
Synthetic Biodegradable Microparticle and Nanoparticle Vaccines against the Respiratory Syncytial Virus



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
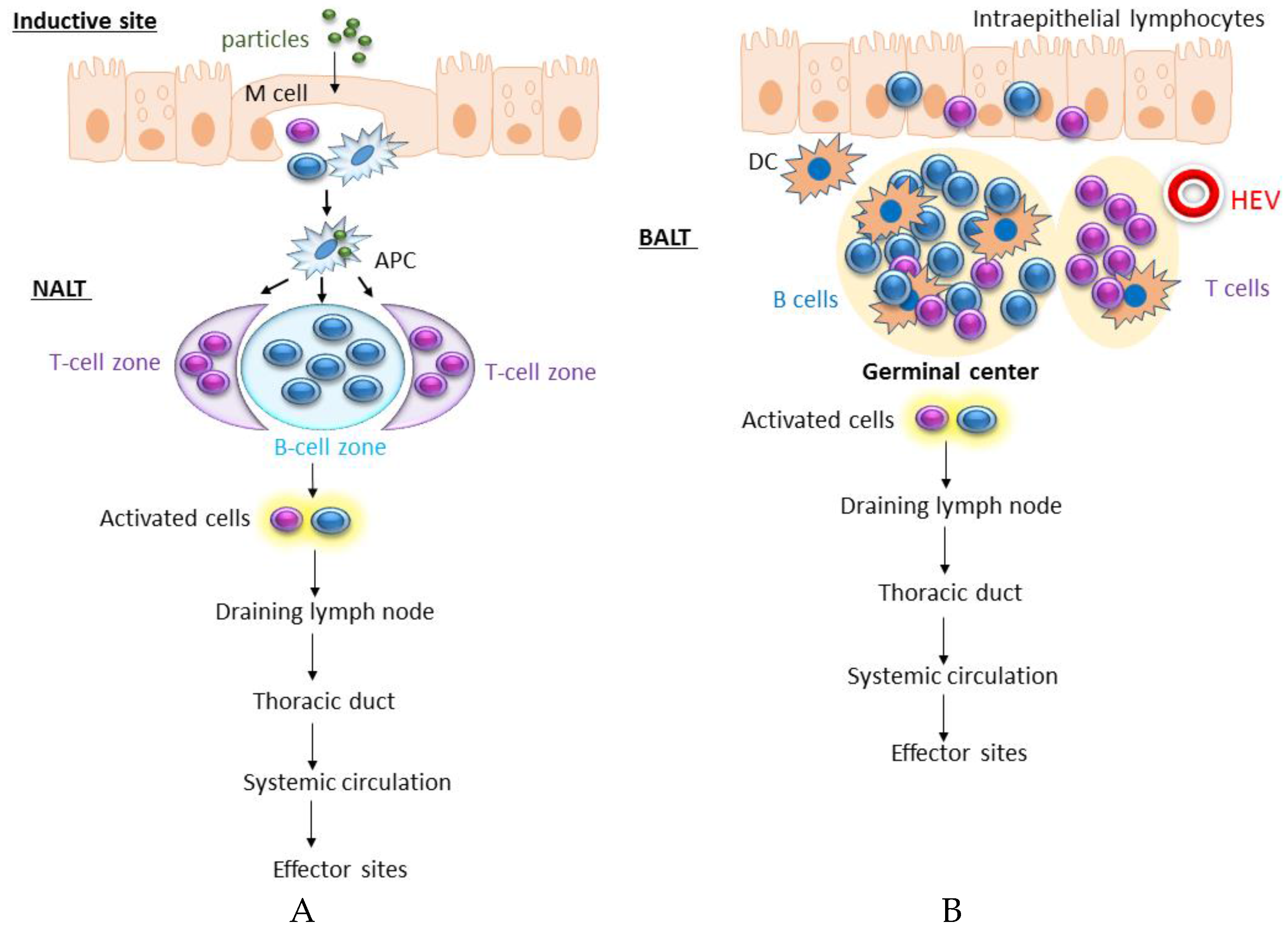
2. Lymphoid Tissue and Immune Response in the Respiratory Tract
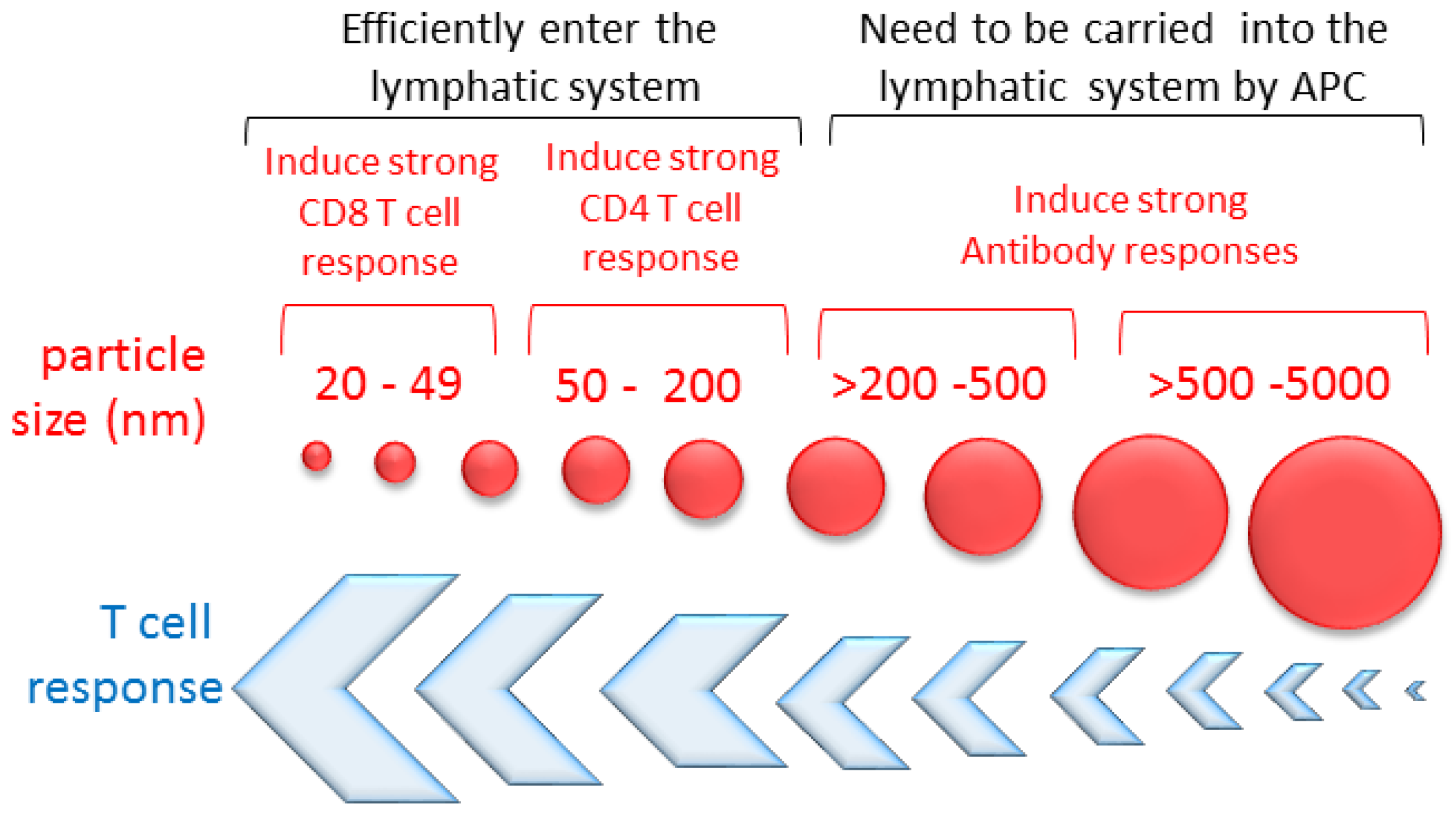
3. The Effect of the Particle Size
4. The Effect of Antigen Presenting Cells
5. The Route of Administration
6. The Particle Material
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pavia, A.T. Viral infections of the lower respiratory tract: Old viruses, new viruses, and the role of diagnosis. Clin. Infect. Dis. 2011, 52, S284–S289. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M. Progress and challenges in rsv prophylaxis and vaccine development. J. Infect. Dis. 2013, 208, S177–S183. [Google Scholar] [CrossRef] [PubMed]
- Habibi, M.S.; Patel, S.; Openshaw, P. Hot topics in the prevention of respiratory syncytial virus disease. Expert Rev. Vaccines 2011, 10, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Openshaw, P.J.; Tregoning, J.S. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin. Microbiol. Rev. 2005, 18, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; O′Hagan, D.T. Recent advances in vaccine adjuvants. Pharm. Res. 2002, 19, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D. Vaccine adjuvants: Why and how. Hum. Vaccines Immunother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, T.; Meijerhof, T.; Stegmann, T.; Lederhofer, J.; Wilschut, J.; de Haan, A. Immunogenicity and protective capacity of a virosomal respiratory syncytial virus vaccine adjuvanted with monophosphoryl lipid a in mice. PLoS ONE 2012, 7, e36812. [Google Scholar] [CrossRef] [PubMed]
- Frietze, K.M.; Peabody, D.S.; Chackerian, B. Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol. 2016, 18, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Lee, Y.T.; Kim, K.H.; Park, S.; Kwon, Y.M.; Lee, Y.; Ko, E.J.; Jung, Y.J.; Lee, J.S.; Kim, Y.J.; et al. Combined virus-like particle and fusion protein-encoding DNA vaccination of cotton rats induces protection against respiratory syncytial virus without causing vaccine-enhanced disease. Virology 2016, 494, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Boigard, H.; Bhatia, B.; Fallon, J.T.; Alimova, A.; Gottlieb, P.; Galarza, J.M. Novel respiratory syncytial virus-like particle vaccine composed of the postfusion and prefusion conformations of the f glycoprotein. Clin. Vaccine Immunol. 2016, 23, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, Y.T.; Hwang, H.S.; Kwon, Y.M.; Kim, M.C.; Ko, E.J.; Lee, J.S.; Lee, Y.; Kang, S.M. Virus-like particle vaccine containing the f protein of respiratory syncytial virus confers protection without pulmonary disease by modulating specific subsets of dendritic cells and effector t cells. J. Virol. 2015, 89, 11692–11705. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.X.; Xie, Y.; Ye, Y.P. Iscoms and iscomatrix. Vaccine 2009, 27, 4388–4401. [Google Scholar] [CrossRef] [PubMed]
