
The TRIMendous Role of TRIMs in Virus–Host Interactions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Ubiquitin
1.2. Tripartite Motif (TRIM) Proteins
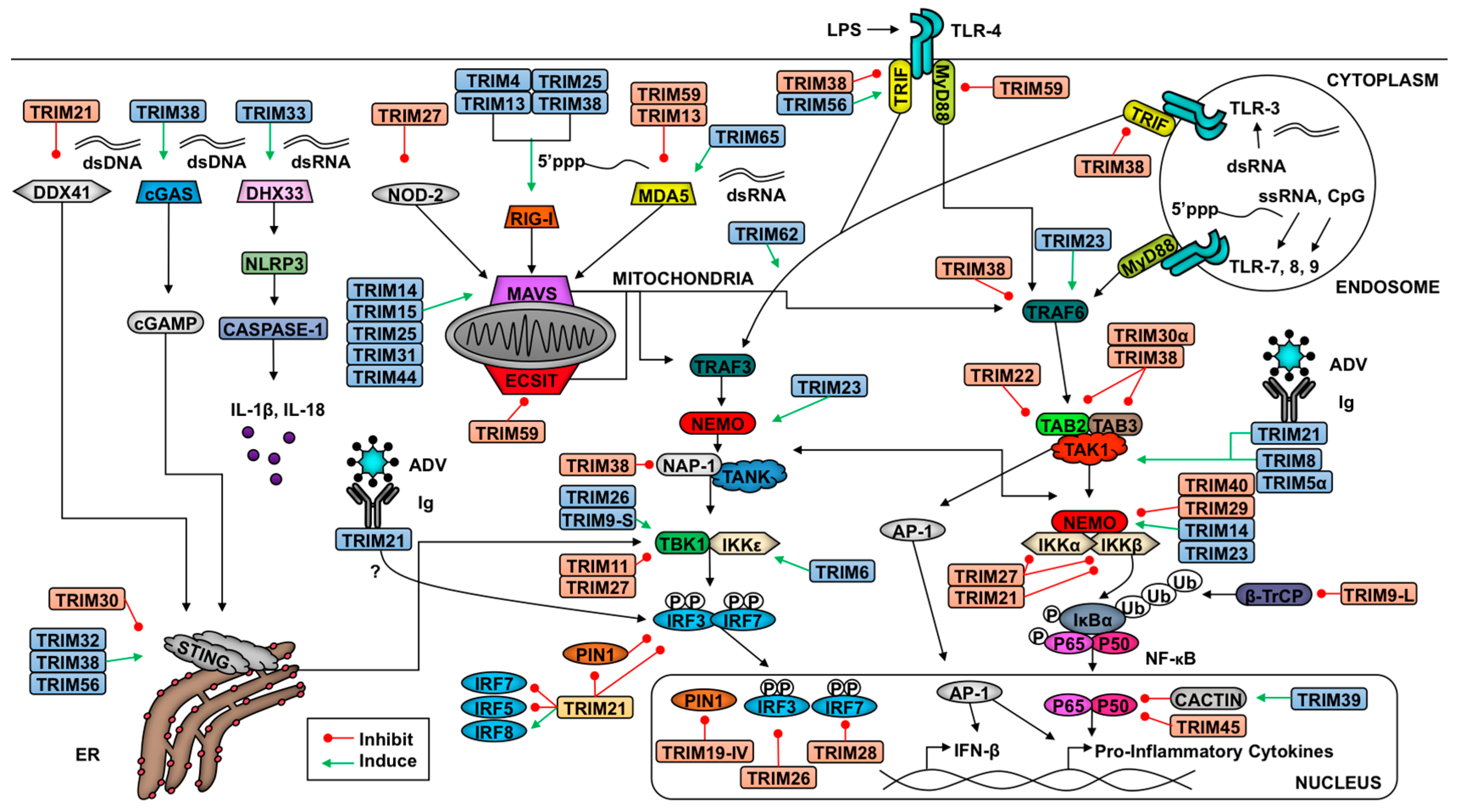
2. TRIM-Mediate Regulation of Antiviral Signaling
2.1. Introduction to Innate Antiviral Responses
2.2. TRIMs and the Retinoic Acid-Inducible Gene I (RIG-I)-like Receptor Pathway
2.3. TRIMs and STING Signaling
2.4. TRIMs and TLR Signaling
2.5. TRIMs and the Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptors (NLR) Pathway
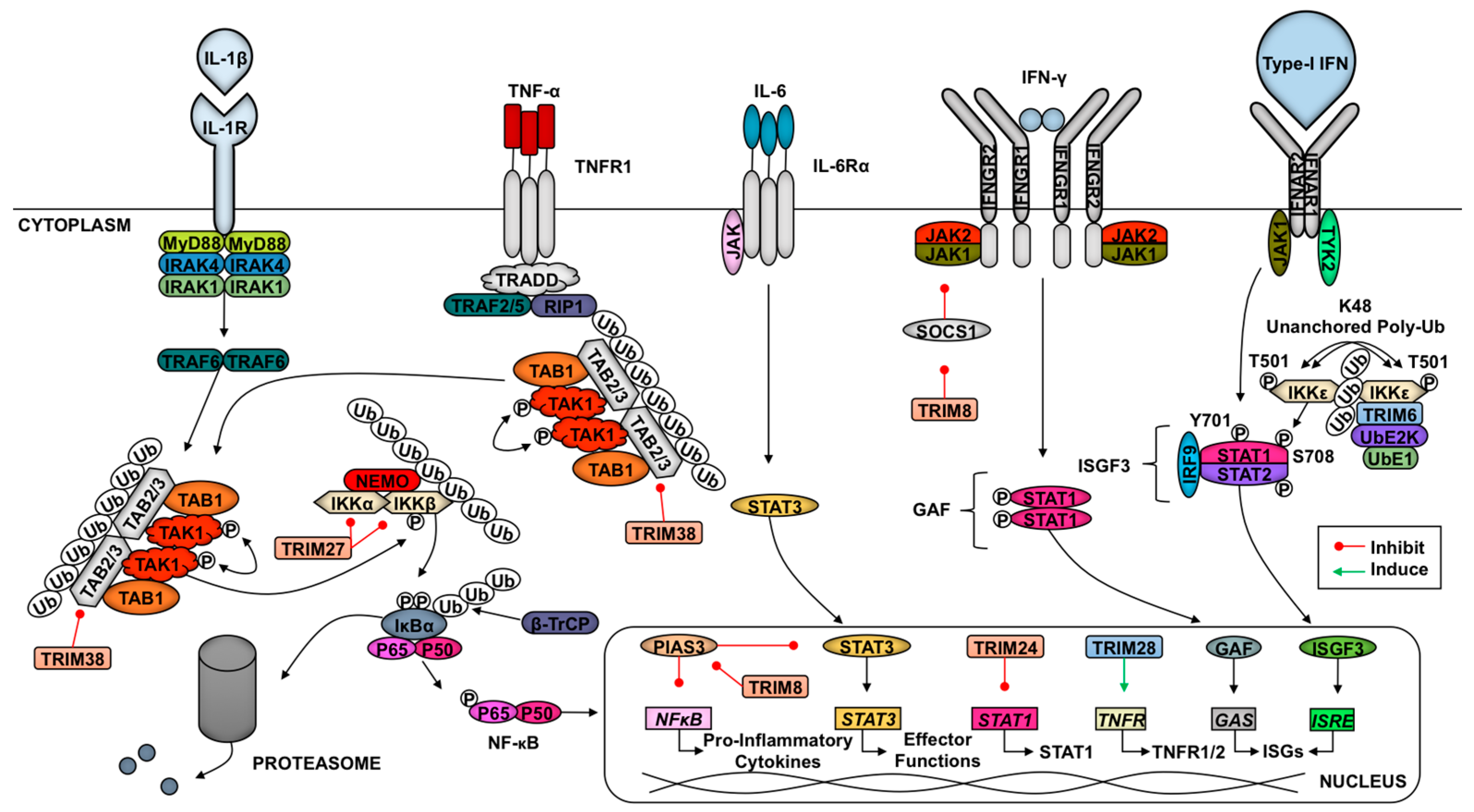
2.6. TRIMs and Cytokine Signaling
2.7. TRIMs and Adaptive Immunity
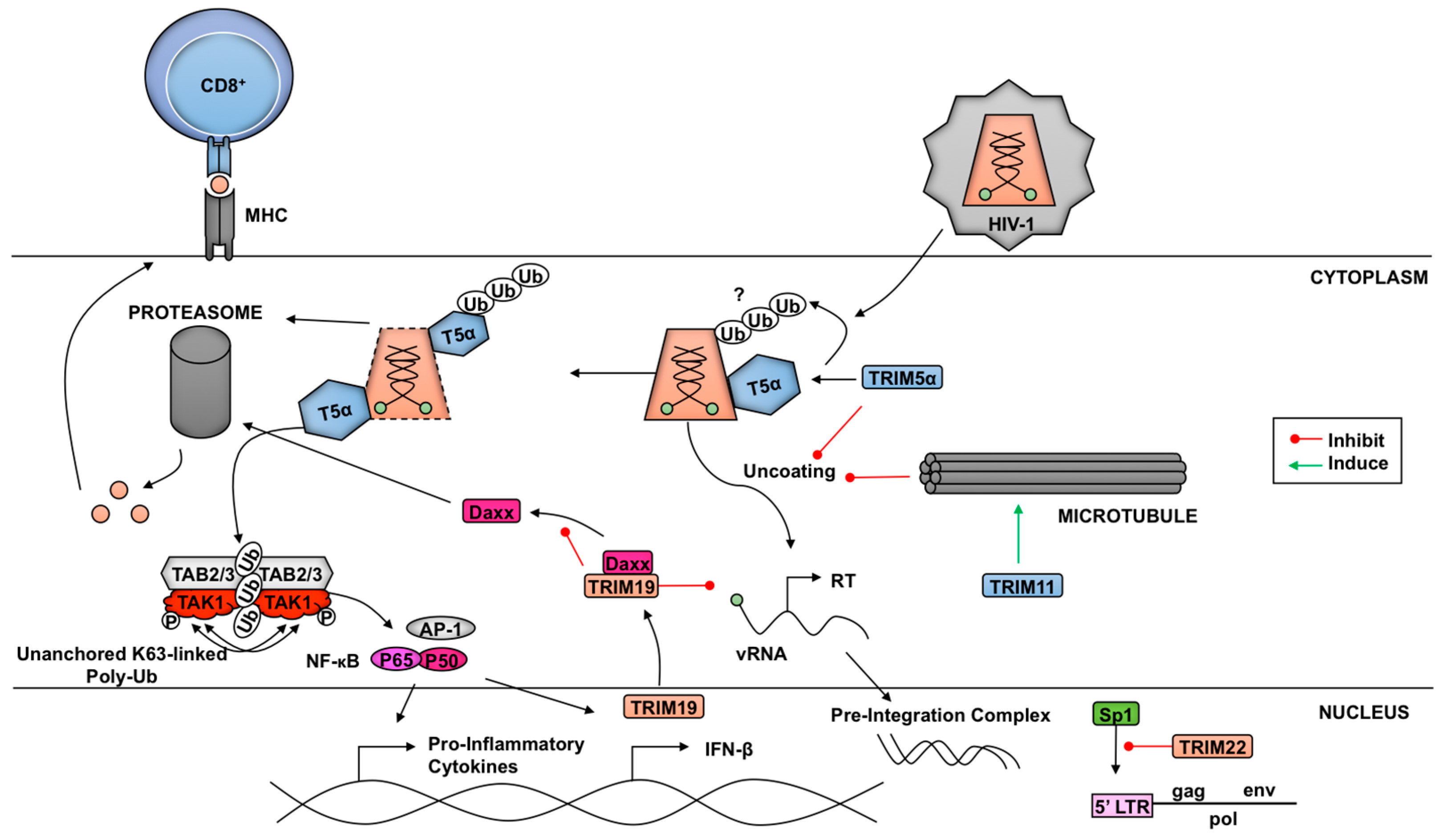
3. TRIM-Mediated Virus Inhibition
3.1. TRIMs and Retroviruses
3.1.1. TRIM5α and Retroviruses
3.1.2. Other TRIMs and Retroviruses
3.2. TRIM21 and Antibody-Dependent Intracellular Neutralization of Viruses
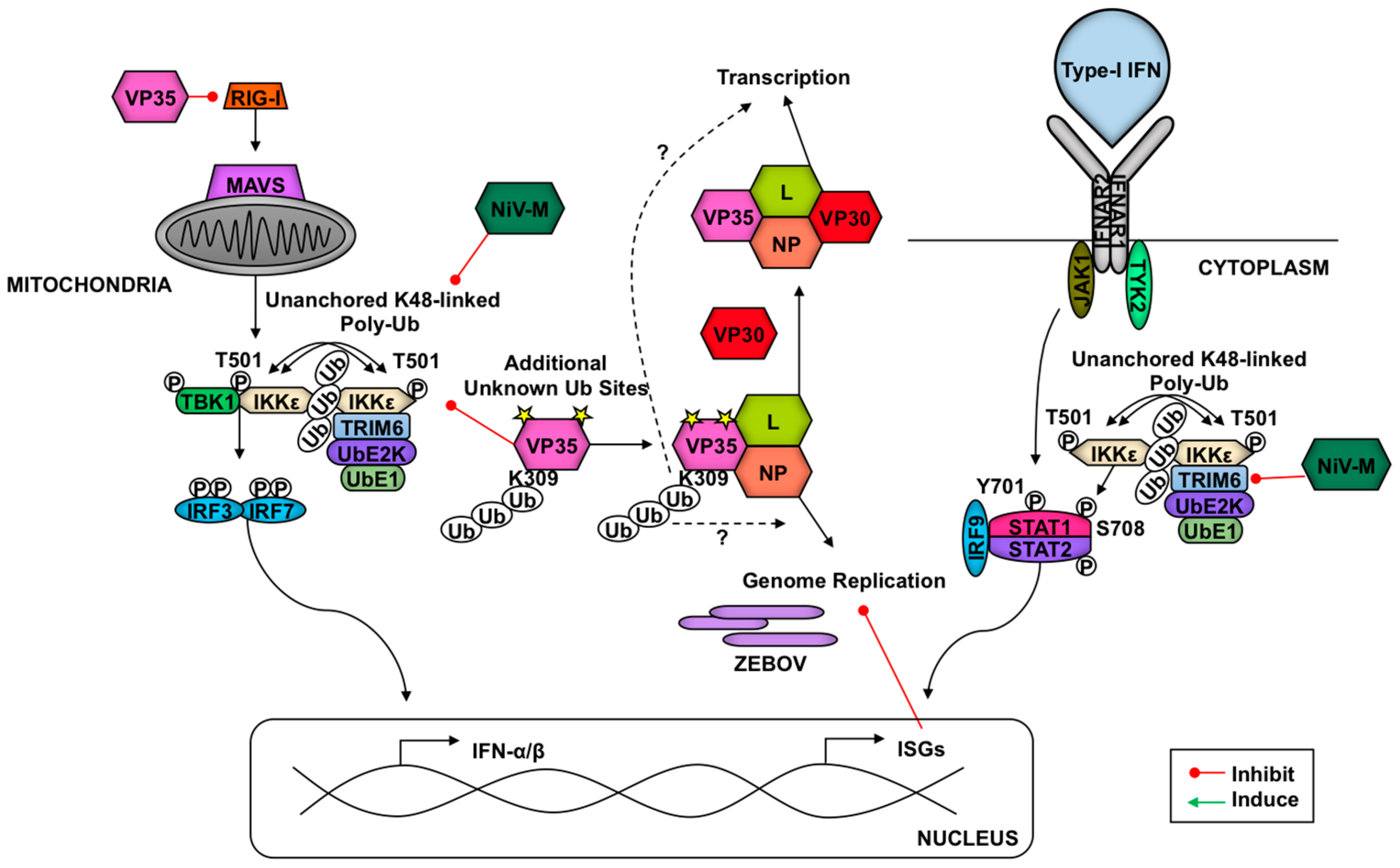
3.3. TRIMs and Negative Sense RNA Viruses
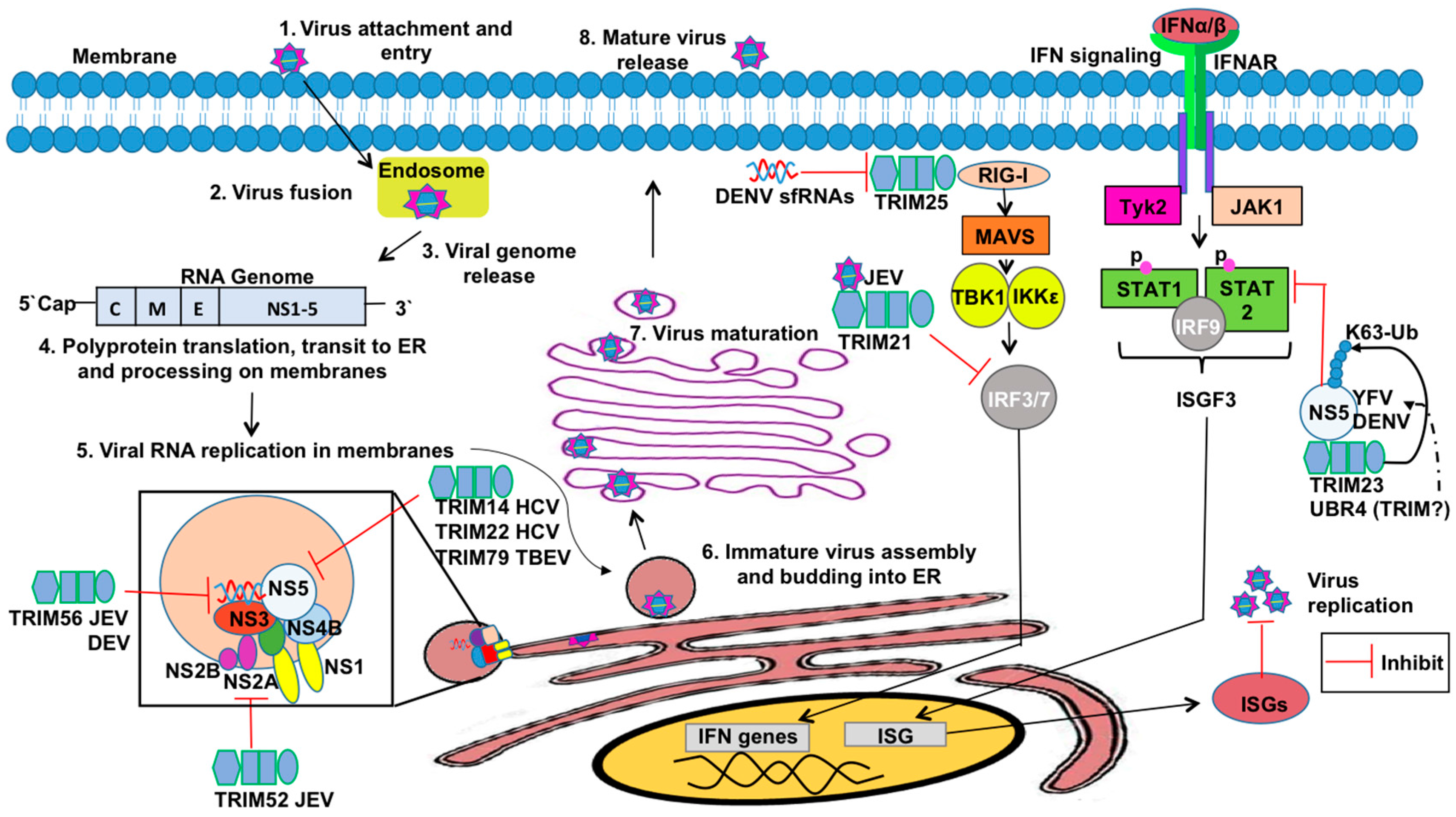
3.4. TRIMs and Positive Sense RNA Viruses
3.5. TRIMs and DNA Viruses
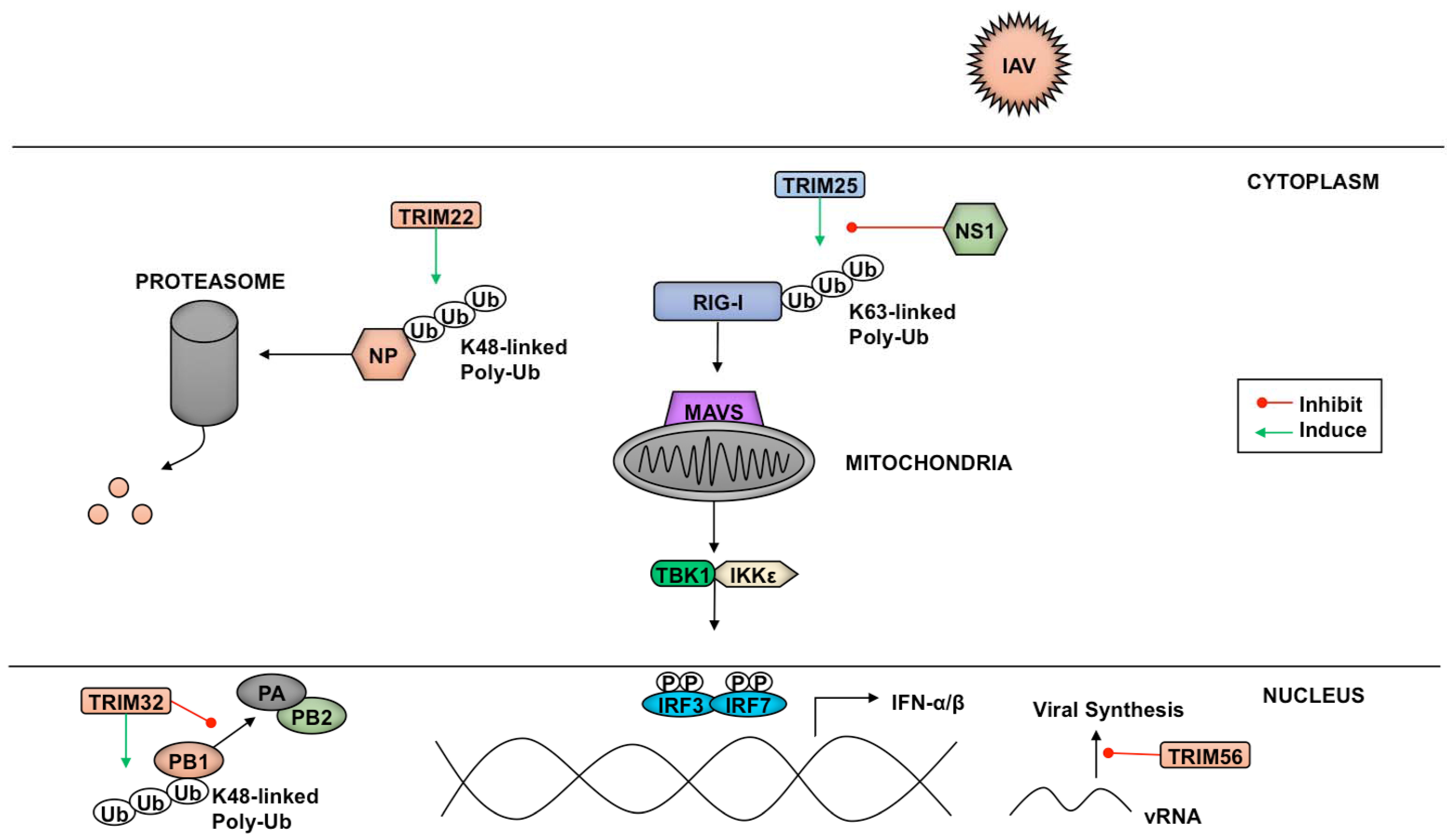
4. Viral Antagonism of TRIMs
4.1. Antagonism of TRIMs by RNA Viruses
4.2. Antagonism of TRIMs by DNA Viruses
5. Hijacking of Antiviral TRIMs as a Novel Mechanism to Directly Enhance Virus Replication
6. Potential Roles of TRIM-mediated Autophagy during Virus Infections
7. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin enzymes in the regulation of immune responses. Crit. Rev. Biochem. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yuan, B.; Zhu, W.; Zhang, R.; Li, L.; Hao, X.; Chen, S.; Hou, F. Ube2d3 and ube2n are essential for RIG-I-mediated mavs aggregation in antiviral innate immunity. Nat. Commun. 2017, 8, 15138. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Garcia-Sastre, A.; Versteeg, G.A. Trimmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Benke, S.; Garcia-Sastre, A.; Rajsbaum, R. Intrimsic immunity: Positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 2014, 25, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015, 16, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Versteeg, G.A.; Schmid, S.; Maestre, A.M.; Belicha-Villanueva, A.; Martinez-Romero, C.; Patel, J.R.; Morrison, J.; Pisanelli, G.; Miorin, L.; et al. Unanchored k48-linked polyubiquitin synthesized by the e3-ubiquitin ligase TRIM6 stimulates the interferon-ikkepsilon kinase-mediated antiviral response. Immunity 2014, 40, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Mallery, D.L.; Watkinson, R.E.; Dickson, C.F.; James, L.C. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc. Natl. Acad. Sci. USA 2015, 112, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Quinlan, B.D.; Chan, W.T.; Luna, J.M.; Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008, 4, e16. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Rajsbaum, R.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Valdiviezo, J.; Shi, M.; Inn, K.S.; Fernandez-Sesma, A.; Jung, J.; Garcia-Sastre, A. The E3-ligase trim family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 2013, 38, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.L.; Malyukova, A.; Holien, J.K.; Koach, J.; Parker, M.W.; Kavallaris, M.; Marshall, G.M.; Cheung, B.B. TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS ONE 2012, 7, e37470. [Google Scholar] [CrossRef] [PubMed]
- Massiah, M.A.; Simmons, B.N.; Short, K.M.; Cox, T.C. Solution structure of the RBCC/TRIM B-box1 domain of human mid1: B-box with a ring. J. Mol. Biol. 2006, 358, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef] [PubMed]
- Dawidziak, D.M.; Sanchez, J.G.; Wagner, J.M.; Ganser-Pornillos, B.K.; Pornillos, O. Structure and catalytic activation of the TRIM23 ring E3 ubiquitin ligase. Proteins 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Weinert, C.; Morger, D.; Djekic, A.; Grutter, M.G.; Mittl, P.R. Crystal structure of TRIM20 c-terminal coiled-coil/b30.2 fragment: Implications for the recognition of higher order oligomers. Sci. Rep. 2015, 5, 10819. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Qin, X.-r.; Hayashi, F.; Kigawa, T.; Finzi, A.; Sarnak, Z.; Lienlaf, M.; Yokoyama, S.; Sodroski, J. A b-box 2 surface patch important for TRIM5α self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 2009, 83, 10737–10751. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sodroski, J. The TRIM5alpha b-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 2008, 82, 11495–11502. [Google Scholar] [CrossRef] [PubMed]
- Streich, F.C., Jr.; Ronchi, V.P.; Connick, J.P.; Haas, A.L. Tripartite motif ligases catalyze polyubiquitin chain formation through a cooperative allosteric mechanism. J. Biol. Chem. 2013, 288, 8209–8221. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. Ring dimerization links higher-order assembly of TRIM5alpha to synthesis of k63-linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-i-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245–E254. [Google Scholar] [CrossRef] [PubMed]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Cho, H.; Inn, K.S.; Yang, A.; Zhao, Z.; Liang, Q.; Versteeg, G.A.; Amini-Bavil-Olyaee, S.; Wong, L.Y.; Zlokovic, B.V.; et al. Negative regulation of NF-κB activity by brain-specific tripartite motif protein 9. Nat. Commun. 2014, 5, 4820. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Suita, N.; Okazaki, I.M.; Shibayama, S.; Honjo, T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat. Immunol. 2012, 13, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Taoka, M.; Shoji, I.; Kato, H.; Sato, T.; Hatakeyama, S.; Isobe, T.; Hachiya, N. 14-3-3 proteins sequester a pool of soluble TRIM32 ubiquitin ligase to repress autoubiquitylation and cytoplasmic body formation. J. Cell Sci. 2013, 126, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Li, X.; Javanbakht, H.; Song, B.; Welikala, S.; Stremlau, M.; Sodroski, J. Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 2006, 349, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T. The cellular level of TRIM31, an RBCC protein overexpressed in gastric cancer, is regulated by multiple mechanisms including the ubiquitin-proteasome system. Cell Biol. Int. 2011, 35, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Meroni, G. Genomics and evolution of the trim gene family. Adv. Exp. Med. Biol. 2012, 770, 1–9. [Google Scholar] [PubMed]
