G.A.; Bridget, D.; Monsalvo, A.C.; Laham, F.F.; Acosta, P.; Delgado, M.F.; Polack, F.P.; Irusta, P.M. Conserved cysteine residues within the attachment g glycoprotein of respiratory syncytial virus play a critical role in the enhancement of cytotoxic t-lymphocyte responses. Virus Genes 2011, 42, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Simard, C.; Nadon, F.; Seguin, C.; Thien, N.N.; Binz, H.; Basso, J.; Laliberte, J.F.; Trudel, M. Subgroup specific protection of mice from respiratory syncytial virus infection with peptides encompassing the amino acid region 174–187 from the g glycoprotein: The role of cysteinyl residues in protection. Vaccine 1997, 15, 423–432. [Google Scholar] [CrossRef]
- Haynes, L.M.; Jones, L.P.; Barskey, A.; Anderson, L.J.; Tripp, R.A. Enhanced disease and pulmonary eosinophilia associated with formalin-inactivated respiratory syncytial virus vaccination are linked to g glycoprotein cx3c-cx3cr1 interaction and expression of substance p. J. Virol. 2003, 77, 9831–9844. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Fu, Z.F.; Alvarez, R.; Henderson, C.; Tripp, R.A. Respiratory syncytial virus (rsv) infects neuronal cells and processes that innervate the lung by a process involving rsv g protein. J. Virol. 2006, 80, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-iii interferon, not type-i, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, C.; Pierangeli, A.; Fabiani, M.; Spano, L.; Nicolai, A.; Papoff, P.; Moretti, C.; Midulla, F.; Antonelli, G.; Scagnolari, C. Interferon lambda 1–3 expression in infants hospitalized for rsv or hrv associated bronchiolitis. J. Infect. 2014, 68, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Villenave, R.; Broadbent, L.; Douglas, I.; Lyons, J.D.; Coyle, P.V.; Teng, M.N.; Tripp, R.A.; Heaney, L.G.; Shields, M.D.; Power, U.F. Induction and antagonism of antiviral responses in respiratory syncytial virus-infected pediatric airway epithelium. J. Virol. 2015, 89, 12309–12318. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.; Dubey, R.; Saini, N. Cooperative and individualistic functions of the micrornas in the mir-23a~27a~24–2 cluster and its implication in human diseases. Mol. Cancer 2010, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Seike, M.; Soeno, C.; Mizutani, H.; Kitamura, K.; Minegishi, Y.; Noro, R.; Yoshimura, A.; Cai, L.; Gemma, A. Mir-23a regulates tgf-beta-induced epithelial-mesenchymal transition by targeting e-cadherin in lung cancer cells. Int. J. Oncol. 2012, 41, 869–875. [Google Scholar] [PubMed]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human mirnas and indications for an involvement of mirna in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Bakre, A.; Wu, W.; Hiscox, J.; Spann, K.; Teng, M.N.; Tripp, R.A. Human respiratory syncytial virus non-structural protein ns1 modifies mir-24 expression via transforming growth factor-beta. J. Gen. Virol. 2015, 96, 3179–3191. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors tlr4 and cd14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Shingai, M.; Azuma, M.; Ebihara, T.; Sasai, M.; Funami, K.; Ayata, M.; Ogura, H.; Tsutsumi, H.; Matsumoto, M.; Seya, T. Soluble g protein of respiratory syncytial virus inhibits toll-like receptor 3/4-mediated ifn-beta induction. Int. Immunol. 2008, 20, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Oshansky, C.M.; Barber, J.P.; Crabtree, J.; Tripp, R.A. Respiratory syncytial virus f and g proteins induce interleukin 1alpha, cc, and cxc chemokine responses by normal human bronchoepithelial cells. J. Infect. Dis. 2010, 201, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Barber, J.; Tripp, R.A. Respiratory syncytial virus (rsv) attachment and nonstructural proteins modify the type i interferon response associated with suppressor of cytokine signaling (socs) proteins and ifn-stimulated gene-15 (isg15). Virol. J. 2008, 5, 116. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.P.; de Groot, B.L.; Berendsen, H.J.; van Oirschot, J.T. Structural homology of the central conserved region of the attachment protein g of respiratory syncytial virus with the fourth subdomain of 55-kda tumor necrosis factor receptor. Virology 1998, 243, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.J.; Hayward, S.L.; Crowe, J.E., Jr. Respiratory syncytial virus regulates human micrornas by using mechanisms involving beta interferon and nf-kappab. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Murphy, B.R. New generation live vaccines against human respiratory syncytial virus designed by reverse genetics. Proc. Am. Thorac. Soc. 2005, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
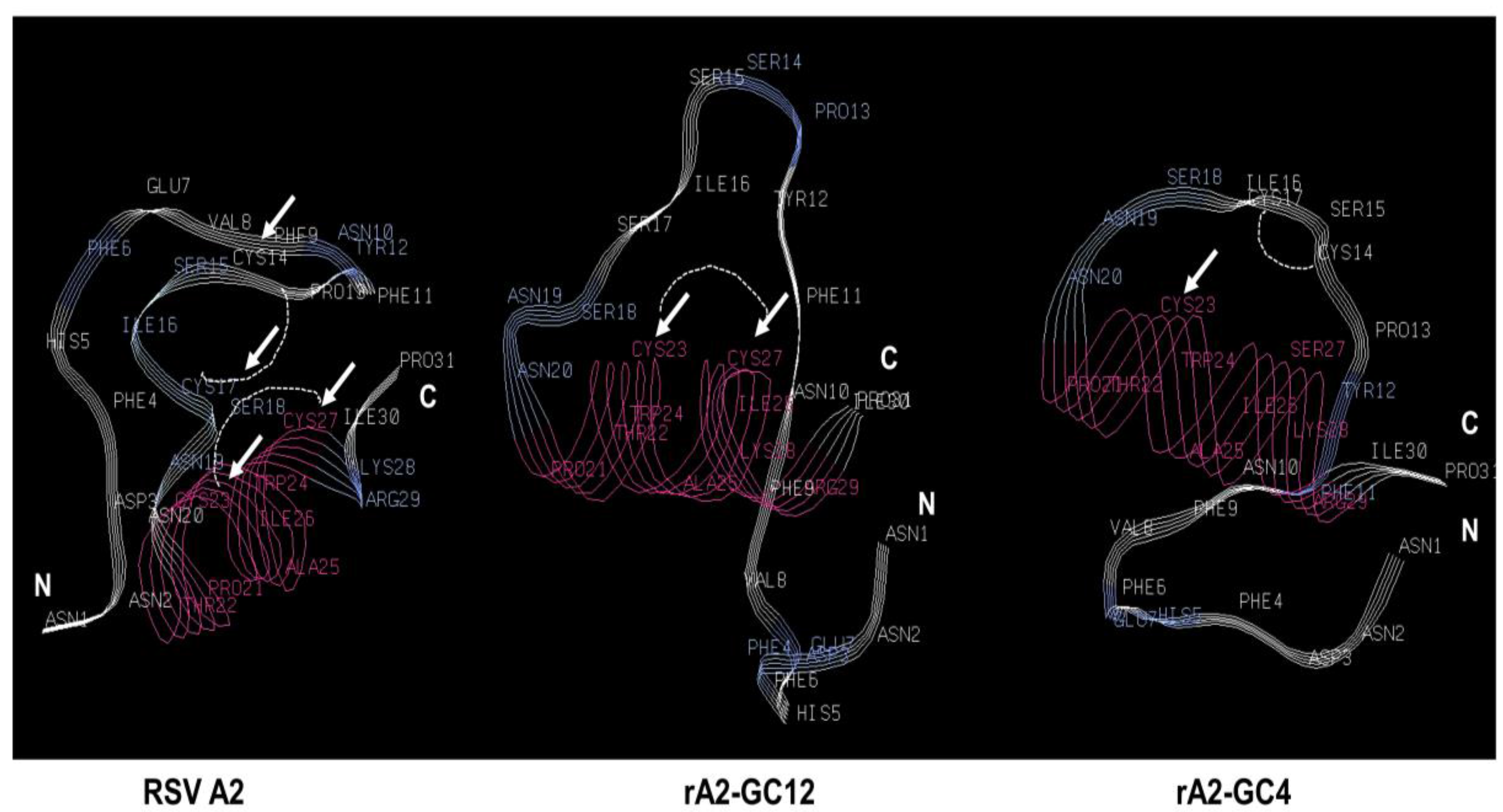{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Sequence |
|---|---|
| 173 176 182 186 | |
| RSV-A2 | NNDFHFEVFNFYPCSICSNNPTCWAICKRIP |
| rA2-GC12 (C173/176S) | NNDFHFEVFNFYPSSISSNNPTCWAICKRIP |
| rA2-GC4 (C186S) | NNDFHFEVFNFYPCSICSNNPTCWAISKRIP |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakre, A.A.; Harcourt, J.L.; Haynes, L.M.; Anderson, L.J.; Tripp, R.A. The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection. Vaccines 2017, 5, 16. https://doi.org/10.3390/vaccines5030016
Bakre AA, Harcourt JL, Haynes LM, Anderson LJ, Tripp RA. The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection. Vaccines. 2017; 5(3):16. https://doi.org/10.3390/vaccines5030016
Chicago/Turabian StyleBakre, Abhijeet A., Jennifer L. Harcourt, Lia M. Haynes, Larry J. Anderson, and Ralph A. Tripp. 2017. "The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection" Vaccines 5, no. 3: 16. https://doi.org/10.3390/vaccines5030016
APA StyleBakre, A. A., Harcourt, J. L., Haynes, L. M., Anderson, L. J., & Tripp, R. A. (2017). The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection. Vaccines, 5(3), 16. https://doi.org/10.3390/vaccines5030016