- Drane, D.; Gittleson, C.; Boyle, J.; Maraskovsky, E. Iscomatrix adjuvant for prophylactic and therapeutic vaccines. Expert Rev. Vaccines 2007, 6, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, C.; Zuidema, J.; Crommelin, D.J.; Storm, G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid compostion and lipid dose. Biochim. Biophys. Acta 1997, 1328, 261–272. [Google Scholar] [CrossRef]
- Adair, B.M. Nanoparticle vaccines against respiratory viruses. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Bienenstock, J.; McDermott, M.R. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005, 206, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.Y.; Phipps, S. Regulation of inducible balt formation and contribution to immunity and pathology. Mucosal Immunol. 2010, 3, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, M.; Kimura, S.; Takahashi-Iwanaga, H.; Hisamoto, M.; Iwanaga, T.; Iida, J. Rankl regulates differentiation of microfold cells in mouse nasopharynx-associated lymphoid tissue (nalt). Cell Tissue Res. 2016, 364, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wagner, N.; Pham, L.N.; Magno, V.; Shan, Z.; Butcher, E.C.; Michie, S.A. Lymphocyte homing to bronchus-associated lymphoid tissue (balt) is mediated by l-selectin/pnad, alpha4beta1 integrin/vcam-1, and lfa-1 adhesion pathways. J. Exp. Med. 2003, 197, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Guy-Grand, D.; Griscelli, C.; Vassalli, P. The gut-associated lymphoid system: Nature and properties of the large dividing cells. Eur. J. Immunol. 1974, 4, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Takahashi, Y.; Yokoi, Y.; Ato, M.; Kodama, Y.; Hachimura, S.; Kurosaki, T.; Kobayashi, K. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. USA 2012, 109, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Viuff, B.; Tjornehoj, K.; Larsen, L.E.; Rontved, C.M.; Uttenthal, A.; Ronsholt, L.; Alexandersen, S. Replication and clearance of respiratory syncytial virus: Apoptosis is an important pathway of virus clearance after experimental infection with bovine respiratory syncytial virus. Am. J. Pathol. 2002, 161, 2195–2207. [Google Scholar] [CrossRef]
- Kirby, A.C.; Coles, M.C.; Kaye, P.M. Alveolar macrophages transport pathogens to lung draining lymph nodes. J. Immunol. 2009, 183, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Braciale, T.J.; Sun, J.; Kim, T.S. Regulating the adaptive immune response to respiratory virus infection. Nat. Rev. Immunol. 2012, 12, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Angelin-Duclos, C.; Cattoretti, G.; Lin, K.I.; Calame, K. Commitment of b lymphocytes to a plasma cell fate is associated with blimp-1 expression in vivo. J. Immunol. 2000, 165, 5462–5471. [Google Scholar] [CrossRef] [PubMed]
