
The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy

Abstract
:1. Introduction
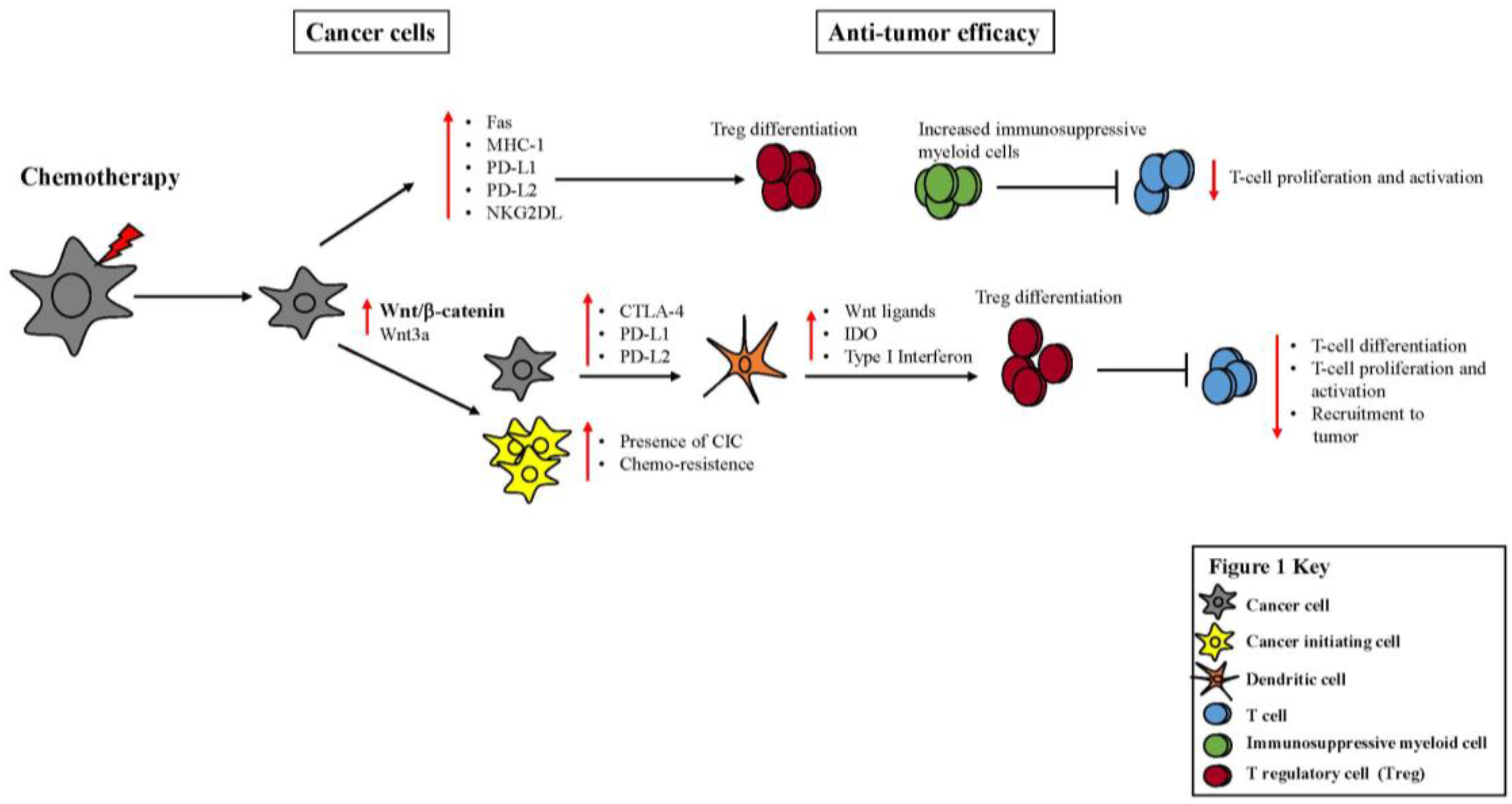
2. Impact of Chemotherapy on Anti-Tumor Immunity and Cancer Cell Immunogenicity
2.1. Chemotherapy Induces Immunogenic Cell Death and Potentiates the Immune Response through Upregulation of MHC, Tumor-Associated Antigens and Co-Stimulatory Ligands on Immune Cells
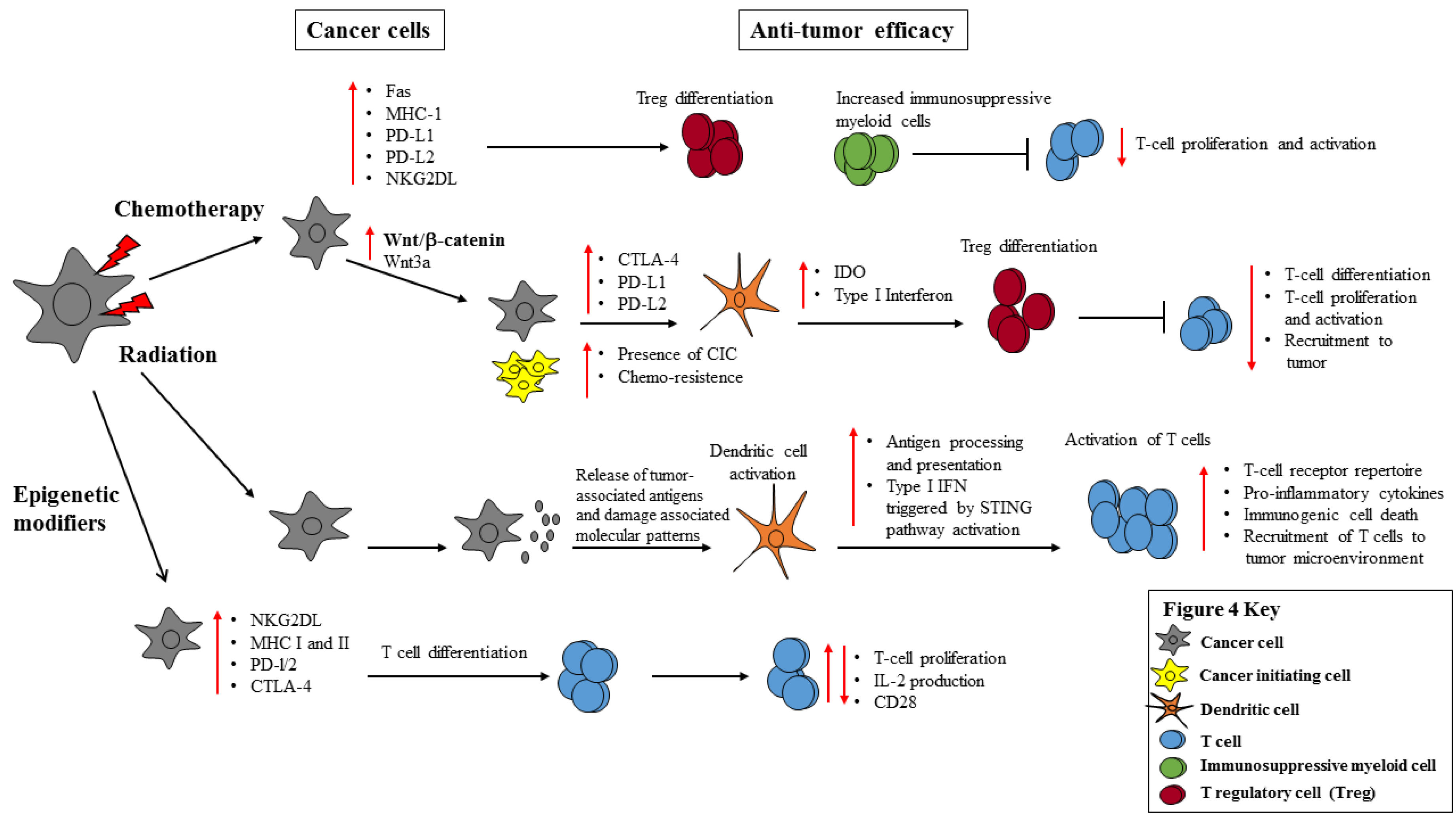
2.2. Chemotherapy Induces the Expression of Inhibitory Checkpoint Molecules, NKG2D Ligands and Pathways
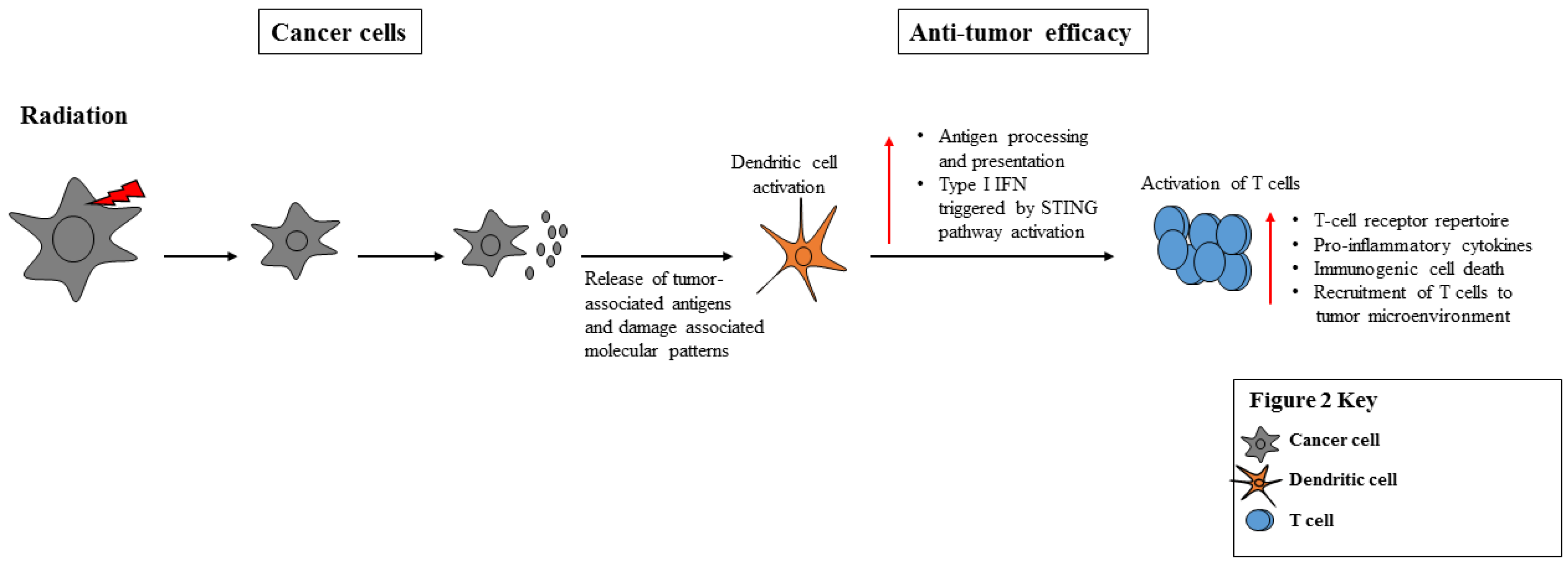
3. Impact of Radiation and Wnt Signaling on Anti-Tumor Immunity and Cancer Cell Immunogenicity
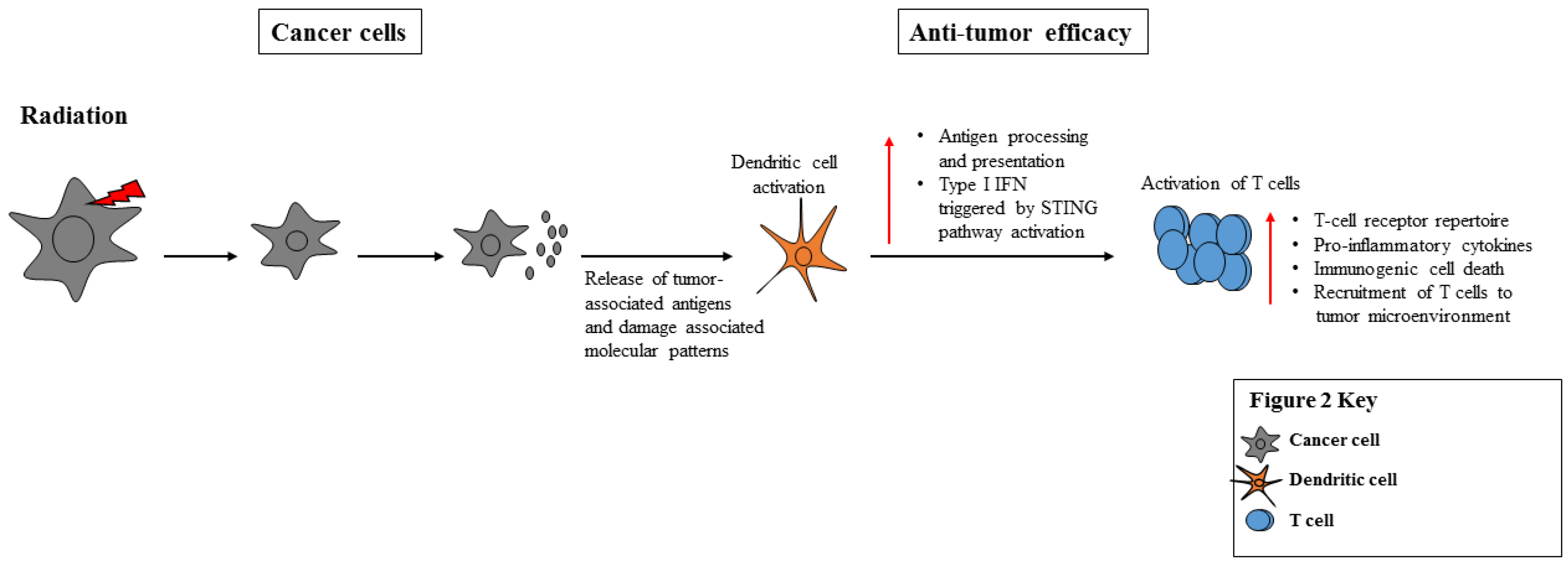
3.1. Radiation Stimulates the Immune System through ICD, Release of TAAs, TLR Engagement and APC Activation
3.2. Wnt Signaling Activates or Inhibits the Immune System by Regulating the Expression of CD137 or IDO
