Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators

{kind=link}
Abstract
:1. The Advent of Adjuvants—A Brief History
What Is an Adjuvant? The Futile Attempt to Categorize
2. Why Use Adjuvants? The Fundamental Rationale and How It Has Changed over Time
Attenuation and Its Impact on the Immune Response
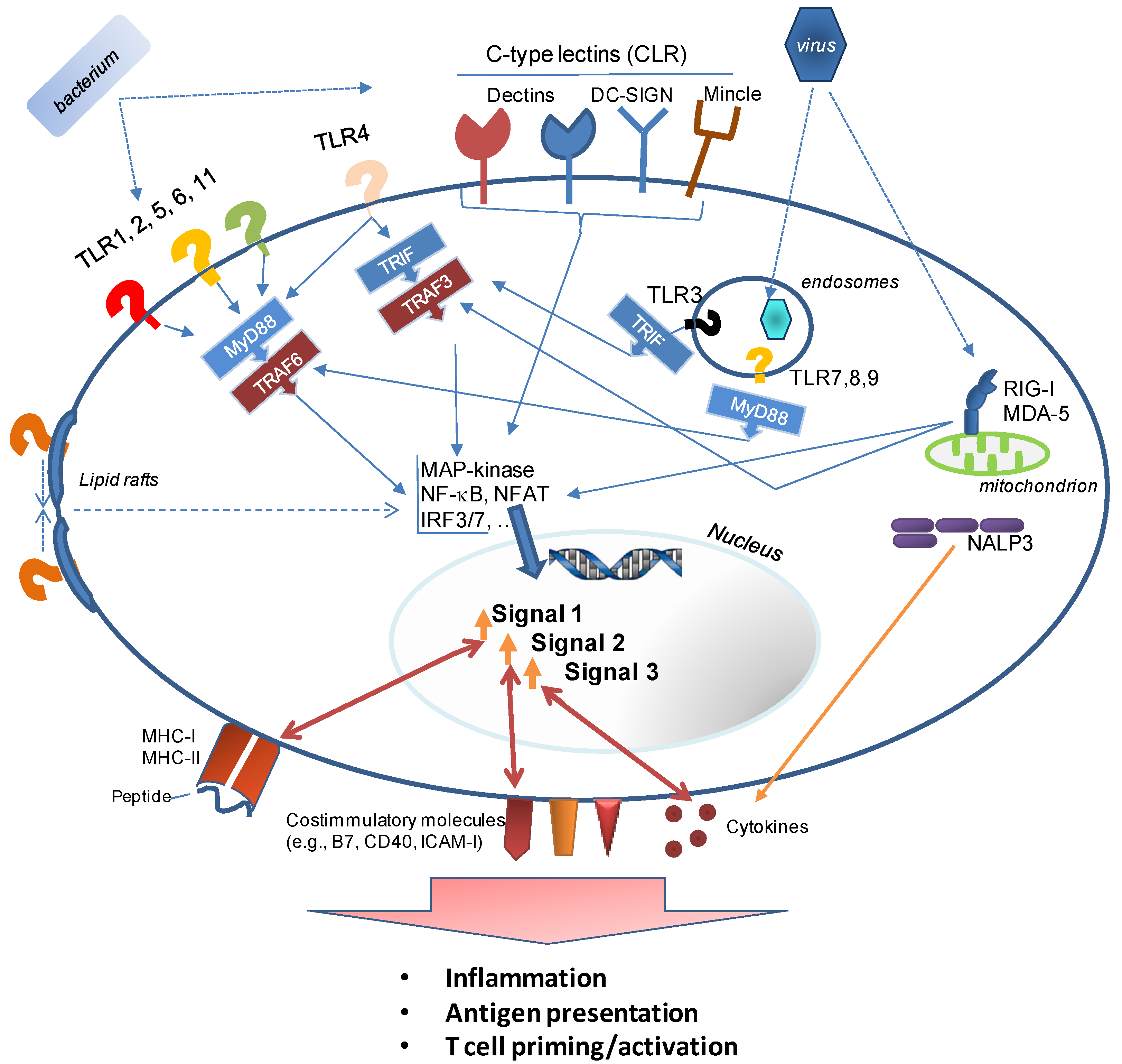
3. How Innate Immunity Controls Lymphocyte Responses
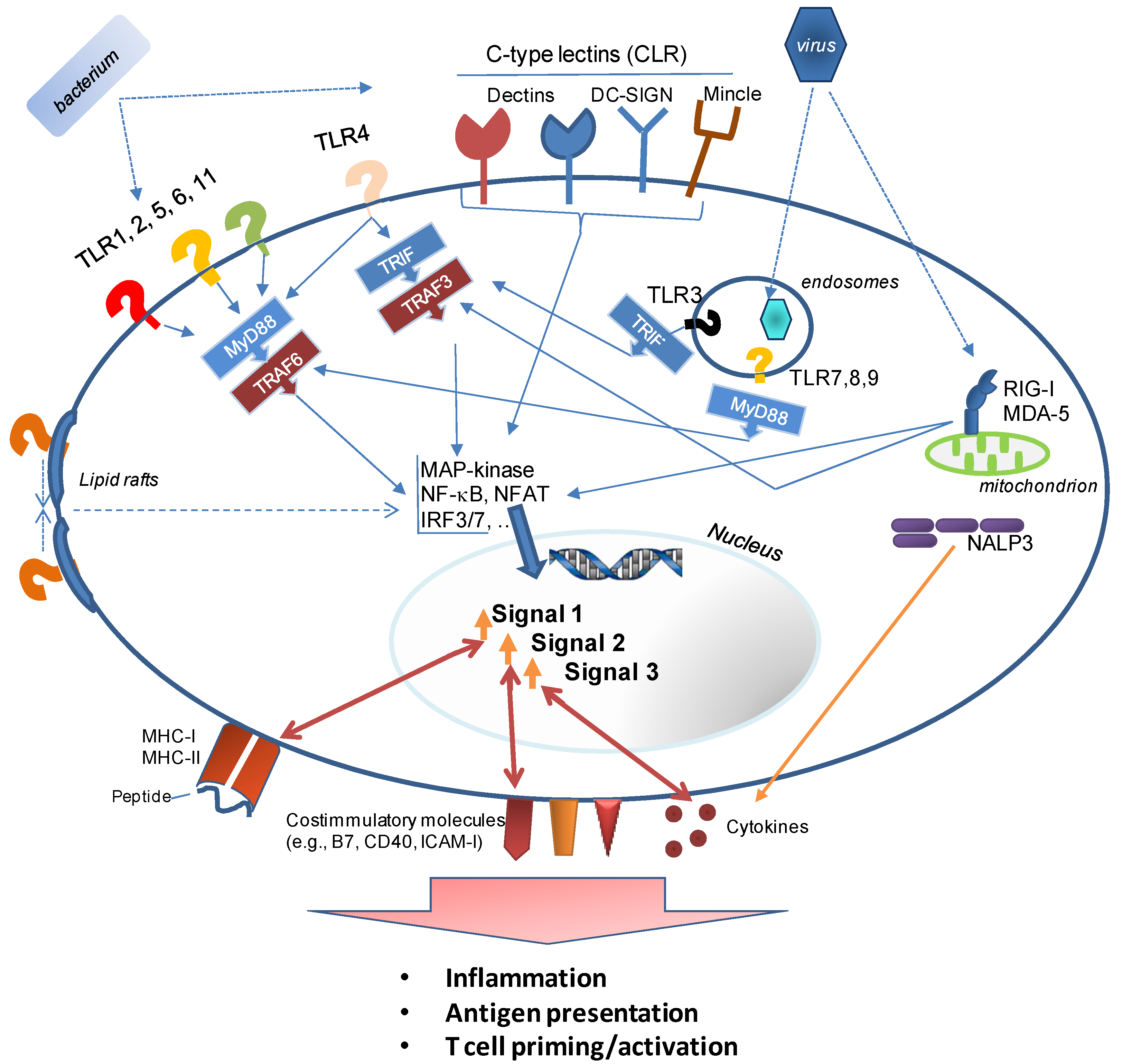
3.1. Formulation, Formulation, Formulation
- (1)
- The proper attachment of antigens to carriers (nano- or microcarriers such as PLGA beads or aluminum-based crystalline adjuvants); their incorporation into liposomes, virosomes or bacterial ghosts (empty bacterial shells which display bacterial PAMPs [32]); or conjugation to macromolecules (protein, lipid, PEG [33]). These procedures assure co-delivery (mostly to APCs) or protection from extracellular degradation and extended availability (depot);
- (2)
- The correct “assembly” of adjuvant components which, individually, have either no adjuvant activity (e.g., vegetable oil and detergents in the case of nanoemulsions [34], or squalene and detergents in the case of MF59™/AddaVax™ [35,36]), or are very reactogenic (as in the case of hemolytic saponins such as QS21 [37] or Quil A, which are detoxified by their association with cholesterol and phospholipid, forming the adjuvant ISCOMATRIX [38]);
- (3)
- The correct particle size of carriers such as cochleates, which consist of multiple layers of lipid membranes [39], or PLGA microspheres [40]. Much, however, remains to be learned about the optimal size of a vaccine carrier/adjuvant. It depends on parameters such as the route of immunization since different populations of APCs are targeted when the vaccine is injected into different tissues (reviewed in [41]). Not only the particle size, but also the material properties of the carrier appear to affect their immunogenicity [42], making it difficult to establish rules that apply to all particulate vaccine formulations;
- (4)