- Waffarn, E.E.; Baumgarth, N. Protective B cell responses to flu—No fluke! J. Immunol. 2011, 186, 3823–3829. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M.; Richard, Y. Marginal zone B-cells, a gatekeeper of innate immunity. Front. Immunol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Singleton, R.; Etchart, N.; Hou, S.; Hyland, L. Inability to evoke a long-lasting protective immune response to respiratory syncytial virus infection in mice correlates with ineffective nasal antibody responses. J. Virol. 2003, 77, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.; Alvarez, R.; Jones, L.P.; Henderson, C.; Anderson, L.J.; Tripp, R.A. Respiratory syncytial virus g protein and g protein cx3c motif adversely affect CX3CR1+ T cell responses. J. Immunol. 2006, 176, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Nidhi; Rashid, M.; Kaur, V.; Hallan, S.S.; Sharma, S.; Mishra, N. Microparticles as controlled drug delivery carrier for the treatment of ulcerative colitis: A brief review. Saudi Pharm. J. 2016, 24, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Benne, N.; van Duijn, J.; Kuiper, J.; Jiskoot, W.; Slutter, B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J. Control. Release 2016, 234, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Cayatte, C.; Guzman, B.; Schneider-Ohrum, K.; Matuszak, R.; Snell, A.; Rajani, G.M.; McCarthy, M.P.; Muralidhara, B. Impact of formulation and particle size on stability and immunogenicity of oil-in-water emulsion adjuvants. Hum. Vaccines Immunother. 2015, 11, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Mottram, P.L.; Leong, D.; Crimeen-Irwin, B.; Gloster, S.; Xiang, S.D.; Meanger, J.; Ghildyal, R.; Vardaxis, N.; Plebanski, M. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial virus. Mol. Pharm. 2007, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.; Wakatsuki, Y.; Tabata, Y.; Kawasaki, H.; Usui, T.; Yoshida, M.; Itoh, T.; Habu, S.; Kita, T. Oral immunization with size-purified microsphere beads as a vehicle selectively induces systemic tolerance and sensitization. Vaccine 2000, 19, 579–588. [Google Scholar] [CrossRef]
- Tabata, Y.; Inoue, Y.; Ikada, Y. Size effect on systemic and mucosal immune responses induced by oral administration of biodegradable microspheres. Vaccine 1996, 14, 1677–1685. [Google Scholar] [CrossRef]
- Huang, X.; Li, L.; Liu, T.; Hao, N.; Liu, H.; Chen, D.; Tang, F. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano 2011, 5, 5390–5399. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tiruppathi, C.; Cho, J.; Minshall, R.D.; Malik, A.B. Delivery of nanoparticle: Complexed drugs across the vascular endothelial barrier via caveolae. IUBMB Life 2011, 63, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L. Secrets of caveolae- and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim. Biophys. Acta 2005, 1746, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Qaddoumi, M.G.; Gukasyan, H.J.; Davda, J.; Labhasetwar, V.; Kim, K.J.; Lee, V.H. Clathrin and caveolin-1 expression in primary pigmented rabbit conjunctival epithelial cells: Role in plga nanoparticle endocytosis. Mol. Vis. 2003, 9, 559–568. [Google Scholar] [PubMed]