3.3. The Impact of Radiation on Cancer Cell Immunogenicity
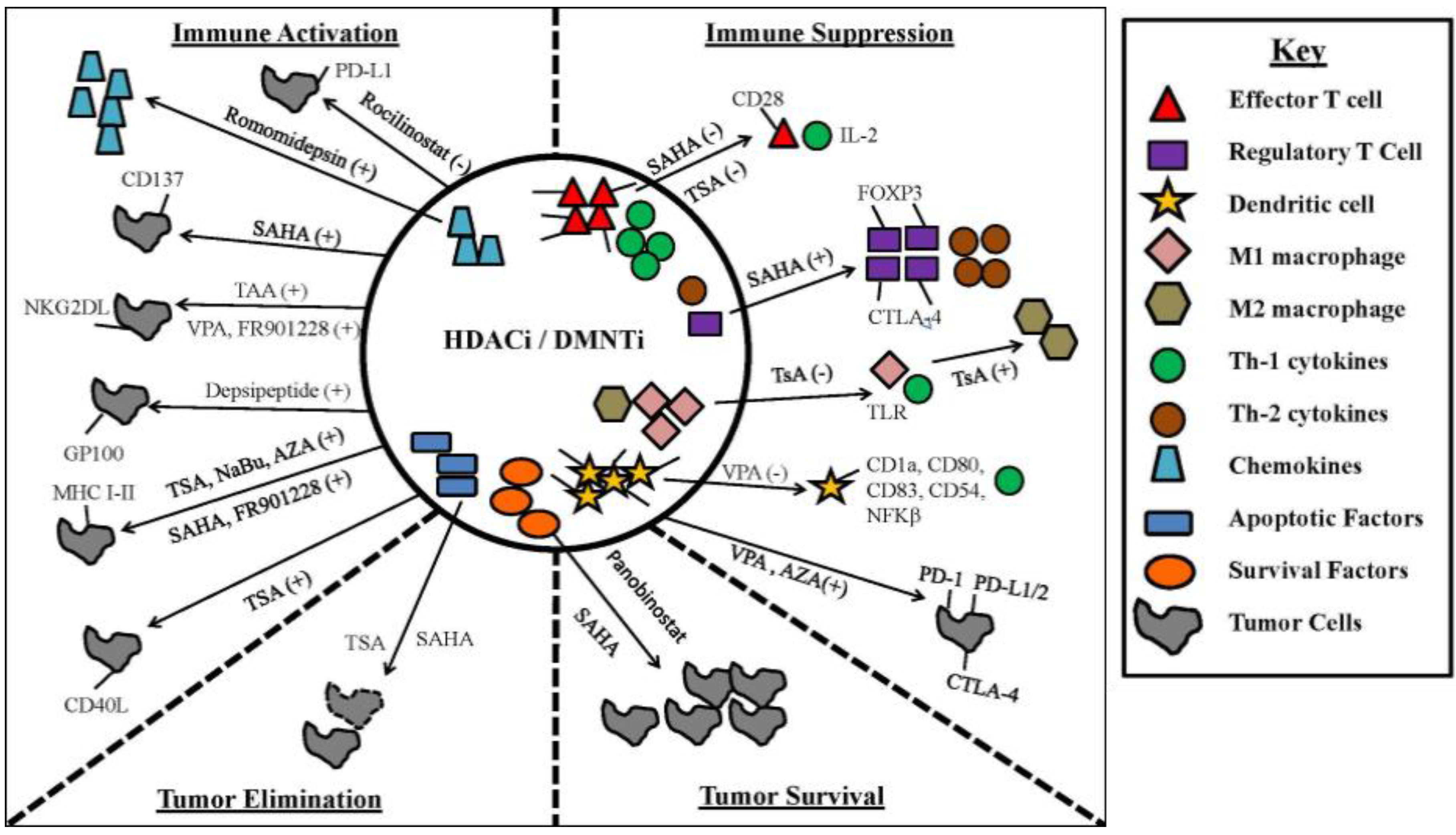
4. Impact of Epigenetic Modifiers on Anti-Tumor Immunity, Cancer Cell Immunogenicity and Inhibitory Ligand Expression
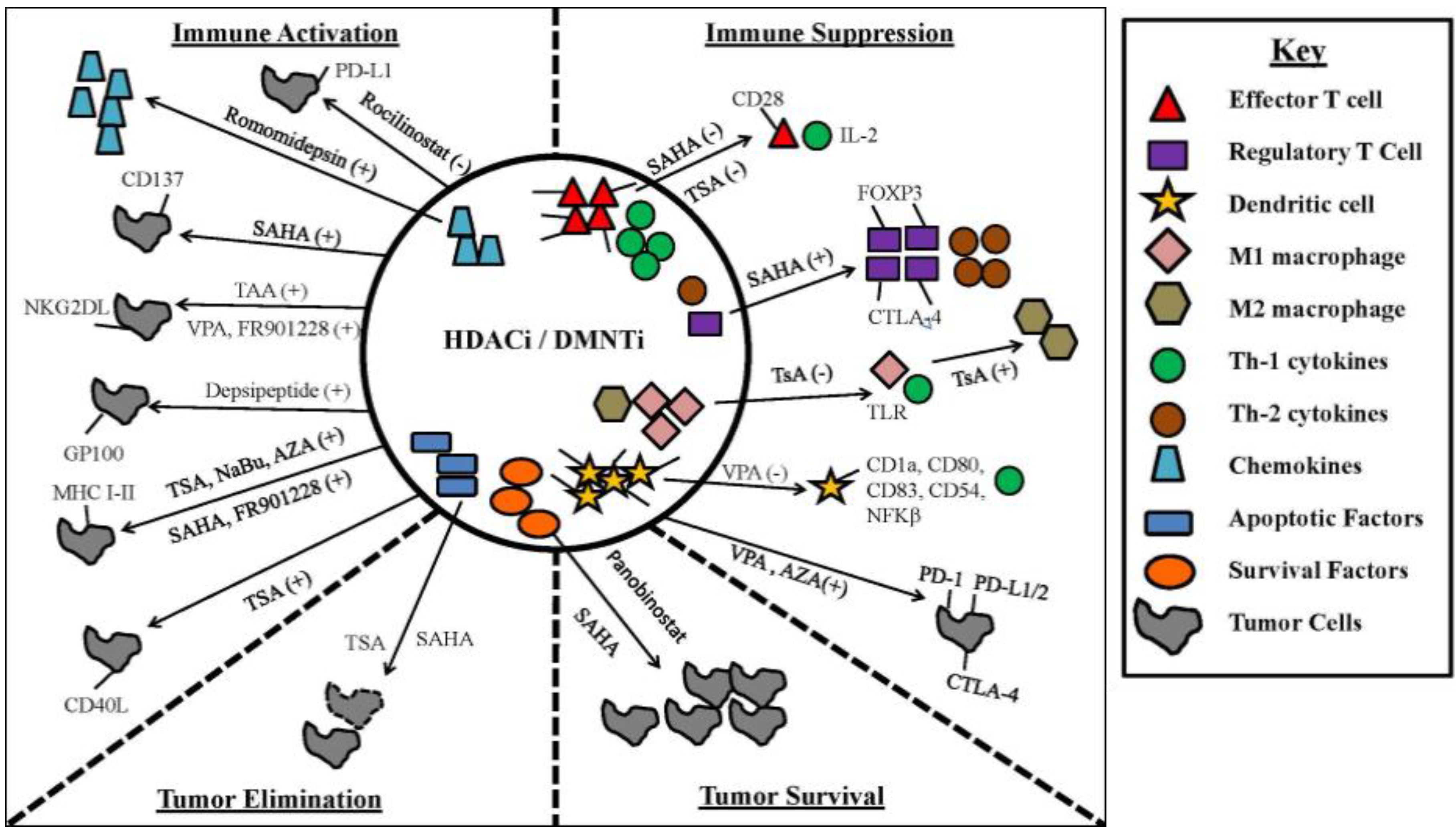
4.1. Epigenetic Modifiers Potentiate the Immune Response through TLR Engagement, TAA and Co-Stimulatory Molecules on Immune Cells
4.2. Epigenetic Modifiers Induce Tumor Expression of Inhibitory Immune Checkpoint Molecules
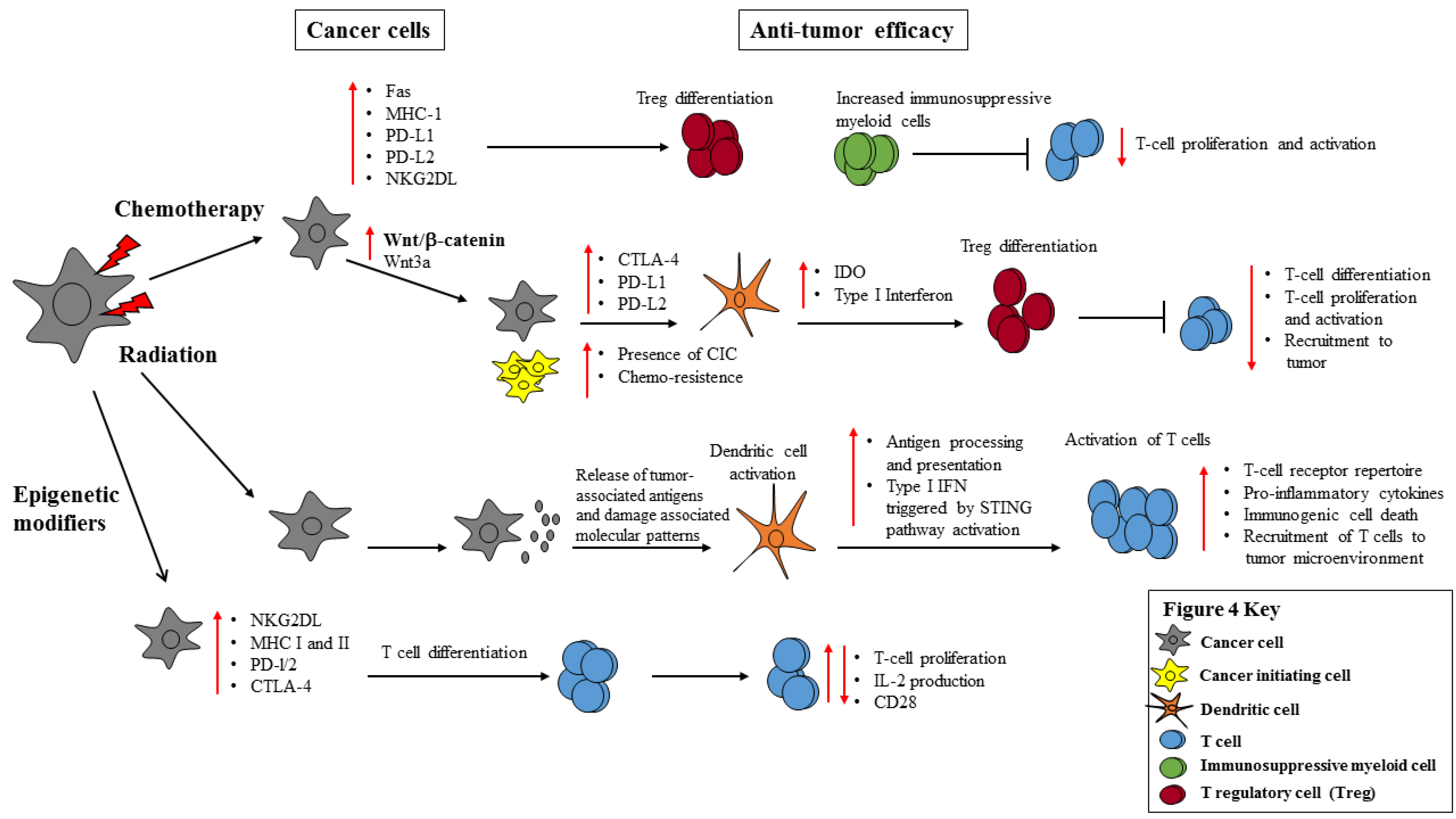
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cell Transfer |
| APCs | Antigen-Presenting Cells |
| AZA | Azacitidine |
| cGAS | Cyclic-GMP-AMP Synthase |
| CICs | Cancer-Initiating Cells |
| CRT | Calreticulin |
| DAMPs | Damage-Associated Molecular Patterns |
| DC | Dendritic Cell |
| DNMTi | DNA Methyltransferase Inhibitor |
| GSK-3 | Glycogen Synthase Kinase-3 |