- Han, K.; Lou, D.I.; Sawyer, S.L. Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 2011, 7, e1002388. [Google Scholar] [CrossRef] [PubMed]
- Boudinot, P.; van der Aa, L.M.; Jouneau, L.; Du Pasquier, L.; Pontarotti, P.; Briolat, V.; Benmansour, A.; Levraud, J.P. Origin and evolution of TRIM proteins: New insights from the complete trim repertoire of zebrafish and pufferfish. PLoS ONE 2011, 6, e22022. [Google Scholar] [CrossRef] [PubMed]
- Malfavon-Borja, R.; Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. An evolutionary screen highlights canonical and noncanonical candidate antiviral genes within the primate TRIM gene family. Genome Biol. Evol. 2013, 5, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- He, D.D.; Lu, Y.; Gittelman, R.; Jin, Y.; Ling, F.; Joshua, A. Positive selection of the TRIM family regulatory region in primate genomes. Proc. Biol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog. 2007, 3, e197. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human trim gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Xu, W.; Wang, Y.; Zhong, L.; Xiong, S. Induction of TRIM22 by IFN-gamma involves jak and pc-plc/pkc, but not mapks and PI3K/AKT/mtor pathways. J. Interferon Cytokine Res. 2013, 33, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Sjostrand, M.; Ambrosi, A.; Brauner, S.; Sullivan, J.; Malin, S.; Kuchroo, V.K.; Espinosa, A.; Wahren-Herlenius, M. Expression of the immune regulator tripartite-motif 21 is controlled by IFN regulatory factors. J. Immunol. 2013, 191, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Xie, X.Q.; Yang, Q.; Liao, C.Y.; Ye, W.; Lin, H.; Shu, H.B. TRIM38 negatively regulates TLR3/4-mediated innate immune and inflammatory responses by two sequential and distinct mechanisms. J. Immunol. 2015, 195, 4415–4425. [Google Scholar] [CrossRef] [PubMed]
- Narayan, K.; Waggoner, L.; Pham, S.T.; Hendricks, G.L.; Waggoner, S.N.; Conlon, J.; Wang, J.P.; Fitzgerald, K.A.; Kang, J. TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J. Virol. 2014, 88, 10748–10757. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.X.; Hong, X.; Liao, B.B.; Shi, S.Z.; Lai, X.F.; Zheng, H.Y.; Xie, L.; Wang, Y.; Wang, X.L.; Xin, H.B.; et al. Expression profiling of TRIM protein family in thp1-derived macrophages following TLR stimulation. Sci. Rep. 2017, 7, 42781. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Manocha, G.D.; Mishra, R.; Sharma, N.; Kumawat, K.L.; Basu, A.; Singh, S.K. Regulatory role of TRIM21 in the type-I interferon pathway in japanese encephalitis virus-infected human microglial cells. J. Neuroinflamm. 2014, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez-Romero, C.; tenOever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bharaj, P.; Wang, Y.E.; Dawes, B.E.; Yun, T.E.; Park, A.; Yen, B.; Basler, C.F.; Freiberg, A.N.; Lee, B.; Rajsbaum, R. The matrix protein of nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the ikkepsilon kinase-mediated type-I IFN antiviral response. PLoS Pathog. 2016, 12, e1005880. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza a virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Garcia-Sastre, A. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 2013, 21, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Mandell, M.; Deretic, V. Precision autophagy directed by receptor regulators––Emerging examples within the TRIM family. J. Cell Sci. 2016, 129, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, Z.J. Expanding role of ubiquitination in NF-κb signaling. Cell Res. 2010, 21, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J. Convergence of the NF-κb and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007, 18, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Runge, S.; Sparrer, K.M.; Lassig, C.; Hembach, K.; Baum, A.; Garcia-Sastre, A.; Soding, J.; Conzelmann, K.K.; Hopfner, K.P. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 2014, 10, e1004081. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5’-triphosphate rna is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. Rig-i-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. RIG-I-like receptor regulation in virus infection and immunity. Curr. Opin. Virol. 2015, 12, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Wu, B.; Yao, H.; Walz, T.; Hur, S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 2013, 51, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Nistal-Villan, E.; Inn, K.S.; Garcia-Sastre, A.; Jung, J.U. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 2010, 84, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Konishi, H.; Shimotohno, K. Ubch8 regulates ubiquitin and ISG15 conjugation to RIG-I. Mol. Immunol. 2008, 45, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. Mavs forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. Mavs recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2013, 2, e00785. [Google Scholar] [CrossRef] [PubMed]