- Chakraborty, A.; Jana, N.R. Clathrin to lipid raft-endocytosis via controlled surface chemistry and efficient perinuclear targeting of nanoparticle. J. Phys. Chem. Lett. 2015, 6, 3688–3697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Roche, P.A. Macropinocytosis in phagocytes: Regulation of mhc class-II-restricted antigen presentation in dendritic cells. Front. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- ten Broeke, T.; Wubbolts, R.; Stoorvogel, W. Mhc class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Baleeiro, R.B.; Rietscher, R.; Diedrich, A.; Czaplewska, J.A.; Lehr, C.M.; Scherliess, R.; Hanefeld, A.; Gottschaldt, M.; Walden, P. Spatial separation of the processing and MHC class I loading compartments for cross-presentation of the tumor-associated antigen her2/neu by human dendritic cells. Oncoimmunology 2015. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Clark, K.; Benacerraf, B.; Rock, K.L. Efficient major histocompatibility complex class i presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA 1993, 90, 4942–4946. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Noh, Y.W.; Lim, Y.T. Polymer nanoparticles for cross-presentation of exogenous antigens and enhanced cytotoxic t-lymphocyte immune response. Int. J. Nanomed. 2016, 11, 3753–3764. [Google Scholar]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S. Antigen presentation: From cell biology to physiology. Immunol. Rev. 2016, 272, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Patino, T.; Soriano, J.; Barrios, L.; Ibanez, E.; Nogues, C. Surface modification of microparticles causes differential uptake responses in normal and tumoral human breast epithelial cells. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xiong, F.; He, J.; Dai, X.; Wang, G. Surface-functionalized, pH-responsive poly(lactic-co-glycolic acid)-based microparticles for intranasal vaccine delivery: Effect of surface modification with chitosan and mannan. Eur. J. Pharm. Biopharm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.N. The importance of injecting vaccines into muscle. Different patients need different needle sizes. Br. Med. J. 2000, 321, 1237–1238. [Google Scholar] [CrossRef]