| HDACs | Histone Deacetylases |
| HDACi | Histone Deacetylase Inhibitor |
| HMGB1 | High-Mobility Group Protein B1 |
| HSP | Heat Shock Proteins |
| ICD | Immunogenic Cell Death |
| IDO | Indolamine 2,3-Dioxygenase |
| IRF3 | Interferon Regulatory Factor 3 |
| MSI | Microsatellite Instability |
| PLD | PEGylated Liposomal Doxorubicin |
| PRRs | Pattern Recognition Receptors |
| SAHA | Suberanilohydroxamic Acid |
| STING | Stimulator of Interferon Genes |
| TAA | Tumor-Associated Antigens |
| TCGA | The Cancer Genome Atlas |
| TCR | T Cell Receptor |
| TIL | Tumor-Infiltrating Lymphocytes |
| TLR | Toll-Like Receptor |
| Tregs | T Regulatory Cells |
| TsA | Trichostatin A |
| TSCM | Stem Memory T Cell |
| VPA | Valproic Acid |
References
- Burton, A.L.; Roach, B.A.; Mays, M.P.; Chen, A.F.; Ginter, B.A.; Vierling, A.M.; Scoggins, C.R.; Martin, R.C.; Stromberg, A.J.; Hagendoorn, L.; et al. Prognostic significance of tumor infiltrating lymphocytes in melanoma. Am. Surg. 2011, 77, 188–192. [Google Scholar] [PubMed]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02–98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human melanoma. Available online: http://cancerimmunolres.aacrjournals.org/content/canimmarch/9/1/3.short (accessed on 5 August 2016).
- Salgado, R.; Denkert, C.; Campbell, C.; Savas, P.; Nuciforo, P.; Aura, C.; de Azambuja, E.; Eidtmann, H.; Ellis, C.E.; Baselga, J.; et al. Tumor-Infiltrating Lymphocytes and Associations With Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated With Lapatinib and Trastuzumab: A Secondary Analysis of the NeoALTTO Trial. JAMA Oncol. 2015, 1, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in head and neck cancers. Available online: http://archive.cancerimmunity.org/v8p16/081043.htm (accessed on 31 July 2016).