- Xie, P. Traf molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the iκb kinase complex by traf6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.K.; Hong, M.; Shaito, A.; Chiu, Y.H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 2004, 15, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.P.; Sun, L.; Chen, X.; Pineda, G.; Jiang, X.; Adhikari, A.; Zeng, W.; Chen, Z.J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009, 461, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NF-κB subunits by phosphorylation. Cells 2016. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.L.; Gioia, R.; Gatot, J.S.; Patrascu, F.; Carpentier, I.; Chapelle, J.P.; O’Neill, L.; Beyaert, R.; Piette, J.; Chariot, A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 2008, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Juang, Y.T.; Lowther, W.; Kellum, M.; Au, W.C.; Lin, R.; Hiscott, J.; Pitha, P.M. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc. Natl. Acad. Sci. USA 1998, 95, 9837–9842. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, Q.; Mao, A.P.; Hu, M.M.; Shu, H.B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for k63-linked ubiquitination. J. Mol. Cell Biol. 2014, 6, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 ring-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Seya, T. Ubiquitin-mediated modulation of the cytoplasmic viral RNA sensor RIG-I. J. Biochem. 2011, 151, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease usp15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Shu, H.B. Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell Mol. Immunol. 2017, 14, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Liao, C.Y.; Yang, Q.; Xie, X.Q.; Shu, H.B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 2017, 214, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Watanabe, M.; Hatakeyama, S. TRIM59 interacts with ecsit and negatively regulates NF-κB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 2012, 422, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Evensen, O.; Munang’andu, H.M. De novo transcriptome analysis shows that sav-3 infection upregulates pattern recognition receptors of the endosomal toll-like and RIG-I-like receptor signaling pathways in macrophage/dendritic like to-cells. Viruses 2016, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Miranzo-Navarro, D.; Magor, K.E. Activation of duck rig-i by TRIM25 is independent of anchored ubiquitin. PLoS ONE 2014, 9, e86968. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villan, E.; Garcia-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza a virus ns1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.Q.; Cheng, Y.; Yang, H.L.; Zhu, Q.; Yu, D.; Liu, Y.P. Molecular characterization, tissue distribution and expression analysis of TRIM25 in gallus gallus domesticus. Gene 2015, 561, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Blaine, A.H.; Miranzo-Navarro, D.; Campbell, L.K.; Aldridge, J.R.; Webster, R.G.; Magor, K.E. Duck TRIM27-l enhances mavs signaling and is absent in chickens and turkeys. Mol. Immunol. 2015, 67, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. Mavs ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor mavs through lys63-linked polyubiquitination. Nat. Immunol. 2016, 18, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of TRIM44 promotes an antiviral response by stabilizing visa. J. Immunol. 2013, 190, 3613–3619. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Liu, Q.; Tian, S.; Xie, W.; Cui, J.; Wang, R.F. TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type i interferon by recruiting GSK3β to TBK1. Cell Res. 2016, 26, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between cytoplasmic RIG-I and sting sensing pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Song, B.; Park, C.; Kwon, K.S. TRIM11 negatively regulates ifnβ production and antiviral activity by targeting TBK1. PLoS ONE 2013, 8, e63255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Yuan, C.; Qi, J.; Meng, H.; Gao, C. Tripartite motif-containing protein 38 negatively regulates TLR3/4- and RIG-I-mediated IFN-β production and antiviral response by targeting NAP1. J. Immunol. 2012, 188, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Funami, K.; Saeki, Y.; Tanaka, K.; Okawa, K.; Takeuchi, O.; Akira, S.; Murakami, Y.; Shimotohno, K. Polyubiquitin conjugation to nemo by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. USA 2010, 107, 15856–15861. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Zhang, J.; Liu, L.L.; Pan, Z.Y.; Nie, Y.; Zhang, H.Y.; Wang, Y.Y. Autoubiquitination of TRIM26 links TBK1 to nemo in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol. 2015, 8, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Wynne, C.; Lazzari, E.; Smith, S.; McCarthy, E.M.; Ni Gabhann, J.; Kallal, L.E.; Higgs, R.; Greco, A.; Cryan, S.A.; Biron, C.A.; et al. TRIM68 negatively regulates IFN-β production by degrading TRK fused gene, a novel driver of IFN-β downstream of anti-viral detection systems. PLoS ONE 2014, 9, e101503. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. Trim30 α negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yao, W.; Huang, F.; Sun, B.; Yang, R. The human antiviral factor TRIM11 is under the regulation of HIV-1 VPR. PLoS ONE 2014, 9, e104269. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.J.; Zhang, Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 2016, 17, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Watanabe, M.; Nakamaru, Y.; Takagi, D.; Takahashi, H.; Fukuda, S.; Hatakeyama, S. TRIM39 negatively regulates the NFκB-mediated signaling pathway through stabilization of cactin. Cell Mol. Life Sci. 2015, 73, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. Sting regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. Sting is a direct innate immune sensor of cyclic di-gmp. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of sting bound to cyclic di-gmp reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 2012, 19, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Omura, H.; Oikawa, D.; Nakane, T.; Kato, M.; Ishii, R.; Ishitani, R.; Tokunaga, F.; Nureki, O. Structural and functional analysis of ddx41: A bispecific immune receptor for DNA and cyclic dinucleotide. Sci. Rep. 2016, 6, 34756. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, K.; Zhang, Z.; Teles, R.M.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase ddx41 recognizes the bacterial secondary messengers cyclic di-gmp and cyclic di-amp to activate a type I interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bao, M.; Lu, N.; Weng, L.; Yuan, B.; Liu, Y.J. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 2012, 14, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lian, Q.; Yang, B.; Yan, S.; Zhou, H.; He, L.; Lin, G.; Lian, Z.; Jiang, Z.; Sun, B. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting sting. PLoS Pathog. 2015, 11, e1005012. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting mita/sting protein for k63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Dalmaroni, M.J.; Gerswhin, M.E.; Adamopoulos, I.E. The critical role of toll-like receptors--from microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2015, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Higgs, R.; Ni Gabhann, J.; Ben Larbi, N.; Breen, E.P.; Fitzgerald, K.A.; Jefferies, C.A. The E3 ubiquitin ligase ro52 negatively regulates IFN-β production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 2008, 181, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Higgs, R.; Lazzari, E.; Wynne, C.; Ni Gabhann, J.; Espinosa, A.; Wahren-Herlenius, M.; Jefferies, C.A. Self protection from anti-viral responses--ro52 promotes degradation of the transcription factor IRF7 downstream of the viral toll-like receptors. PLoS ONE 2010, 5, e11776. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, E.; Korczeniewska, J.; Ni Gabhann, J.; Smith, S.; Barnes, B.J.; Jefferies, C.A. Tripartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PLoS ONE 2014, 9, e103609. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Zhang, M.; Yuan, C.; Gao, C. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J. Immunol. 2012, 188, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Zhou, Z.; Lei, X.; Liu, X.; He, B.; Wang, J.; Hung, T. TRIM38 negatively regulates TLR3-mediated IFN-β signaling by targeting trif for degradation. PLoS ONE 2012, 7, e46825. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, N.L.; Wang, J.; Liu, B.; Lester, S.; Li, K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 2012, 287, 36404–36413. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Forman, M.; Arav-Boger, R. Activation of nucleotide oligomerization domain 2 (NOD2) by human cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS ONE 2014, 9, e92704. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 2012, 3, 328. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Mitoma, H.; Trichot, C.; Bao, M.; Liu, Y.; Zhang, Z.; Liu, Y.J. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J. Immunol. 2014, 193, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mao, K.; Zeng, Y.; Chen, S.; Tao, Z.; Yang, C.; Sun, S.; Wu, X.; Meng, G.; Sun, B. Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J. Immunol. 2010, 185, 7699–7705. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [PubMed]
- Zurek, B.; Schoultz, I.; Neerincx, A.; Napolitano, L.M.; Birkner, K.; Bennek, E.; Sellge, G.; Lerm, M.; Meroni, G.; Soderholm, J.D.; et al. TRIM27 negatively regulates NOD2 by ubiquitination and proteasomal degradation. PLoS ONE 2012, 7, e41255. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Sripada, L.; Prajapati, P.; Singh, R.; Singh, A.K. Nucleo-cytoplasmic trafficking of TRIM8, a novel oncogene, is involved in positive regulation of TNF induced NF-κB pathway. PLoS ONE 2012, 7, e48662. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Matsunaga, Y.; Katayama, Y.; Nakayama, K.I.; Hatakeyama, S. TRIM8 modulates STAT3 activity through negative regulation of PIAS3. J. Cell Sci. 2010, 123, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Yang, Q.; Zhang, J.; Liu, S.M.; Zhang, Y.; Lin, H.; Huang, Z.F.; Wang, Y.Y.; Zhang, X.D.; Zhong, B.; et al. TRIM38 inhibits TNFα- and IL-1β-triggered NF-κB activation by mediating lysosome-dependent degradation of TAB2/3. Proc. Natl. Acad. Sci. USA 2014, 111, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Huang, Y.; Dai, X.; Liu, Y.; Liu, Z.; Wang, N.; Zhang, P. Tripartite motif-containing 28 bridges endothelial inflammation and angiogenic activity by retaining expression of TNFR-1 and -2 and VEGFR2 in endothelial cells. FASEB J. 2017, 31, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferons and viral infections. Biofactors 2009, 35, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tisserand, J.; Khetchoumian, K.; Thibault, C.; Dembele, D.; Chambon, P.; Losson, R. Tripartite motif 24 (TRIM24/TIF1α) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor α (RARα) inhibition. J. Biol. Chem. 2011, 286, 33369–33379. [Google Scholar] [CrossRef] [PubMed]