- Appropriate buffer species (“salting”) with a particular pH and ionic strength [24], since even minor changes in the type of salt used to formulate a vaccine may significantly influence adjuvanticity [43]. The selection of buffers may also impact the stability of the vaccine formulation and, therefore, the immunogenicity of the vaccine;
- (5)
- The spatial arrangement of antigens and/or PAMPs on a particle [44,45] determines how these molecules interact with—and how they cross-link—receptors on APCs such as dendritic cells, macrophages or B cells. The spacing of molecules on a carrier can be precisely controlled for example by using novel programmable DNA nanostructures. On such scaffolds, the impact of incremental changes in molecular distances between molecules (epitope density) can be studied, an approach not adequately used yet to determine the optimal spacing of antigens or PAMPs on a particulate vaccine formulation [46]; and
- (6)
- The ratio of antigen and adjuvant: in most studies, fixed ratios between the two components of the vaccine, adjuvant and antigen, are being used. For each antigen, however, a different ratio may be optimal;
- (7)
- Attenuation of the pathogen: when using killed or partially attenuated pathogens as vaccines, the impact of the attenuation procedure on endogenous adjuvants is rarely discussed or considered. In the case of respiratory syncytial virus (RSV), formalin-based inactivation of the virus unexpectedly created a vaccine that enhanced, rather than prevented disease in RSV-naïve children. The proposed mechanism was a drastically reduced adjuvant effect of viral TLR-ligands, which had been degraded by formalin (reviewed in [47]). The subsequently poor TLR ligation resulted in suboptimal immune responses characterized by low-avidity, non-neutralizing antibodies and T cell-mediated immunopathology [48]. This severe defect of this attenuated RSV vaccine can be overcome by formulating the vaccine with exogenous TLR ligands [49]. It might also be possible to avoid it by employing a milder attenuation protocol, such as the use of hydrogen peroxide instead of formalin. Hydrogen peroxide only causes minimal damage to antigenic structures and thus better preserves the immunogenicity of pathogen-derived antigens [50].