- Habibi, M.S.; Jozwik, A.; Makris, S.; Dunning, J.; Paras, A.; DeVincenzo, J.P.; de Haan, C.A.; Wrammert, J.; Openshaw, P.J.; Chiu, C.; et al. Impaired antibody-mediated protection and defective iga b-cell memory in experimental infection of adults with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2015, 191, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Chandrudu, S.; Toth, I. Strategies for intranasal delivery of vaccines. Drug Deliv. Transl. Res. 2013, 3, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Mangal, S.; Khambete, H.; Tyagi, R.K. Mucosal delivery of vaccines: Role of mucoadhesive/biodegradable polymers. Recent Pat. Drug Deliv. Formul. 2010, 4, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, F.; Earley, B.; Cassidy, J.P.; Markey, B.; Doherty, S.; Welsh, M.D. Comparing the immune response to a novel intranasal nanoparticle PLGA vaccine and a commercial BPI3V vaccine in dairy calves. BMC Vet. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Firdous, J.; Islam, M.A.; Park, S.M.; Cheon, I.S.; Shim, B.S.; Yoon, H.S.; Song, M.; Chang, J.; Choi, Y.J.; Park, Y.M.; et al. Induction of long-term immunity against respiratory syncytial virus glycoprotein by an osmotic polymeric nanocarrier. Acta Biomater. 2014, 10, 4606–4617. [Google Scholar] [CrossRef] [PubMed]
- Kraan, H.; Vrieling, H.; Czerkinsky, C.; Jiskoot, W.; Kersten, G.; Amorij, J.P. Buccal and sublingual vaccine delivery. J. Control. Release 2014, 190, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Jalilian, F.A.; Yusoff, K.; Suhaimi, S.; Amini, R.; Sekawi, Z.; Jahanshiri, F. Development of two salmonella-based oral vaccines against human respiratory syncytial virus. J. Biol. Regul. Homeost. Agents 2015, 29, 7–18. [Google Scholar]
- Fu, Y.H.; Jiao, Y.Y.; He, J.S.; Giang, G.Y.; Zhang, W.; Yan, Y.F.; Ma, Y.; Hua, Y.; Zhang, Y.; Peng, X.L.; et al. Sublingual administration of a helper-dependent adenoviral vector expressing the codon-optimized soluble fusion glycoprotein of human respiratory syncytial virus elicits protective immunity in mice. Antiviral Res. 2014, 105, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tree, J.A.; Bembridge, G.; Hou, S.; Taylor, G.; Fashola-Stone, E.; Melero, J.; Cranage, M.P. An assessment of different DNA delivery systems for protection against respiratory syncytial virus infection in the murine model: Gene-gun delivery induces igg in the lung. Vaccine 2004, 22, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- de Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of cpg enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad. Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [PubMed]
- Bobbala, S.; Hook, S. Is there an optimal formulation and delivery strategy for subunit vaccines? Pharm. Res. 2016, 33, 2078–2097. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt Cda, S.; Silva, L.B.; Pereira, P.A.; Gelfuso, G.M.; Faccioli, L.H. Microspheres prepared with different co-polymers of poly (lactic-glycolic acid) (plga) or with chitosan cause distinct effects on macrophages. Colloids Surf. B 2015, 136, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An update on potential biomedical and pharmaceutical applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Boyapalle, S.; Wong, T.; Opoku-Nsiah, K.; Bedi, R.; Crannell, W.C.; Perry, A.F.; Nguyen, H.; Sampayo, V.; Devareddy, A.; et al. Mucosal delivery of a double-stapled rsv peptide prevents nasopulmonary infection. J. Clin. Investig. 