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/gerp ring finger protein interacts with socs-1. J. Biol. Chem. 2002, 277, 37315–37322. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase ikkepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.L.; Friedman, B.A.; Schmid, S.; Gertz, J.; Myers, R.M.; Tenoever, B.R.; Maniatis, T. Iκb kinase ε (IKKε) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. USA 2011, 108, 21170–21175. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, A.; Mechti, N. Control of foxo4 activity and cell survival by TRIM22 directs TLR3-stimulated cells toward IFN type I gene induction or apoptosis. J. Interferon Cytokine Res. 2015, 35, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Gongora, C.; Tissot, C.; Cerdan, C.; Mechti, N. The interferon-inducible STAF50 gene is downregulated during T cell costimulation by CD2 and CD28. J. Interferon Cytokine Res. 2000, 20, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Obad, S.; Olofsson, T.; Mechti, N.; Gullberg, U.; Drott, K. Regulation of the interferon-inducible p53 target gene TRIM22 (STAF50) in human T lymphocyte activation. J. Interferon Cytokine Res. 2007, 27, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, J.; Okoye, I.S.; Guidi, R.; Kannan, Y.; Coomes, S.M.; Czieso, S.; Mengus, G.; Davidson, I.; Wilson, M.S. T-cell-intrinsic TIF1α/TRIM24 regulates IL-1r expression on Th2 cells and Th2 cell-mediated airway allergy. Proc. Natl. Acad. Sci. USA 2016, 113, E568–E576. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Srivastava, S.; Sun, Y.; Li, Z.; Wu, H.; Zuvela-Jelaska, L.; Li, J.; Salamon, R.S.; Backer, J.M.; Skolnik, E.Y. Tripartite motif containing protein 27 negatively regulates CD4 T cells by ubiquitinating and inhibiting the class II PI3K-c2β. Proc. Natl. Acad. Sci. USA 2011, 108, 20072–20077. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Hur, J.Y.; Lee, M.S.; Zhang, Q.; Choi, W.Y.; Kim, L.K.; Lee, W.B.; Oh, G.T.; Kim, Y.J. Tripartite motif-containing protein 30 modulates TCR-activated proliferation and effector functions in CD4+ T cells. PLoS ONE 2014, 9, e95805. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.W.; Nisole, S.; Lynch, C.; Stoye, J.P. TRIM5α protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. USA 2004, 101, 10786–10791. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Lynch, C.; Stoye, J.P.; Yap, M.W. A TRIM5-cyclophilin a fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 2004, 101, 13324–13328. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin a retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Christensen, D.E.; Nelson, C.; Tan, C.P.; Schaller, T.; Lehner, P.J.; Sundquist, W.I.; Towers, G.J. TRIM5α requires ube2w to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 2015, 34, 2078–2095. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, R.; Mukherjee, S.; Smolin, N.; Pauszek, R.F.; Bradley, M.; Sastri, J.; Robia, S.L.; Millar, D.; Campbell, E.M. Dynamic conformational changes in the rhesus TRIM5α dimer dictate the potency of HIV-1 restriction. Virology 2017, 500, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Chandrasekaran, V.; Carter, S.D.; Woodward, C.L.; Christensen, D.E.; Dryden, K.A.; Pornillos, O.; Yeager, M.; Ganser-Pornillos, B.K.; Jensen, G.J. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife 2016, 5, e16269. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Roganowicz, M.D.; Skorupka, K.; Alam, S.L.; Christensen, D.; Doss, G.; Wan, Y.; Frank, G.A.; Ganser-Pornillos, B.K.; Sundquist, W.I. Mechanism of B-box 2 domain-mediated higher-order assembly of the retroviral restriction factor TRIM5α. Elife 2016, 5, e16309. [Google Scholar] [CrossRef] [PubMed]
- Keown, J.R.; Goldstone, D.C. Crystal structure of the TRIM5α bbox2 domain from rhesus macaques describes a plastic oligomerisation interface. J. Struct. Biol. 2016, 195, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Sastri, J.; Campbell, E.M. Recent insights into the mechanism and consequences of TRIM5α retroviral restriction. AIDS Res. Hum. Retroviruses 2011, 27, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5αprotein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5α restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Perez-Caballero, D.; Bieniasz, P.D. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 2013, 9, e1003214. [Google Scholar] [CrossRef] [PubMed]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moyano, E.; Ruiz, A.; Kløverpris, H.N.; Rodriguez-Plata, M.T.; Peña, R.; Blondeau, C.; Selwood, D.L.; Izquierdo-Useros, N.; Moris, A.; Clotet, B. Nonhuman TRIM5 variants enhance recognition of HIV-1-infected cells by CD8+ T cells. J. Virol. 2016, 90, 8552–8562. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ramos, M.; Stoye, J.P. Capsid-binding retrovirus restriction factors: Discovery, restriction specificity and implications for the development of novel therapeutics. J. Gen. Virol. 2013, 94, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Grütter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Weingart, J.; Sette, P.; Opp, S.; Sastri, J.; O’Connor, S.K.; Talley, S.; Diaz-Griffero, F.; Hirsch, V.; Bouamr, F. TRIM5α-mediated ubiquitin chain conjugation is required for inhibition of HIV-1 reverse transcription and capsid destabilization. J.Virol. 