- Example 1 is the detoxification of LPS, resulting in Monophosphoryl lipid A (MPL®) [51] (or its commercially available equivalent, MPLA), a TLR4 agonist, which is safe for use in humans and a component of AS04™. The latter is used in the human Papilloma Virus vaccine Cervarix® (the first FDA-approved vaccine with an adjuvant other than alum). The lower toxicity is a result of much weaker signaling through the MyD88 signaling pathway of TLR4. This signaling cascade activates transcription factors (predominantly NF-κB) associated with inflammatory gene products. Signaling through TLR4’s second signaling cascade, the TRAM/TRIF (Toll IL-1 receptor domain-containing adaptor-inducing IFNβ) pathway is preserved after binding of MPL [52]. TRIF-signaling is associated with a Type I IFN response which is required for the induction of a strong adaptive immune response. However, it should be noted that while the induction of Type I IFN is frequently cited as an indicator of adjuvanticity, at least two adjuvants—the clinically used oil-in-water emulsion MF59 and the TLR-agonist Pam3CSK4—are poor inducers of IFN-related innate immune pathways [53].A novel, fully synthetic, mimetic of Lipid A (aminoalkyl glucosaminide4-phosphate (AGP) [54]) has been developed based on the insights into TLR4 signaling pathways gained with MPL. AGP combine the advantages of a highly defined, pure synthetic molecule with the safety of an adjuvant that not only binds to a defined innate immune receptor (TLR4), but also preferentially triggers a beneficial innate immune response profile (TRIF-signaling and Type I IFN production) while avoiding a signaling cascade which results in excessive inflammation (MyD88 signaling). While this development is very encouraging, it is important to evaluate the immune responses induced by the modified and “safer” adjuvants to determine whether any benefits of the “stronger” adjuvants had been lost. In the case of MPL, it appears that both LPS and its derivative promote strong CD4+ T cell expansion, but long-term retention of these cells was only supported efficiently by LPS [55].
- Example 2 is the “decoration” of antigens with small-molecule adjuvants. This is a formulation approach which drastically reduces the amount of adjuvant being delivered and thus curbs the inflammatory response while selectively and efficiently triggering the activation of PRR on those APCs which encounter the antigen. This approach was used for a synthetic TLR7 agonist, imidazoquinoline [56], and resulted in the induction of a robust, high-affinity antibody response. The direct conjugation of antigen to another TLR-agonist also supported the induction of CD8+ T cell responses (further discussed below), likely by enhancing cross-presentation [57]. While this approach is only limited by the creativity and skill of the medicinal chemist, the product has to be evaluated very carefully to ensure that immune responses to the “decorated” antigen are not negatively affected by PAMPs attached to crucial T or B cell epitopes. Masking of a crucial epitope for broadly neutralizing antibodies was indeed observed after a gp120 HIV vaccine was decorated with TLR-agonists [58]. This problem may be avoided by conjugating both adjuvant and antigen to a carrier (such as nanolipoprotein particles [59]).
3.2. Adaptive Correlates of Adjuvanticity—What Is the Best Readout?
4. What Does an Adjuvant Do and How Does It Affect Adaptive Immune Responses?
4.1. Correlates of Adjuvanticity—Soluble Factors?
4.2. Correlates of Adjuvanticity—Leukocyte Recruitment?
4.3. Correlates of Adjuvanticity—The T Cell Perspective
4.4. Polarization of T Cell Responses towards Cellular or Humoral Responses
4.5. Alum—The Stumbling Block for Better Vaccines?
4.6. Wanted—Adjuvants That Induce CD8+ T Cells
- TLR9 is an intracellular sensor of dsDNA characterized by a central, non-methylated CpG and flanking sequence motifs. Synthetic CpG oligonucleotides (ODN) have been reported to induce strong cytotoxic T cell and Th1 responses [84], and also support a robust antibody response. Not only are CpG motifs species-specific (complicating their translation from small-animal models to the clinic), but different motifs target different populations of APCs [85] and trigger different response profiles [86].