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, suppressors and antigen presenters: How lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Vacchelli, E.; Aranda, F.; Castoldi, F.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Fucikova, J.; Galon, J.; Spisek, R. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Galluzzi, L.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Kroemer, G. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; Johnston, B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: Impact on clinical studies and considerations for combined therapies. Oncotarget 2015, 6, 41600–41619. [Google Scholar] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Kimura, Y.; Latz, E. Inflammasome activation in response to dead cells and their metabolites. Curr. Opin. Immunol. 2014, 30, 91–98. [Google Scholar] [PubMed]
- Rock, K.L.; Lai, J.J.; Kono, H. Innate and adaptive immune responses to cell death. Immunol. Rev. 2011, 243, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; Reis e Sousa, C. Adaptive immunity after cell death. Trends Immunol. 2013, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Aymeric, L.; Locher, C.; Mattarollo, S.R.; Delahaye, N.F.; Pereira, P.; Boucontet, L.; Apetoh, L.; Ghiringhelli, F.; Casares, N. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J. Exp. Med. 2011, 208, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Séror, C.; Métivier, D.; Perfettini, J.L.; Zitvogel, L. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009, 8, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent B cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D′Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8 T cell responses and immune memory. Oncoimmunology 2015. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS ONE 2012, 7, e31732. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Hsu, Y.T.; Wu, C.C.; Yang, Y.C.; Wang, C.; Wu, T.C.; Hung, C.F. Immune mechanism of the antitumor effects generated by bortezomib. J. Immunol. 2012, 189, 3209–3220. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Santori, F.R.; Ng, B.; Liebes, L.; Formenti, S.C.; Vukmanovic, S. Select forms of tumor cell apoptosis induce dendritic cell maturation. J. Leukocyte Biol. 2005, 77, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, H.; Bruchard, M.; Berger, H.; Derangère, V.; Odoul, L.; Euvrard, R.; Ladoire, S.; Chalmin, F.; Végran, F.; Rébé, C.; et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS ONE 2013, 8, e65181. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, Y.; Zhou, Q.; Weiss, J.M.; Howard, O.Z.; McPherson, J.M.; Wakefield, L.M.; Oppenheim, J.J. Effective chemoimmunotherapy with anti-TGFbeta antibody and cyclophosphamide in a mouse model of breast cancer. PLoS ONE 2014, 9, e85398. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell. Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Munster, P.N. Epigenetic modifiers in immunotherapy: A focus on checkpoint inhibitors. Immunotherapy 2016, 8, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. How the immune system works to protect the host from infection: A personal view. Proc. Natl. Acad. Sci. USA 2001, 98, 7461–7468. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Castro-Vega, I.; Redondo, M. Role of gene methylation in antitumor immune response: Implication for tumor progression. Cancers 2011, 3, 1672–1690. [Google Scholar] [CrossRef] [PubMed]
- Pellicciotta, I.; Yang, C.P.H.; Goldberg, G.L.; Shahabi, S. Epothilone B enhances Class. I HLA and HLA-A2 surface molecule expression in ovarian cancer cells. Gynecol Oncol. 2011, 122, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fu, L. The effect of chemotherapy on programmed cell death 1/programmed cell death 1 ligand axis: Some chemotherapeutical drugs may finally work through immune response. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Schutsky, K.; Powell, Jr.; Daniel, J.; (Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA). Unpublished observations: Chemotherapy and HDACi induce PD-L1 and Class I expression on ovarian cancer tumor cells. 2016. [Google Scholar]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Gou, H.L.; Huang, J.; Shi, H.S.; Chen, X.C.; Wang, Y.S. Chemo-immunotherapy with oxaliplatin and interleukin-7 inhibits colon cancer metastasis in mice. PLoS ONE 2014, 9, e85789. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Kadima, A.N.; El-Naggar, S.A.; Rubinstein, M.P.; Chen, Y.; Gillanders, W.E.; Cole, D.J. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T cell response to peptide vaccination: Creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J. Immunother. 2007, 30, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Skov, S.; Pedersen, M.T.; Andresen, L.; Straten, P.T.; Woetmann, A.; Odum, N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005, 65, 11136–11145. [Google Scholar] [PubMed]
- Prlic, M.; Lefrancois, L.; Jameson, S.C. Multiple choices: Regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J. Exp. Med. 2002, 195, F49–F52. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ernst, B.; Kieper, W.C.; LeRoy, E.; Sprent, J.; Surh, C.D. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 2002, 195, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015, 36, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z.; et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Investig. 2007, 117, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Wallen, H.; Thompson, J.A.; Reilly, J.Z.; Rodmyre, R.M.; Cao, J.; Yee, C. Fludarabine modulates immune response and extends in vivo survival of adoptively transferred CD8 T cells in patients with metastatic melanoma. PLoS ONE 2009, 4, e4749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, Y.; Li, J.; Yang, F.; Wu, H.; Dai, F.; Hu, M.; Lu, X.; Peng, Y.; Liu, M.; et al. Antitumor action of a novel histone deacetylase inhibitor, YF479, in breast cancer. Neoplasia 2014, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Wargo, J.A.; Robbins, P.F.; Li, Y.; Zhao, Y.; El-Gamil, M.; Caragacianu, D.; Zheng, Z.; Hong, J.A.; Downey, S.; Schrump, D.S.; et al. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol. Immunother. 2009, 58, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, A.J.; West, A.; Banks, K.M.; Haynes, N.M.; Teng, M.W.; Smyth, M.J.; Johnstone, R.W. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Mobus, V.J.; Asphal, W.; Knapstein, P.G.; Kreienberg, R. Effects of interferon gamma on the proliferation and modulation of cell-surface structures of human ovarian carcinoma cell lines. J. Cancer Res. Clin. Oncol. 1993, 120, 27–34. [Google Scholar] [PubMed]