2016, 90, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Lascano, J.; Uchil, P.D.; Mothes, W.; Luban, J. TRIM5 retroviral restriction activity correlates with the ability to induce innate immune signaling. J. Virol. 2016, 90, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Ourmanov, I.; Riddick, N.; Matsuda, K.; Whitted, S.; Plishka, R.J.; Buckler-White, A.; Starost, M.F.; Hirsch, V.M. TRIM5α restriction affects clinical outcome and disease progression in simian immunodeficiency virus-infected rhesus macaques. J. Virol. 2015, 89, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Letvin, N.L.; Mascola, J.R.; Sun, Y.; Gorgone, D.A.; Buzby, A.P.; Xu, L.; Yang, Z.-y.; Chakrabarti, B.; Rao, S.S.; Schmitz, J.E. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science 2006, 312, 1530–1533. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the spry domain of human TRIM5αleads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Si, Z.; Vandegraaff, N.; O’Huigin, C.; Song, B.; Yuan, W.; Xu, C.; Perron, M.; Li, X.; Marasco, W.A.; Engelman, A.; et al. Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7454–7459. [Google Scholar] [CrossRef] [PubMed]
- Ylinen, L.M.; Keckesova, Z.; Webb, B.L.; Gifford, R.J.; Smith, T.P.; Towers, G.J. Isolation of an active lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J. Virol. 2006, 80, 7332–7338. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, P.; Crespo, H.; Glaria, I.; Lujan, L.; Contreras, A.; Rosati, S.; de Andres, D.; Amorena, B.; Towers, G.J.; Reina, R. Ovine TRIM5α can restrict visna/maedi virus. J. Virol. 2012, 86, 9504–9509. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.J.; Nisole, S.; Carthagena, L.; Coutant, F.; Souque, P.; Brussel, A.; Estaquier, J.; Charneau, P. Lack of endogenous TRIM5α-mediated restriction in rhesus macaque dendritic cells. Blood 2008, 112, 3772–3776. [Google Scholar] [CrossRef] [PubMed]
- Portilho, D.M.; Fernandez, J.; Ringeard, M.; Machado, A.K.; Boulay, A.; Mayer, M.; Müller-Trutwin, M.; Beignon, A.-S.; Kirchhoff, F.; Nisole, S. Endogenous TRIM5α function is regulated by sumoylation and nuclear sequestration for efficient innate sensing in dendritic cells. Cell Rep. 2016, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yao, W.; Tokunaga, K.; Yang, R.; Sun, B. An HIV-1 capsid binding protein TRIM11 accelerates viral uncoating. Retrovirology 2016, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Pawlica, P.; Le Sage, V.; Poccardi, N.; Tremblay, M.J.; Mouland, A.J.; Berthoux, L. Functional evidence for the involvement of microtubules and dynein motor complexes in TRIM5α-mediated restriction of retroviruses. J. Virol. 2014, 88, 5661–5676. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza a virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Dharan, A.; Fricke, T.; Diaz-Griffero, F.; Campbell, E.M. HIV-1 uncoating is facilitated by dynein and kinesin 1. J. Virol. 2014, 88, 13613–13625. [Google Scholar] [CrossRef] [PubMed]
- Pawlica, P.; Berthoux, L. Cytoplasmic dynein promotes HIV-1 uncoating. Viruses 2014, 6, 4195–4211. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Garcia-Sastre, A. Virology. Unanchored ubiquitin in virus uncoating. Science 2014, 346, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Hara, Y.; Takagi, C.; Yamamoto, T.S.; Ueno, N. MID1 and MID2 are required for xenopus neural tube closure through the regulation of microtubule organization. Development 2010, 137, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Turrini, F.; Marelli, S.; Kajaste-Rudnitski, A.; Lusic, M.; Lint, C.; Das, A.T.; Harwig, A.; Berkhout, B.; Vicenzi, E. HIV-1 transcriptional silencing caused by TRIM22 inhibition of sp1 binding to the viral promoter. Retrovirology 2015, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Tabah, A.A.; Tardif, K.; Mansky, L.M. Anti-HIV-1 activity of TRIM 37. J. Gen. Virol. 2014, 95, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Masroori, N.; Merindol, N.; Berthoux, L. The interferon-induced antiviral protein pml (TRIM19) promotes the restriction and transcriptional silencing of lentiviruses in a context-specific, isoform-specific fashion. Retrovirology 2016, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Kahle, T.; Volkmann, B.; Eissmann, K.; Herrmann, A.; Schmitt, S.; Wittmann, S.; Merkel, L.; Reuter, N.; Stamminger, T.; Gramberg, T. TRIM19/pml restricts HIV infection in a cell type-dependent manner. Viruses 2015, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Dutrieux, J.; Maarifi, G.; Portilho, D.M.; Arhel, N.J.; Chelbi-Alix, M.K.; Nisole, S. Pml/TRIM19-dependent inhibition of retroviral reverse-transcription by daxx. PLoS Pathog. 2015, 11, e1005280. [Google Scholar] [CrossRef] [PubMed]
- Bidgood, S.R.; Tam, J.C.; McEwan, W.A.; Mallery, D.L.; James, L.C. Translocalized IGA mediates neutralization and stimulates innate immunity inside infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 13463–13468. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed]
- Keeble, A.H.; Khan, Z.; Forster, A.; James, L.C. TRIM21 is an igg receptor that is structurally, thermodynamically, and kinetically conserved. Proc. Natl. Acad. Sci. USA 2008, 105, 6045–6050. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; Tam, J.C.; Watkinson, R.E.; Bidgood, S.R.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, R.E.; Tam, J.C.; Vaysburd, M.J.; James, L.C. Simultaneous neutralization and innate immune detection of a replicating virus by TRIM21. J. Virol. 2013, 87, 7309–7313. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, R.E.; McEwan, W.A.; Tam, J.C.; Vaysburd, M.; James, L.C. TRIM21 promotes cgas and RIG-I sensing of viral genomes during infection by antibody-opsonized virus. PLoS Pathog. 2015, 11, e1005253. [Google Scholar] [CrossRef] [PubMed]