2014, 124, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S. Mucosal gene expression vaccine: A novel vaccine strategy for respiratory syncytial virus. Pediatr. Infect. Dis. J. 2003, 22, S100–S103. [Google Scholar] [CrossRef] [PubMed]
- Boyoglu, S.; Vig, K.; Pillai, S.; Rangari, V.; Dennis, V.A.; Khazi, F.; Singh, S.R. Enhanced delivery and expression of a nanoencapsulated DNA vaccine vector for respiratory syncytial virus. Nanomedicine 2009, 5, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, E.; Tiwari, P.M.; Waffo, A.B.; Miller, M.E.; Vig, K.; Dennis, V.A.; Singh, S.R. A nonviral phema+chitosan nanosphere-mediated high-efficiency gene delivery system. Int. J. Nanomed. 2013, 8, 1403–1415. [Google Scholar]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; San Juan-Vergara, H.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal sirna nanoparticles targeting the viral ns1 gene. Nat. Med. 2005, 11, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Salatin, S.; Barar, J.; Barzegar-Jalali, M.; Adibkia, K.; Milani, M.A.; Jelvehgari, M. Hydrogel nanoparticles and nanocomposites for nasal drug/vaccine delivery. Arch. Pharm. Res. 2016, 39, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chattopadhyay, M.; Sen, K.K.; Saha, M.K. Development and characterization of alginate coated low molecular weight chitosan nanoparticles as new carriers for oral vaccine delivery in mice. Carbohydr. Polym. 2015, 121, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly (lactic-co-glycolic) acid (plga)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Helson, R.; Olszewska, W.; Singh, M.; Megede, J.Z.; Melero, J.A.; O′Hagan, D.; Openshaw, P.J. Polylactide-co-glycolide (plg) microparticles modify the immune response to DNA vaccination. Vaccine 2008, 26, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Power, U.F.; Robert, A.; Haeuw, J.F.; Helffer, K.; Perez, A.; Asin, M.A.; Corvaia, N.; Libon, C. The respiratory syncytial virus g protein conserved domain induces a persistent and protective antibody response in rodents. PLoS ONE 2012, 7, e34331. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, O.V.; Adair, B.M.; Welsh, M.; Earley, B. Immunogenetic responses in calves to intranasal delivery of bovine respiratory syncytial virus (brsv) epitopes encapsulated in poly (d,l-lactide-co-glycolide) microparticles. Res. Vet. Sci. 2013, 95, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Vallhov, H.; Gabrielsson, S.; Stromme, M.; Scheynius, A.; Garcia-Bennett, A.E. Mesoporous silica particles induce size dependent effects on human dendritic cells. Nano Lett. 2007, 7, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiang, H.; Zhao, Q.; Wang, S.; Zou, M.; Cheng, G. Enhanced mucosal and systemic immune responses obtained by porous silica nanoparticles used as an oral vaccine adjuvant: Effect of silica architecture on immunological properties. Int. J. Pharm. 