- Xu, X.; Rao, G.S.; Groh, V.; Spies, T.; Gattuso, P.; Kaufman, H.L.; Plate, J.; Prinz, R.A. Major histocompatibility complex class I-related chain A/B (MICA/B) expression in tumor tissue and serum of pancreatic cancer: Role of uric acid accumulation in gemcitabine-induced MICA/B expression. BMC Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Bae, J.H.; Lee, S.H.; Lee, E.Y.; Chung, B.S.; Kim, S.H.; Kang, C.D. Induction of NKG2D ligands and subsequent enhancement of NK cell-mediated lysis of cancer cells by arsenic trioxide. J. Immunother. 2008, 31, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Ghosh, D.; Gujja, S. Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages. Vaccines 2015, 3, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic cell. Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Efimova, E.V.; Hamzeh, K.W.; Darga, T.E.; Mauceri, H.J.; Fu, Y.X.; Kron, S.J.; Weichselbaum, R.R. Radiation-inducible immunotherapy for cancer: Senescent tumor cells as a cancer vaccine. Mol. Ther. 2012, 20, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Zhang, L.; Yang, Y.; Zuo, W.; Bi, Y.; Gao, W.; Deng, B.; Sun, J.; Shao, Q.; Qu, X. New insights of CTLA-4 into its biological function in breast cancer. Curr. Cancer Drug Targets 2010, 10, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Pistillo, M.P.; Tazzari, P.L.; Palmisano, G.L.; Pierri, I.; Bolognesi, A.; Ferlito, F.; Capanni, P.; Polito, L.; Ratta, M.; Pileri, S.; et al. CTLA-4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood 2003, 101, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Lu, X.; Yu, M.; Lemos, H.; Huang, L.; Chandler, P.; Liu, K.; Walters, M.; Krasinski, A.; Mack, M.; et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014, 74, 3441–3453. [Google Scholar] [CrossRef] [PubMed]
- Onyema, O.O.; Decoster, L.; Njemini, R.; Forti, L.N.; Bautmans, I.; De Waele, M.; Mets, T. Chemotherapy-induced changes and immunosenescence of CD8+ T cells in patients with breast cancer. Anticancer Res. 2015, 35, 1481–1489. [Google Scholar] [PubMed]
- Hwang, W.T.; Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Alagkiozidis, I.; Alagkiozidis, I.; Facciabene, A.; Tsiatas, M.; Carpenito, C.; Benencia, F.; Adams, S.; Jonak, Z.; June, C.H.; Powell, D.J., Jr.; et al. Time-dependent cytotoxic drugs selectively cooperate with IL-18 for cancer chemo-immunotherapy. J. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, F.; Simpkins, F.; Flores, A.; Chu, C.; Berek, J.S.; Lucci, J., 3rd; Murray, S.; Bauman, J.; Struemper, H.; Germaschewski, F.; et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: A phase I dose-escalation study. Cancer Immunol. Res. 2013, 1, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, D.M.; Liang, M.; Fu, J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol. Immunol. 2008, 45, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wang, H.; Meng, F.; Li, J.; Zhang, S. Combined Trabectedin and anti-PD1 antibody produces a synergistic antitumor effect in a murine model of ovarian cancer. J. Transl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Song, Q.; Lu, X.; Gong, W.; Zhao, J.; Min, P.; Yi, X. Paclitaxel induced B7-H1 expression in cancer cells via the MAPK pathway. J. Chemother. 2011, 23, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin induces programmed death-1-ligand 1(PD-L1) over-expression in hepatoma H22 cells via Erk /MAPK signaling pathway. Cell Mol. Biol. 2010, 56, OL1366–OL1372. [Google Scholar] [PubMed]
- Wu, L.; Chen, Z.; Zhang, J.; Xing, Y. Effect of miR-513a-5p on etoposide-stimulating B7-H1 expression in retinoblastoma cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, L.; Zou, L.; Yang, P.; Wu, R.; Mao, Y.; Zhou, H.; Li, R.; Wang, K.; Wang, W.; et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum. Immunol. 2014, 75, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, F.; Mao, Y.; Zhou, H.; Sun, J.; Li, R.; Liu, C.; Chen, W.; Hua, D.; Zhang, X. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum. Genet. 2013, 132, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Rodic, N.; Anders, R.A.; Eshleman, J.R.; Lin, M.T.; Xu, H.; Kim, J.H.; Beierl, K.; Chen, S.; Luber, B.S.; Wang, H.; et al. PD-L1 expression in melanocytic lesions does not correlate with the BRAF V600E mutation. Cancer Immunol. Res. 2015, 3, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.; Drake, C. Immunotherapy earns its spot in the ranks of cancer therapy. J. Exp. Med. 2012, 209, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Leggatt, G.R.; Gabrielli, B. Histone deacetylase inhibitors in the generation of the anti-tumour immune response. Immunol. Cell Biol. 2012, 90, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Khallouf, H.; Märten, A.; Serba, S.; Teichgräber, V.; Büchler, M.W.; Jäger, D.; Schmidt, J. 5-Fluorouracil and interferon-alpha immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J. Immunother. 2012, 35, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/beta-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, A.B.; Joseph, P.; Kovalenko, O.; Singh, S.; Armstrong, A.; Redline, R.; Resnick, K.; Zanotti, K.; Waggoner, S.; DiFeo, A. Critical role of Wnt/beta-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 2015, 6, 23720–23734. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.V.; Chien, A.J.; Yee, C.; Moon, R.T. CTLA-4 is a direct target of Wnt/beta-catenin signaling and is expressed in human melanoma tumors. J. Investig. Dermatol. 2008, 128, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Pilones, K.A.; Vanpouille-Box, C.; Demaria, S. Combination of radiotherapy and immune checkpoint inhibitors. Semin. Radiat. Oncol. 2015, 25, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Pilones, K.; Emerson, R.; Formenti, S.; Robins, H.; Demaria, S. Unique changes in the TCR repertoire of tumorinfiltrating lymphocytes underlie the synergy of radiotherapy with CTLA-4 blockade. J. Immunother. Cancer 2014. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Palmisano, G.L.; Martelli, A.M.; Kato, T.; Tazzari, P.L.; Pierri, I.; Clavio, M.; Dozin, B.; Balbi, G.; Megna, M.; et al. CTLA-4 expressed by chemoresistant, as well as untreated, myeloid leukaemia cells can be targeted with ligands to induce apoptosis. Br. J. Haematol. 2007, 136, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Queirolo, P.; Boero, S.; Salvi, S.; Piccioli, P.; Boccardo, S.; Minghelli, S.; Morabito, A.; Fontana, V.; Pietra, G.; et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J. Transl. Med. 2013. [Google Scholar] [CrossRef]
- Zamarin, D.; Postow, M.A. Immune checkpoint modulation: Rational design of combination strategies. Pharmacol. Ther. 2015, 150, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Asea, A. Radiation-induced effects and the immune system in cancer. Front. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Yee, C.; Lee, K.M. The effect of radiation on the immune response to cancers. Int. J. Mol. Sci. 2014, 15, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Immunotherapy of cancer with 4–1BB. Mol. Cancer Ther. 2012, 11, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.A.; Sarnaik, A.A.; Chen, J.Q.; Creasy, C.; Kale, C.; Robinson, J.; Weber, J.; Hwu, P.; Pilon-Thomas, S.; Radvanyi, L. Manipulating the tumor microenvironment ex vivo for enhanced expansion of tumor-infiltrating lymphocytes for adoptive cell therapy. Clin. Cancer Res. 2015, 21, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.A.; Wu, R.C.; Sukhumalchandra, P.; Molldrem, J.J.; Sarnaik, A.; Pilon-Thomas, S.; Weber, J.; Hwu, P.; Radvanyi, L. Co-stimulation through 4–1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T cell therapy. PLoS ONE 2013, 8, e60031. [Google Scholar] [CrossRef] [PubMed]