- TLR5 is a membrane-based receptor for bacterial flagellin. Vaccine constructs consisting of antigen-flagellin fusions induce balanced Th1 and Th2 responses [87]. Flagellin has already been tested as an adjuvant for a novel influenza vaccine in humans [88] and in addition to its exploration as a vaccine adjuvant for a variety of infectious disease vaccines in animal models, it has been able to enhance papilloma virus-specific CD8+ T cell responses in a therapeutic cancer vaccine model [89] or CD8+ T cells associated with protective immunity in a malaria model [90].
- TLR7 and TLR8 are intracellular sensors of single stranded RNA and ligands for these PRR have been used in the clinic for topical treatment of various types of skin cancer [91]. Numerous agonists have been developed (reviewed in [92]), such as the TLR7-selective and Th1/Th17 polarizing guanosine-analog Loxoribine [93]. Signals through TLR7 have been found to promote cross-presentation by dendritic cells (DC), thus enhancing the induction of CD8+ T cells [94]. Testing of TLR7 or TLR8 agonists is complicated by the fact that the cellular distribution of TLR7 is significantly different between mice and humans, and the ligand specificity of TLR8 is drastically different between the two species (leading to the initial conclusion that mouse TLR8 was not functional).
- Numerous reports have documented the usefulness of the TLR3 agonist polyriboinosinic acid-polyribocytidylic acid (poly(I:C)) to induce cellular immune responses. The synthetic RNA molecule allows the induction of CD8+ T cells against soluble proteins in mice [95,96] and has been added to DC-based vaccines [97] or peptide-vaccines [98] for cancer immunotherapy. It induces strong CD8+ T cell responses against the co-delivered HIV Gag protein and this response was shown to be further improved through the addition of ISCOMs. Combining the two types of adjuvant provided a synergistic adjuvant effect [99].
- Among adjuvants with unknown receptor specificity or a defined mechanism-of-action, ISCOM-based formulations have been used to induce antibodies as well as CD8+ T cell responses, either in the form of ISCOMATRIX, which is simply added to the antigen or as ISCOM-based vaccines. In the latter, the antigen is encapsulated within nanocages consisting of saponin, cholesterol, and phospholipid. This type of adjuvant is a component of two veterinary vaccines and has also proven to be efficacious in clinical trials (reviewed in [100]). Not surprisingly, strategies to enhance the association of free antigen with ISCOMATRIX (such as increasing the electrostatic interaction between protein and the nanocage) result in stronger CD4+ and CD8+ T cell responses, as shown, for example, with an HCV vaccine in primates [101].
4.7. Wanted—Adjuvants that Induce Better CD8+ T Cells
- MPL, the derivative of the bacterial TLR4 agonist LPS, has been used alone as well as in combination with other adjuvants such as QS21 in liposomes (AS01) or an emulsion (AS02), alum (AS04), or CpG and QS21 in an emulsion (AS15) with the goal of promoting T cell responses (MPL has been used in millions of doses of vaccines (licensed products as well as experimental vaccines). These vaccines include Fendrix® (HBV), Cervarix® (HPV), RTS,S (malaria; final stages of licensure), Pollinex Quattro® (allergy)). A related LPS derivative, Glucopyranosyl Lipid Adjuvant (GLA) [102], formulated in a stable emulsion (SE), which by itself has adjuvant properties, has been used as an adjuvant for experimental, clinical vaccines against Leishmania, Influenza, TB, and malaria (reviewed in [7]). TLR4 promotes B cell maturation [103], changes the trafficking of B cells into the germinal centers of lymphatic organs [104], and likely regulates affinity maturation [105]. In the context of T cell responses, it should be noted that TLR4-signalling mediates the trapping of activated CD8+ T cells in the liver [106]. Depending on the targeted disease, this could be advantageous when effector T cells accumulate at the site where the pathogen resides (e.g., in the case of Hepatitis or malaria). However, this is based on the assumption that T cell function is retained in the liver which has been described as a lymphoid organ with suppressive rather than stimulatory characteristics (reviewed in [107]).
- Combining CpG ODN with alum improves both antibody and T cell responses [108].
- Phytol is a branched aliphatic alcohol and a constituent of chlorophyll. Synthetic, modified phytols are potent immunostimulatory molecules and can be used to drive Th2 or Th1 responses, depending on the specific chemical modifications [109,110]. These compounds promote humoral immune responses as well as the induction of potent CD8+ CTL responses [111]. Although the mechanism of phytol-based adjuvants is still unknown, and specific cellular receptors which mediate their function(s) remain to be identified, these compounds are a reminder that natural compound libraries likely contain many novel adjuvant candidates waiting to be discovered.