- Foss, S.; Watkinson, R.E.; Grevys, A.; McAdam, M.B.; Bern, M.; Hoydahl, L.S.; Dalhus, B.; Michaelsen, T.E.; Sandlie, I.; James, L.C.; et al. TRIM21 immune signaling is more sensitive to antibody affinity than its neutralization activity. J. Immunol. 2016, 196, 3452–3459. [Google Scholar] [CrossRef] [PubMed]
- Vaysburd, M.; Watkinson, R.E.; Cooper, H.; Reed, M.; O’Connell, K.; Smith, J.; Cruickshanks, J.; James, L.C. Intracellular antibody receptor TRIM21 prevents fatal viral infection. Proc. Natl. Acad. Sci. USA 2013, 110, 12397–12401. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zhang, D.; Qian, P.; Qian, S.; Wu, M.; Chen, H.; Li, X. Swine TRIM21 restricts fmdv infection via an intracellular neutralization mechanism. Antiviral Res. 2016, 127, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Li, J.; Fan, W.; Zheng, W.; Yu, M.; Chen, C.; Sun, L.; Bi, Y.; Ding, C.; Gao, G.F. Robust lys63-linked ubiquitination of RIG-I promotes cytokine eruption in early influenza b virus infection. J. Virol. 2016, 90, 6263–6275. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza a virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza a polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 senses and restricts influenza a virus by ubiquitination of pb1 polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-terminal tail of TRIM56 dictates antiviral restriction of influenza A and B viruses by impeding viral RNA synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.R. Historical perspectives on flavivirus research. Viruses 2017. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of flavivirus infections: Using and abusing the host cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Qashqari, H.; Al-Mars, A.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Alqahtani, M.; Mahmoud, M.; El Sayed Zaki, M.; Fatima, K.; Qadri, I. Understanding the molecular mechanism(s) of hepatitis C virus (HCV) induced interferon resistance. Infect. Genet. Evol. 2013, 19, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5a gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996, 334, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Y.; Li, C.; Wu, Y.; Guo, L.; Peng, C.; Huang, Y.; Cheng, G.; Qin, F.X. TRIM14 inhibits hepatitis C virus infection by spry domain-dependent targeted degradation of the viral NS5a protein. Sci. Rep. 2016, 6, 32336. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, X.; Sun, D.; Yang, L.; Chong, C.; Pan, Y.; Chi, X.; Gao, Y.; Wang, M.; Shi, X.; et al. Interferon α(ifnα)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5a. Cell Mol. Immunol. 2016, 13, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Wu, M.; Qian, S.; Zhou, Y.; Chen, H.; Li, X.; Qian, P. TRIM52 inhibits japanese encephalitis virus replication by degrading the viral NS2A. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits α interferon signaling by reducing stat2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.C.; Yu, C.Y.; Liang, J.J.; Lin, E.; Liao, C.L.; Lin, Y.L. Blocking double-stranded RNA-activated protein kinase PKR by japanese encephalitis virus nonstructural protein 2a. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, N.L.; Wang, J.; Shi, P.Y.; Wang, T.; Miller, M.A.; Li, K. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J. Virol. 2014, 88, 13821–13835. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, B.; Wang, N.; Lee, Y.M.; Liu, C.; Li, K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011, 85, 3733–3745. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.J.; Lubick, K.J.; Freedman, B.A.; Carmody, A.B.; Best, S.M. Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signaling to inhibit dendritic cell function. J. Immunol. 2014, 192, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S. Mechanisms of evasion of the type I interferon antiviral response by flaviviruses. J. Interferon Cytokine Res. 2009, 29, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of interferon-stimulated jak-stat signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Lubick, K.J.; Robertson, S.J.; Broughton, J.P.; Bloom, M.E.; Bresnahan, W.A.; Best, S.M. TRIM79α, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 2011, 10, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (zap). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Pampin, M.; Chelbi-Alix, M.K. Promyelocytic leukemia isoform iv confers resistance to encephalomyocarditis virus via the sequestration of 3d polymerase in nuclear bodies. J. Virol. 2011, 85, 13164–13173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, J.-T.; Wu, J.Z.; Yang, G. Identification and characterization of multiple trim proteins that inhibit hepatitis B virus transcription. PLoS ONE 2013, 8, e70001. [Google Scholar] [CrossRef] [PubMed]
- Hattlmann, C.J.; Kelly, J.N.; Barr, S.D. TRIM22: A diverse and dynamic antiviral protein. Mol. Biol. Int. 2012, 2012, 153415. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located ring domain. Hepatology 2009, 50, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Chen, C.-S.; Wang, W.-H.; Hsu, S.-W.; Tsai, H.-H.; Liu, S.-T.; Chang, L.-K. TRIM5α promotes ubiquitination of RTA from epstein–barr virus to attenuate lytic progression. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Stamminger, T. Emerging role of PML nuclear bodies in innate immune signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wolff, T. Influenza A virus TRIMs the type I interferon response. Cell Host Microbe 2009, 5, 420–421. [Google Scholar]