- Lee do, Y.; Choi, B.K.; Lee, D.G.; Kim, Y.H.; Kim, C.H.; Lee, S.J.; Kwon, B.S. 4–1BB signaling activates the T cell factor 1 effector/beta-catenin pathway with delayed kinetics via ERK signaling and delayed PI3K/AKT activation to promote the proliferation of CD8+ T Cells. PLoS ONE 2013, 8, e69677. [Google Scholar]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Pharmacologic induction of CD8+ T cell memory: Better living through chemistry. Sci. Transl. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V. Tumors STING adaptive antitumor immunity. Immunity 2014, 41, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Lugrin, J.; Le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Beier, U.H.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases and T cell immune responses. Blood 2012, 119, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Ge, G.; Golovina, T.; Mikheeva, T.; Wang, L.; Riley, J.L.; Hancock, W.W. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin. Immunol. 2010, 136, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Edens, R.E.; Dagtas, S.; Gilbert, K.M. Histone deacetylase inhibitors induce antigen specific anergy in lymphocytes: A comparative study. Int. Immunopharmacol. 2006, 6, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- .Matsuoka, H.; Fujimura, T.; Hayashi, M.; Matsuda, K.; Ishii, Y.; Aramori, I.; Mutoh, S. Disruption of HDAC4/N-CoR complex by histone deacetylase inhibitors leads to inhibition of IL-2 gene expression. Biochem. Pharmacol. 2007, 74, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Fujimura, T.; Mori, H.; Aramori, I.; Mutoh, S. Mechanism of HDAC inhibitor FR235222-mediated IL-2 transcriptional repression in Jurkat cells. Int. Immunopharmacol. 2007, 7, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Karagiannis, T.C. Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Beck, J.; Werth, D.; Grünebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin. Cancer Res. 2007, 13, 3933–3941. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, F.; Chipoy, C. Histone deacetylase inhibitors: New drugs for the treatment of inflammatory diseases? Drug Discov. Today 2005, 10, 197–204. [Google Scholar] [CrossRef]
- Heninger, E.; Krueger, T.E.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015. [Google Scholar] [CrossRef]
- Sun, N.; Zang, W.; Li, W. Bioinformatics analysis reveals potential candidate drugs for psychological stress in ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1362–1366. [Google Scholar] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC inhibitors enhance T cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Mattarollo, S.R.; Shortt, J.; Cluse, L.A.; Christiansen, A.J.; Smyth, M.J.; Johnstone, R.W. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013, 73, 7265–7276. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Liapis, V.; Bouralexis, S.; Welldon, K.; Hay, S.; Thai le, M.; Labrinidis, A.; Tilley, W.D.; Findlay, D.M.; Evdokiou, A. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int. J. Cancer 2006, 119, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Buttitta, G.; Emanuele, S.; di Fiore, R.; Martinez, R.; Rolfo, C.; Vento, R.; Tesoriere, G. The synergistic effect of SAHA and parthenolide in MDA-MB231 breast cancer cells. J. Cell Physiol. 2015, 230, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Pardee, A.B. Suberoylanilide hydroxamic acid as a potential therapeutic agent for human breast cancer treatment. Mol. Med. 2000, 6, 849–866. [Google Scholar] [PubMed]
- Bellarosa, D.; Bressan, A.; Bigioni, M.; Parlani, M.; Maggi, C.A.; Binaschi, M. SAHA/Vorinostat induces the expression of the CD137 receptor/ligand system and enhances apoptosis mediated by soluble CD137 receptor in a human breast cancer cell line. Int. J. Oncol. 2012, 41, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Sikorski, R.; Sampath, P.; Thorne, S.H. Modulation of NKG2D-ligand cell surface expression enhances immune cell therapy of cancer. J. Immunother. 2011, 34, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Magner, W.J.; Tomasi, T.B. An epigenetic vaccine model active in the prevention and treatment of melanoma. J. Transl. Med. 2007. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Tomasi, T.B. Histone deacetylase regulation of immune gene expression in tumor cells. Immunol. Res. 2008, 40, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Sato, A.; Chun, N.A.; Hara, M.; Naito, Y.; Kobayashi, Y.; Kano, Y.; Ohtsuki, M.; Furukawa, Y.; Kobayashi, E. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J. Investig. Dermatol. 2008, 128, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 2000, 165, 7017–7024. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Smyth, M.J.; Johnstone, R.W. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology 2014. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, J.; Gressmann, S.; Becker, S.; Wittig, S.; Schmudde, M.; Beck, J.F. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother. Pharmacol. 2010, 66, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Gueugnon, F.; Boutin, B.; Guillot, F.; Blanquart, C.; Rogel, A.; Padieu, M.; Pouliquen, D.; Fonteneau, J.F.; Grégoire, M. A 5-aza-2'-deoxycytidine/valproate combination induces cytotoxic T cell response against mesothelioma. Eur. Respir. J. 2011, 38, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Wachowska, M.; Gabrysiak, M.; Muchowicz, A.; Bednarek, W.; Barankiewicz, J.; Rygiel, T.; Boon, L.; Mroz, P.; Hamblin, M.R.; Golab, J. 5-Aza-2'-deoxycytidine potentiates antitumour immune response induced by photodynamic therapy. Eur. J. Cancer 2014, 50, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: Surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Lyberi, M.; Chatzilymperis, G.; Nezos, A.; Kamper, E. CD40/CD40L signaling and its implication in health and disease. Biofactors 2009, 35, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell. Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Chueh, A.C.; Tse, J.W.; Tögel, L.; Mariadason, J.M. Mechanisms of Histone Deacetylase Inhibitor-Regulated Gene Expression in Cancer Cells. Antioxid. Redox Signal. 2015, 23, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Lehe, C.; Barhoush, E.; Al-Romaih, K.; Tulbah, A.; Al-Alwan, M.; Hendrayani, S.F.; Manogaran, P.; Alaiya, A.; Al-Tweigeri, T.; et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Punt, C.J.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.; Nierkens, S.; Schreibelt, G.; de Boer, A.; Van Herpen, C.M.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.J.; Maharaj, K.K.; Sahakian, E.; Xing, L.; PerezVillarroel, P.; Knox, T.; Quayle, S.; Jones, S.S.; Villagra, A.; Sotomayor, E.M.; et al. Histone deacetylase 6 (HDAC6) as a regulator of immune checkpoint molecules in chronic lymphocytic leukemia (CLL). Blood 2014, 124, 3311. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. Cancer Genome Atlas Research Network The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [PubMed]