4.8. What Does It Take to Activate CD8+ T Cells?
4.9. Inducing the Strongest CD8+ T Cell Response—A Good Idea?
5. The Confusing (Molecular) Mechanism of Action of Well-Known Adjuvants
5.1. What Is an “Adjuvant Effect” on a Molecular/Cellular Level?
5.2. Alum—The Never Ending Story
5.3. Nalp3 Inflammasome—The Missing Link?
5.4. Death by Alum
5.5. New Players in the Model of Alum’s Mechanism
6. Beyond Macrophages and Dendritic Cells: Targeting Other Contributors to Vaccine-Induced Immunity by Specialized Adjuvants
6.1. Adjuvants Targeting Natural Killer and Natural Killer T Cells
6.2. Adjuvants Targeting Mast Cells
7. Beyond Adjuvants: How to Properly Deliver Adjuvanted Vaccines
7.1. Ensuring the Presence of Specific Immune Cells at the Pathogen’s Point of Entry
7.2. Mucosal Vaccination—Why Not?
7.3. Altering Immune Responses through Vaccination Regimens
7.4. Altering Immune Responses through Vaccination Intervals
8. Off-the-Shelf Vaccines Are Not for Everyone: Using Adjuvants for Targeted Solutions
8.1. Heterogeneity of the Adaptive Immune Response in the Human Population
8.2. Heterogeneity of the Innate Immune Response in the Human Population
8.3. Heterogeneity Based on Age and Immune Status of the Human Population
8.4. Heterogeneity of the Global Population
9. Conclusions
9.1. Are We There Yet?
9.2. Current Trends in Adjuvant Research
9.2.1. Heterologous Prime-Boost
9.2.2. Combination Adjuvants and the Future of Alum
9.2.3. Overall Combination Adjuvants
9.2.4. Comparative Adjuvant Research
9.2.5. Novel Targets
9.2.6. Mechanism of Action
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Hilleman, M.R. Vaccines in historic evolution and perspective: A narrative of vaccine discoveries. Vaccine 2000, 18, 1436–1447. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Tsai, T.F.; del Giudice, G. New technologies for influenza vaccines. Hum. Vaccines Immunother. 2012, 8, 45–58. [Google Scholar] [CrossRef]
- Nussenzweig, R.S.; Vanderberg, J.; Most, H.; Orton, C. Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei. Nature 1967, 216, 160–162. [Google Scholar] [CrossRef]
- Wijayalath, W.; Cheesman, S.; Tanabe, K.; Handunnetti, S.; Carter, R.; Pathirana, S. Strain-specific protective effect of the immunity induced by live malarial sporozoites under chloroquine cover. PLoS One 2012, 7, e45861. [Google Scholar]
- Schwartz, J.L. The first rotavirus vaccine and the politics of acceptable risk. Milbank Q. 2012, 90, 278–310. [Google Scholar] [CrossRef]
- Alving, C.R.; Peachman, K.K.; Rao, M.; Reed, S.G. Adjuvants for human vaccines. Curr. Opin. Immunol. 2012, 24, 310–315. [Google Scholar] [CrossRef]
- Mbow, M.L.; de Gregorio, E.; Valiante, N.M.; Rappuoli, R. New adjuvants for human vaccines. Curr. Opin. Immunol. 2010, 22, 411–416. [Google Scholar] [CrossRef]
- Gebril, A.; Alsaadi, M.; Acevedo, R.; Mullen, A.B.; Ferro, V.A. Optimizing efficacy of mucosal vaccines. Expert Rev. Vaccines 2012, 11, 1139–1155. [Google Scholar] [CrossRef]
- Kobiyama, K.; Jounai, N.; Aoshi, T.; Tozuka, M.; Takeshita, F.; Coban, C.; Ishii, K.J. Innate Immune Signaling by, and Genetic Adjuvants for DNA Vaccination. Vaccines 2013, 1, 278–292. [Google Scholar] [CrossRef]
- Baz, M.; Samant, M.; Zekki, H.; Tribout-Jover, P.; Plante, M.; Lanteigne, A.M.; Hamelin, M.E.; Mallett, C.; Papadopoulou, B.; Boivin, G. Effects of different adjuvants in the context of intramuscular and intranasal routes on humoral and cellular immune responses induced by detergent-split A/H3N2 influenza vaccines in mice. Clin. Vaccine Immunol. 2012, 19, 209–218. [Google Scholar] [CrossRef]
- Tetsutani, K.; Ishii, K.J. Adjuvants in influenza vaccines. Vaccine 2012, 30, 7658–7661. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Leitner, W.W.; Tsokos, G.C. Complement 3d: From molecular adjuvant to target of immune escape mechanisms. Clin. Immunol. 2006, 121, 177–185. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Pizza, M.; Giuliani, M.M.; Fontana, M.R.; Monaci, E.; Douce, G.; Dougan, G.; Mills, K.H.G.; Rappuoli, R.; del Giudice, G. Mucosal vaccines: Non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 2001, 19, 2534–2541. [Google Scholar] [CrossRef]
- Immunization Action Coalition. Available online: http://www.immunize.org/timeline (accessed on 20 March 2014).