2012, 436, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Mathaes, R.; Winter, G.; Engert, J. Encapsulation of antigen-loaded silica nanoparticles into microparticles for intradermal powder injection. Eur. J. Pharm. Sci. 2014, 63, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Francica, J.R.; Lynn, G.M.; Laga, R.; Joyce, M.G.; Ruckwardt, T.J.; Moribito, K.M.; Chen, M.; Chaudhuri, R.; Zhang, B.; Sastry, M.; et al. Thermo-responsive polymer nanoparticles co-deliver rsv f trimers with a TLR-7/8 adjuvant. Bioconjug. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Björnmalm, M.; Caruso, F. Assembly of layer-by-layer particles and their interactions with biological systems. Chem. Mater. 2014, 26, 452–460. [Google Scholar] [CrossRef]
- Jorquera, P.A.; Choi, Y.; Oakley, K.E.; Powell, T.J.; Boyd, J.G.; Palath, N.; Haynes, L.M.; Anderson, L.J.; Tripp, R.A. Nanoparticle vaccines encompassing the respiratory syncytial virus (RSV) G protein CX3C chemokine motif induce robust immunity protecting from challenge and disease. PLoS ONE 2013, 8, e74905. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.J.; Mistillis, M.; Palath, N.; Tang, J.; Jacobs, A.; Cardenas, W.; Boyd, J.; Prausnitz, M. Immunization with synthetic lbl microparticle vaccine administered using a microneedle patch elicits humoral and cellular immune responses and protects mice from challenge with respiratory syncytial virus. J. Immunol. 2016, 196, 76–78. [Google Scholar]
- Gu, H.; Li, T.; Han, L.; Zhu, P.; Zhang, P.; Zhang, S.; Sun, S.; Duan, Y.; Xing, L.; Zhao, Z.; et al. Protection conferred by virus-like particle vaccines against respiratory syncytial virus infection in mice by intranasal vaccination. Hum. Vaccines Immunother. 2015, 11, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Quan, F.S.; Kim, Y.; Lee, S.; Yi, H.; Kang, S.M.; Bozja, J.; Moore, M.L.; Compans, R.W. Viruslike particle vaccine induces protection against respiratory syncytial virus infection in mice. J. Infect. Dis. 2011, 204, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Cullen, L.M.; Blanco, J.C.; Morrison, T.G. Cotton rat immune responses to virus-like particles containing the pre-fusion form of respiratory syncytial virus fusion protein. J. Transl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, T.; Hurwitz, J.L.; Coleclough, C.; Prouser, C.; Krishnamurthy, S.; Zhan, X.; Boyd, K.; Scroggs, R.A.; Brown, B.; Nagai, Y.; et al. Recombinant sendai virus expressing the g glycoprotein of respiratory syncytial virus (rsv) elicits immune protection against rsv. J. Virol. 2004, 78, 6043–6047. [Google Scholar] [CrossRef] [PubMed]