- Sigalotti, L.; Coral, S.; Fratta, E.; Lamaj, E.; Danielli, R.; Di Giacomo, A.M.; Altomonte, M.; Maio, M. Epigenetic modulation of solid tumors as a novel approach for cancer immunotherapy. Semin. Oncol. 2005, 32, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Simova, J.; Polláková, V.; Indrová, M.; Mikyšková, R.; Bieblová, J.; Stěpánek, I.; Bubeník, J.; Reiniš, M. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br. J. Cancer 2011, 105, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.; Indrova, M.; Lubyova, B.; Pribylova, H.; Bieblova, J.; Hejnar, J.; Simova, J.; Jandlova, T.; Bubenik, J.; Reinis, M. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology 2008, 123, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Tanzarella, S.; Lionello, I.; Mendez, R.; Traversari, C.; Ruiz-Cabello, F.; Garrido, F. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2′-deoxycytidine treatment. Int. J. Cancer 2001, 94, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Fonsatti, E.; Nicolay, H.J.; Sigalotti, L.; Calabrò, L.; Pezzani, L.; Colizzi, F.; Altomonte, M.; Guidoboni, M.; Marincola, F.M.; Maio, M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2′-deoxycytidine in cutaneous melanoma: Immunotherapeutic implications. Clin. Cancer Res. 2007, 13, 3333–3338. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I. Examples of Agents Which Induce ICD | Cancers | Triggers | CRT | ATP | HMGb1 | Type I IFNs | ICD | Immune Impact | Ref. |
| Anthracyclines | Breast, Colon, Sarcoma, Leukemia | ER Stress/Autophagy | CD8+ T activation, proinflammatory | ||||||
| i. Doxorubicin | Yes | Yes | Yes | Yes | Yes | cytokine release, secondary necrosis | [20,21,22,23,24,26,27,28,29] | ||
| ii. Epirubicin | Yes | Yes | Yes | Und | Yes | likely enhancing antigen presentation | [21,22,23,24,29] | ||
| iii. Idarubicin | Yes | Und | Yes | Und | Yes | [20,29] | |||
| iv. Mitoxantrone | Yes | Und | Yes | Und | Yes | [20,22,23,27,29,30,31] | |||
| Bortezomib | Lymphoma, Myeloma | ER Stress, HSP Exposure | Yes | Und | Yes | Yes | Yes | DC maturation/activation | [34,35,36,37] |
| Bleomycin | Colon | ER Stress/Autophagy | Yes | Yes | Yes | Yes | Yes | CD8+ T activation, and proinflammatory | [23,32,33,39] |
| cytokine release | |||||||||
| Cisplatin | Colon | - | No | Und | Yes | Und | No | Limited | [22,40] |
| Cyclophosphamide | Lymphoma, Glioma | ER Stress | Yes | Yes | Yes | Yes | Yes | CD8+ T, NK, macrophage activation | [22,25,27,30,40,53] |
| Oxaliplatin | Colon, Mouse sarcoma | ER Stress/ Autophagy | Yes | Und | Yes | Und | Possible | TLR4 engagement, DC activation | |
| Vorinostat (HDACi) | Colon, Central Nervous System | Yes | Yes | Yes | Und | Likely | DC, B cell activation | [63,64,65] | |
| IIa. Examples of Agents Which Do/Do Not Enhance MHC (Antigen Presentation) | Cancers | Increased HLA Expression | Cytokine Effects | ||||||
| Carboplatin (Cisplatin) | Ovarian, Lung | No (Ovarian); Yes (Lung) | [51,66] | ||||||
| Cyclophosphamide | Kidney, Breast, Prostate, Colon | Yes | [52] | ||||||
| Gemcitabine | Ovarian, Breast, Kidney, Prostate, Colon | Yes | [52] | ||||||
| HDACi (FR901228) | Leukemia, Lymphoma, Breast, Cervical | No | [13] | ||||||
| Interferons (Type I or II) | Ovarian, Melanoma | Yes | [50,51] | ||||||
| Microtubule destabilizers | Ovarian | Increased IFNα | |||||||
| i. Epothilone B | Yes | IL-1β, IL-6, IL-12 | [48] | ||||||
| ii. Taxol | Yes | [48] | |||||||
| iii. Vinblastine | Yes | [48] | |||||||
| Oxaliplatin | Kidney, Breast, Prostate, Colon | Yes | [52] | ||||||
| Paxclitaxel or Paxclitaxel-Carboplatin | Ovarian | Yes | [42] | ||||||
| IIb. Examples of Agents Which Enhance NKG2D (Antigen Presentation) | Cancers | Immune Impact | Specific NKG2D Ligand | ||||||
| Arsenic Trioxide | Leukemia, Breast | NK/HSP activation | MICA, MICB, ULBP1/2 | [67] | |||||
| 5′-Flurouracil | Pancreas | Synergy with Type I IFNs | Mult-1, Rae-1 | [68] | |||||
| Gemcitabine | Pancreas | NK activation | MICA | [69] | |||||
| III. Lymphodepleting Agents Improve T Cell Function (T cell Activation, Persistence) | Cancers | ||||||||
| Cyclophosphamide | Melanoma, Several Others | Increased Type I IFNs, TLR/DC activation, Treg depletion, increased Th17 cells, TRAIL activation, improved persistence of administered T cells | [23,54] | ||||||
| Fludarabine | Melanoma | Increased IL-7, IL-15, improved persistence of administered T cells | [61] | ||||||
| Irradiation | Melanoma | Increased IL-7, IL-15, homeostatic space and persistence of administered T cells, Treg depletion, release of immunostimulatory gut microflora | [10,60] | ||||||
| IV. Radiation as a Immunostimulatory Treatment (Antigen release, presentation, T cell/ and APC Priming) | Multiple Cancers | Increased Release of TAAs, improved antigen processing/presentation | [70,71,72,73,74,75] | ||||||
| Proinflammatory cytokine secretion | |||||||||
| ICD | |||||||||
| Recruitment of immune cells to tumor microenvironment |
| Chemotherapeutic | Category | Tumor Type | PD-L1 Protein | PD-L1 RNA | In Vivo | PD-L2 Protein | PD-L2 RNA | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Carboplatin | Alkylating Agent | Ovarian | + (S/I) | + | + (M) | OBS | OBS | NFKb | [2] |
| Carboplatin | Alkylating Agent | Ovarian | + (S) | + | + (M) | + (S) | + | JAK/STAT, Antiviral Defense | [51] |
| Cisplatin | Alkylating Agent | Liver | + (S) | OBS | OBS | OBS | OBS | MEK-ERK-MAPK | [94] |
| Cisplatin | Alkylating Agent | Breast | NC | OBS | OBS | OBS | OBS | - | [28] |
| Cisplatin | Alkylating Agent | Melanoma | − (S) | OBS | OBS | OBS | OBS | STAT6 Inhibition | [28] |
| Docetaxel | Alkylating Agent | Breast | NC | OBS | OBS | OBS | OBS | - | [28] |
| Gemcitabine | Antimetabolite | Ovarian | + (S/I) | + | + (M) | OBS | OBS | NFKb | [2] |
| 5-Fluorouracil | Antimetabolite | Breast | + (S) | OBS | OBS | OBS | OBS | JAK/STAT, MAPK, PI3K/AKT | [4] |
| Paclitaxel | Antimicrotubule | Breast | + (S) | OBS | OBS | OBS | OBS | JAK/STAT, MAPK, PI3K/AKT | [4] |
| Paclitaxel | Antimicrotubule | Ovarian | + (S/I) | + | + (M) | OBS | OBS | NFKb | [2] |
| Paclitaxel | Antimicrotubule | Colon | + (S/I) | + | OBS | OBS | OBS | MEK-ERK-MAPK | [6] |
| Paclitaxel | Antimicrotubule | Liver | + (S/I) | + | OBS | OBS | OBS | MEK-ERK-MAPK | [6] |
| Azacytidine a | DMNTi | Lung | + (S) | + (S) | OBS | NC | NC | STAT, Antiviral Defense | [17] |
| Decitabine a | DNMTi | Leukemia | + (S/I) | + | + (P) | + (S/I) | + | NE | [16] |
| HDACi (s) | HDACi Class I | Melanoma | + (S) | + | + (P/M) | + (S) | + | Acetylation of PD-L1/2 Promotor | [27] |
| Valproic Acid | HDACi Class I, II | Ovarian | + (S) | + | OBS | OBS | OBS | JAK/STAT | [51] |
| Rocilinostat | HDACi Class VI | Leukemia | − (S) | OBS | − (P) | OBS | OBS | NE | [30] |
| Doxorubicin | Topoisomerase (−) | Breast | − (S/I) + (N) | OBS | +/− | OBS | OBS | PI3K/AKT, non-PI3K/AKT | [28] |
| Etoposide | Topoisomerase (−) | Breast | + (S) | OBS | OBS | OBS | OBS | JAK/STAT, MAPK, PI3K/AKT | [4] |
| Etoposide | Topoisomerase (−) | Occular | + (S/I) | + | OBS | OBS | OBS | miR | [8] |
| Mitoxantrone | Topoisomerase (−) | Breast | NC | OBS | OBS | OBS | OBS | - | [28] |
| Trabectedin | Undefined Cytoxin | Ovarian | + (S/I) | OBS | + (M) | OBS | OBS | IFNg release | [5] |
| Arsenic Trioxide | Undefined Cytoxin | Leukemia | + (S/I) | OBS | OBS | OBS | OBS | miR | [9] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacon, J.A.; Schutsky, K.; Powell, D.J. The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy. Vaccines 2016, 4, 43. https://doi.org/10.3390/vaccines4040043
Chacon JA, Schutsky K, Powell DJ. The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy. Vaccines. 2016; 4(4):43. https://doi.org/10.3390/vaccines4040043
Chicago/Turabian StyleChacon, Jessica Ann, Keith Schutsky, and Daniel J. Powell. 2016. "The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy" Vaccines 4, no. 4: 43. https://doi.org/10.3390/vaccines4040043
APA StyleChacon, J. A., Schutsky, K., & Powell, D. J. (2016). The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy. Vaccines, 4(4), 43. https://doi.org/10.3390/vaccines4040043