- Chen, J.; Pompano, R.R.; Santiago, F.W.; Maillat, L.; Sciammas, R.; Sun, T.; Han, H.; Topham, D.J.; Chong, A.S.; Collier, J.H. The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials 2013, 34, 8776–8785. [Google Scholar] [CrossRef]
- Saade, F.; Honda-Okubo, Y.; Trec, S.; Petrovsky, N. A novel hepatitis B vaccine containing Advax, a polysaccharide adjuvant derived from delta inulin, induces robust humoral and cellular immunity with minimal reactogenicity in preclinical testing. Vaccine 2013, 31, 1999–2007. [Google Scholar] [CrossRef]
- Wong, P.T.; Wang, S.H.; Ciotti, S.; Makidon, P.E.; Smith, D.M.; Fan, Y.; Schuler, C.F.; Baker, J.R. Formulation and Characterization of Nanoemulsion Intranasal Adjuvants: Effects of Surfactant Composition on Mucoadhesion and Immunogenicity. Mol. Pharm. 2014, 11, 531–544. [Google Scholar] [CrossRef]
- Belz, G.T.; Carbone, F.R.; Heath, W.R. Cross-presentation of antigens by dendritic cells. Crit. Rev. Immunol. 2002, 22, 439–448. [Google Scholar]
- Murphy, B.R.; Coelingh, K. Principles underlying the development and use of live attenuated cold-adapted influenza A and B virus vaccines. Viral Immunol. 2002, 15, 295–323. [Google Scholar] [CrossRef]
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 2009, 227, 221–233. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef]
- Flach, T.L.; Ng, G.; Hari, A.; Desrosiers, M.D.; Zhang, P.; Ward, S.M.; Seamone, M.E.; Vilaysane, A.; Mucsi, A.D.; Fong, Y.; et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat. Med. 2011, 17, 479–487. [Google Scholar]
- Brito, L.A.; Malyala, P.; O’Hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145. [Google Scholar] [CrossRef]
- Pearse, M.J.; Drane, D. ISCOMATRIX adjuvant for antigen delivery. Adv. Drug Deliv. Rev. 2005, 57, 465–474. [Google Scholar] [CrossRef]
- Tonkin, D.R.; Jorquera, P.; Todd, T.; Beard, C.W.; Johnston, R.E.; Barro, M. Alphavirus replicon-based enhancement of mucosal and systemic immunity is linked to the innate response generated by primary immunization. Vaccine 2010, 28, 3238–3246. [Google Scholar] [CrossRef]
- Klinman, D.M. Adjuvant activity of CpG oligodeoxynucleotides. Int. Rev. Immunol. 2006, 25, 135–154. [Google Scholar] [CrossRef]
- Glenny, A.T.; Pope, C.G.; Waddington, H.; Wallace, U. Immunological notes XVII–XXIV. J. Pathol. Bacteriol. 1926, 29, 31–40. [Google Scholar] [CrossRef]
- Shi, Y.; HogenEsch, H.; Regnier, F.E.; Hem, S.L. Detoxification of endotoxin by aluminum hydroxide adjuvant. Vaccine 2001, 19, 1747–1752. [Google Scholar] [CrossRef]
- Aebig, J.A.; Mullen, G.E.; Dobrescu, G.; Rausch, K.; Lambert, L.; Ajose-Popoola, O.; Long, C.A.; Saul, A.; Miles, A.P. Formulation of vaccines containing CpG oligonucleotides and alum. J. Immunol. Methods 2007, 323, 139–146. [Google Scholar] [CrossRef]
- Cain, D.W.; Sanders, S.E.; Cunningham, M.M.; Kelsoe, G. Disparate adjuvant properties among three formulations of “alum”. Vaccine 2013, 31, 653–660. [Google Scholar] [CrossRef]
- Langemann, T.; Koller, V.J.; Muhammad, A.; Kudela, P.; Mayr, U.B.; Lubitz, W. The Bacterial Ghost platform system: Production and applications. Bioeng. Bugs 2010, 1, 326–336. [Google Scholar] [CrossRef]
- Chan, M.; Hayashi, T.; Mathewson, R.D.; Yao, S.; Gray, C.; Tawatao, R.I.; Kalenian, K.; Zhang, Y.; Hayashi, Y.; Lao, F.S.; et al. Synthesis and characterization of PEGylated toll like receptor 7 ligands. Bioconjug. Chem. 2011, 22, 445–454. [Google Scholar] [CrossRef]
- Makidon, P.E.; Belyakov, I.M.; Blanco, L.P.; Janczak, K.W.; Landers, J.; Bielinska, A.U.; Groom, J.V., 2nd; Baker, J.R., Jr. Nanoemulsion mucosal adjuvant uniquely activates cytokine production by nasal ciliated epithelium and induces dendritic cell trafficking. Eur. J. Immunol. 2012, 42, 2073–2086. [Google Scholar] [CrossRef]
- Ott, G.; Barchfeld, G.L.; Chernoff, D.; Radhakrishnan, R.; van Hoogevest, P.; van Nest, G. MF59. Design and evaluation of a safe and potent adjuvant for human vaccines. Pharm. Biotechnol. 1995, 6, 277–296. [Google Scholar]
- Calabro, S.; Tritto, E.; Pezzotti, A.; Taccone, M.; Muzzi, A.; Bertholet, S.; de Gregorio, E.; O'Hagan, D.T.; Baudner, B.; Seubert, A. The adjuvant effect of MF59 is due to the oil-in-water emulsion formulation, none of the individual components induce a comparable adjuvant effect. Vaccine 2013, 31, 3363–3369. [Google Scholar] [CrossRef]