- Heikel, G.; Choudhury, N.R.; Michlewski, G. The role of TRIM25 in development, disease and RNA metabolism. Biochem. Soc. Trans. 2016, 44, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.; Yamauchi, Y. Ubiquitin in influenza virus entry and innate immunity. Viruses 2016, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, H.; Wang, A.; Sun, L.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y. TRIM25 identification in the chinese goose: Gene structure, tissue expression profiles, and antiviral immune responses in vivo and in vitro. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aparicio, M.T.; Ayllon, J.; Leo-Macias, A.; Wolff, T.; Garcia-Sastre, A. Subcellular localizations of RIG-I, TRIM25, and MAVS complexes. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [PubMed]
- Santiago, F.W.; Covaleda, L.M.; Sanchez-Aparicio, M.T.; Silvas, J.A.; Diaz-Vizarreta, A.C.; Patel, J.R.; Popov, V.; Yu, X.J.; Garcia-Sastre, A.; Aguilar, P.V. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 2014, 88, 4572–4585. [Google Scholar] [CrossRef] [PubMed]
- Fatima, M.; Kumari, R.; Schwamborn, J.C.; Mahadevan, A.; Shankar, S.; Raja, R.; Seth, P. Tripartite containing motif 32 modulates proliferation of human neural precursor cells in HIV-1 neurodegeneration. Cell Death Differ. 2016, 23, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. Ns5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts ubr4 to degrade Stat2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian E3 ubiquitin ligases that contain the ubr box motif and recognize N-degrons. Mol. Cell Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.F. Hendra and nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among paramyxovirinae matrix proteins. PLoS Pathog. 2015, 11, e1004739. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.E.; Park, A.; Lake, M.; Pentecost, M.; Torres, B.; Yun, T.E.; Wolf, M.C.; Holbrook, M.R.; Freiberg, A.N.; Lee, B. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010, 6, e1001186. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E. Hepatitis b virus x protein: Trimming antiviral defences in hepatocytes. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.-H.; Park, E.-S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.-H.; Kim, C.W. Suppression of interferon-mediated anti-hbv response by single CpG methylation in the 5′-utr of TRIM22. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Burton, E.M.; Bhaduri-McIntosh, S. Chloroquine triggers epstein-barr virus replication through phosphorylation of kap1/TRIM28 in burkitt lymphoma cells. PLoS Pathog. 2017, 13, e1006249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, X.-L.; Gu, Q.-H.; Huang, F.; Zheng, W.; Li, Z.-W. Tripartite motif-containing 22 gene-364t/c polymorphism associated with hepatitis B virus infection in chinese han population. Hepat. Mon. 2014. [Google Scholar] [CrossRef] [PubMed]
- Conwell, S.E.; White, A.E.; Harper, J.W.; Knipe, D.M. Identification of TRIM27 as a novel degradation target of herpes simplex virus 1 icp0. J. Virol. 2015, 89, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Bharaj, P.; Atkins, C.; Luthra, P.; Giraldo, M.I.; Dawes, B.E.; Miorin, L.; Johnson, J.R.; Krogan, N.J.; Basler, C.F.; Freiberg, A.N.; et al. The host E3-ubiquitin ligase TRIM6 ubiquitinates the ebola virus VP35 protein and promotes virus replication. J. Virol. 2017, 91, e00833-17. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, W.B.; Loo, Y.M.; Gale, M., Jr.; Hartman, A.L.; Kimberlin, C.R.; Martinez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits α/β interferon production induced by RIG-I signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the ebola virus VP35 protein and the RIG-I activator pact determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Cardenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases ikkepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Muhlberger, E. Filovirus replication and transcription. Future Virol. 2007, 2, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in leukocytes and other cells: Mechanisms, subsystem organization, selectivity, and links to innate immunity. J. Leukoc. Biol. 2016, 100, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-directed selective autophagy regulates immune activation. Autophagy 2017, 13, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Kimura, T.; Jain, A.; Johansen, T.; Deretic, V. TRIM proteins regulate autophagy: TRIM5 is a selective autophagy receptor mediating HIV-1 restriction. Autophagy 2014, 10, 2387–2388. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the trim5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus–Host Interactions. Vaccines 2017, 5, 23. https://doi.org/10.3390/vaccines5030023
Van Tol S, Hage A, Giraldo MI, Bharaj P, Rajsbaum R. The TRIMendous Role of TRIMs in Virus–Host Interactions. Vaccines. 2017; 5(3):23. https://doi.org/10.3390/vaccines5030023
Chicago/Turabian StyleVan Tol, Sarah, Adam Hage, Maria Isabel Giraldo, Preeti Bharaj, and Ricardo Rajsbaum. 2017. "The TRIMendous Role of TRIMs in Virus–Host Interactions" Vaccines 5, no. 3: 23. https://doi.org/10.3390/vaccines5030023
APA StyleVan Tol, S., Hage, A., Giraldo, M. I., Bharaj, P., & Rajsbaum, R. (2017). The TRIMendous Role of TRIMs in Virus–Host Interactions. Vaccines, 5(3), 23. https://doi.org/10.3390/vaccines5030023