- Walpita, P.; Johns, L.M.; Tandon, R.; Moore, M.L. Mammalian cell-derived respiratory syncytial virus-like particles protect the lower as well as the upper respiratory tract. PLoS ONE 2015, 10, e0130755. [Google Scholar] [CrossRef] [PubMed]
- Yusibov, V.; Mett, V.; Mett, V.; Davidson, C.; Musiychuk, K.; Gilliam, S.; Farese, A.; Macvittie, T.; Mann, D. Peptide-based candidate vaccine against respiratory syncytial virus. Vaccine 2005, 23, 2261–2265. [Google Scholar] [CrossRef] [PubMed]
- Yusibov, V.; Streatfield, S.J.; Kushnir, N.; Roy, G.; Padmanaban, A. Hybrid viral vectors for vaccine and antibody production in plants. Curr. Pharm. Des. 2013, 19, 5574–5586. [Google Scholar] [CrossRef] [PubMed]
- Schickli, J.H.; Whitacre, D.C.; Tang, R.S.; Kaur, J.; Lawlor, H.; Peters, C.J.; Jones, J.E.; Peterson, D.L.; McCarthy, M.P.; Van Nest, G.; et al. Palivizumab epitope-displaying virus-like particles protect rodents from rsv challenge. J. Clin. Investig. 2015, 125, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Van Braeckel-Budimir, N.; Haijema, B.J.; Leenhouts, K. Bacterium-like particles for efficient immune stimulation of existing vaccines and new subunit vaccines in mucosal applications. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zhang, Y.; Chai, F.; Tan, Y.; Huo, C.; Pan, Z. Chimeric virus-like particles containing a conserved region of the g protein in combination with a single peptide of the M2 protein confer protection against respiratory syncytial virus infection. Antiviral Res. 2016, 131, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Glenn, G.M.; Fries, L.F.; Thomas, D.N.; Smith, G.; Kpamegan, E.; Lu, H.; Flyer, D.; Jani, D.; Hickman, S.P.; Piedra, P.A. A randomized, blinded, controlled, dose-ranging study of a respiratory syncytial virus recombinant fusion (f) nanoparticle vaccine in healthy women of childbearing age. J. Infect. Dis. 2016, 213, 411–422. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorquera, P.A.; Tripp, R.A. Synthetic Biodegradable Microparticle and Nanoparticle Vaccines against the Respiratory Syncytial Virus. Vaccines 2016, 4, 45. https://doi.org/10.3390/vaccines4040045
Jorquera PA, Tripp RA. Synthetic Biodegradable Microparticle and Nanoparticle Vaccines against the Respiratory Syncytial Virus. Vaccines. 2016; 4(4):45. https://doi.org/10.3390/vaccines4040045
Chicago/Turabian StyleJorquera, Patricia A., and Ralph A. Tripp. 2016. "Synthetic Biodegradable Microparticle and Nanoparticle Vaccines against the Respiratory Syncytial Virus" Vaccines 4, no. 4: 45. https://doi.org/10.3390/vaccines4040045
APA StyleJorquera, P. A., & Tripp, R. A. (2016). Synthetic Biodegradable Microparticle and Nanoparticle Vaccines against the Respiratory Syncytial Virus. Vaccines, 4(4), 45. https://doi.org/10.3390/vaccines4040045