- Stewart-Tull, D.E. The Use of Adjuvants in Experimental Vaccines: IV. ISCOMS. Methods Mol. Med. 1996, 4, 153–155. [Google Scholar]
- Sun, H.X.; Xie, Y.; Ye, Y.P. ISCOMs and ISCOMATRIX. Vaccine 2009, 27, 4388–4401. [Google Scholar] [CrossRef]
- Gil, D.; Bracho, G.; Zayas, C.; del Campo, J.; Acevedo, R.; Toledo, A.; Lastre, M.; Perez, O. Strategy for determination of an efficient Cochleate particle size. Vaccine 2006, 24, S92–S93. [Google Scholar] [CrossRef]
- Thomas, C.; Gupta, V.; Ahsan, F. Particle size influences the immune response produced by hepatitis B vaccine formulated in inhalable particles. Pharm. Res. 2010, 27, 905–919. [Google Scholar] [CrossRef]
- Mastelic, B.; Garcon, N.; del Giudice, G.; Golding, H.; Gruber, M.; Neels, P.; Fritzell, B. Predictive markers of safety and immunogenicity of adjuvanted vaccines. Biologicals 2013, 41, 458–468. [Google Scholar] [CrossRef]
- Cohen, J.A.; Beaudette, T.T.; Tseng, W.W.; Bachelder, E.M.; Mende, I.; Engleman, E.G.; Frechet, J.M. T-cell activation by antigen-loaded pH-sensitive hydrogel particles in vivo: The effect of particle size. Bioconjug. Chem. 2009, 20, 111–119. [Google Scholar] [CrossRef]
- Yan, W.; Huang, L. The effects of salt on the physicochemical properties and immunogenicity of protein based vaccine formulated in cationic liposome. Int. J. Pharm. 2009, 368, 56–62. [Google Scholar] [CrossRef]
- Abdel-Aal, A.B.; Zaman, M.; Fujita, Y.; Batzloff, M.R.; Good, M.F.; Toth, I. Design of three-component vaccines against group A streptococcal infections: Importance of spatial arrangement of vaccine components. J. Med. Chem. 2010, 53, 8041–8046. [Google Scholar] [CrossRef]
- Mancini, R.J.; Tom, J.K.; Esser-Kahn, A.P. Covalently coupled immunostimulant heterodimers. Angew. Chem. Int. Ed. Engl. 2014, 53, 189–192. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Yu, T.; Clifford, C.; Liu, Y.; Yan, H.; Chang, Y. A DNA nanostructure platform for directed assembly of synthetic vaccines. Nano Lett. 2012, 12, 4254–4259. [Google Scholar] [CrossRef]
- Ubol, S.; Halstead, S.B. How Innate Immune Mechanisms Contribute to Antibody-Enhanced Viral Infections. Clin. Vaccine Immunol. 2010, 17, 1829–1835. [Google Scholar] [CrossRef]
- Delgado, M.F.; Coviello, S.; Monsalvo, A.C.; Melendi, G.A.; Hernandez, J.Z.; Batalle, J.P.; Diaz, L.; Trento, A.; Chang, H.Y.; Mitzner, W.; et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 2009, 15, 34–41. [Google Scholar] [CrossRef]
- Hancock, G.E.; Heers, K.M.; Pryharski, K.S.; Smith, J.D.; Tiberio, L. Adjuvants recognized by toll-like receptors inhibit the induction of polarized type 2 T cell responses by natural attachment (G) protein of respiratory syncytial virus. Vaccine 2003, 21, 4348–4358. [Google Scholar] [CrossRef]
- Amanna, I.J.; Raue, H.P.; Slifka, M.K. Development of a new hydrogen peroxide-based vaccine platform. Nat. Med. 2012, 18, 974–979. [Google Scholar] [CrossRef]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef]
- Cekic, C.; Casella, C.R.; Eaves, C.A.; Matsuzawa, A.; Ichijo, H.; Mitchell, T.C. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J. Biol. Chem. 2009, 284, 31982–31991. [Google Scholar]
- Caproni, E.; Tritto, E.; Cortese, M.; Muzzi, A.; Mosca, F.; Monaci, E.; Baudner, B.; Seubert, A.; de Gregorio, E. MF59 and Pam3CSK4 boost adaptive responses to influenza subunit vaccine through an IFN type I-independent mechanism of action. J. Immunol. 2012, 188, 3088–3098. [Google Scholar] [CrossRef]
- Bowen, W.S.; Minns, L.A.; Johnson, D.A.; Mitchell, T.C.; Hutton, M.M.; Evans, J.T. Selective TRIF-dependent signaling by a synthetic toll-like receptor 4 agonist. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Thompson, B.S.; Chilton, P.M.; Ward, J.R.; Evans, J.T.; Mitchell, T.C. The low-toxicity versions of LPS, MPL adjuvant and RC529, are efficient adjuvants for CD4+ T cells. J. Leukoc. Biol. 2005, 78, 1273–1280. [Google Scholar]
- Shukla, N.M.; Lewis, T.C.; Day, T.P.; Mutz, C.A.; Ukani, R.; Hamilton, C.D.; Balakrishna, R.; David, S.A. Toward self-adjuvanting subunit vaccines: Model peptide and protein antigens incorporating covalently bound toll-like receptor-7 agonistic imidazoquinolines. Bioorg. Med. Chem. Lett. 2011, 21, 3232–3236. [Google Scholar] [CrossRef]
- Oh, J.Z.; Kedl, R.M. The capacity to induce cross-presentation dictates the success of a TLR7 agonist-conjugate vaccine for eliciting cellular immunity. J. Immunol. 2010, 185, 4602–4608. [Google Scholar] [CrossRef]
- Feng, Y.; Forsell, M.N.; Flynn, B.; Adams, W.; Lore, K.; Seder, R.; Wyatt, R.T.; Karlsson Hedestam, G.B. Chemical cross-linking of HIV-1 Env for direct TLR7/8 ligand conjugation compromises recognition of conserved antigenic determinants. Virology 2013, 446, 56–65. [Google Scholar] [CrossRef]
- Fischer, N.O.; Rasley, A.; Corzett, M.; Hwang, M.H.; Hoeprich, P.D.; Blanchette, C.D. Colocalized delivery of adjuvant and antigen using nanolipoprotein particles enhances the immune response to recombinant antigens. J. Am. Chem. Soc. 2013, 135, 2044–2047. [Google Scholar] [CrossRef]
- Khurana, S.; Chearwae, W.; Castellino, F.; Manischewitz, J.; King, L.R.; Honorkiewicz, A.; Rock, M.T.; Edwards, K.M.; del Giudice, G.; Rappuoli, R.; et al. Vaccines with MF59 adjuvant expand the antibody repertoire to target protective sites of pandemic avian H5N1 influenza virus. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Shukla, N.M.; Salunke, D.B.; Balakrishna, R.; Mutz, C.A.; Malladi, S.S.; David, S.A. Potent adjuvanticity of a pure TLR7-agonistic imidazoquinoline dendrimer. PLoS One 2012, 7, e43612. [Google Scholar]
- Guiducci, C.; Gong, M.; Cepika, A.M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef] [Green Version]
- Saenz, R.; Souza Cda, S.; Huang, C.T.; Larsson, M.; Esener, S.; Messmer, D. HMGB1-derived peptide acts as adjuvant inducing immune responses to peptide and protein antigen. Vaccine 2010, 28, 7556–7562. [Google Scholar] [CrossRef]
- Martinez-Gil, L.; Ayllon, J.; Ortigoza, M.B.; Garcia-Sastre, A.; Shaw, M.L.; Palese, P. Identification of small molecules with type I interferon inducing properties by high-throughput screening. PLoS One 2012, 7, e49049. [Google Scholar]
- Chan, M.; Hayashi, T.; Mathewson, R.D.; Nour, A.; Hayashi, Y.; Yao, S.; Tawatao, R.I.; Crain, B.; Tsigelny, I.F.; Kouznetsova, V.L.; et al. Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J. Med. Chem. 2013, 56, 4206–4223. [Google Scholar]
- Pashine, A.; Valiante, N.M.; Ulmer, J.B. Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 2005, 11, S63–S68. [Google Scholar] [CrossRef]
- Fritz, J.H.; Le Bourhis, L.; Magalhaes, J.G.; Philpott, D.J. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol. 2008, 29, 41–49. [Google Scholar] [CrossRef]
- Bobanga, I.D.; Petrrosiute, A.; Huang, A.Y. Chemokines as Cancer Vaccine Adjuvants. Vaccines 2013, 1, 444–462. [Google Scholar] [CrossRef]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Abebe, F. Is interferon-gamma the right marker for bacille Calmette-Guerin-induced immune protection? The missing link in our understanding of tuberculosis immunology. Clin. Exp. Immunol. 2012, 169, 213–219. [Google Scholar] [CrossRef]
- Zaitseva, M.; Romantseva, T.; Blinova, K.; Beren, J.; Sirota, L.; Drane, D.; Golding, H. Use of human MonoMac6 cells for development of in vitro assay predictive of adjuvant safety in vivo. Vaccine 2012, 30, 4859–4865. [Google Scholar] [CrossRef]
- Calabro, S.; Tortoli, M.; Baudner, B.C.; Pacitto, A.; Cortese, M.; O’Hagan, D.T.; de Gregorio, E.; Seubert, A.; Wack, A. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine 2011, 29, 1812–1823. [Google Scholar] [CrossRef]
- Lambert, S.L.; Yang, C.F.; Liu, Z.; Sweetwood, R.; Zhao, J.; Cheng, L.L.; Jin, H.; Woo, J. Molecular and Cellular Response Profiles Induced by the TLR4 Agonist-Based Adjuvant Glucopyranosyl Lipid A. PLoS One 2012, 7, e51618. [Google Scholar]
- Yang, C.W.; Strong, B.S.; Miller, M.J.; Unanue, E.R. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J. Immunol. 2010, 185, 2927–2934. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Kool, M.; Willart, M.A.; Hammad, H. Mechanism of action of clinically approved adjuvants. Curr. Opin. Immunol. 2009, 21, 23–29. [Google Scholar] [CrossRef]
- Baumgartner, C.K.; Malherbe, L.P. Regulation of CD4 T-cell receptor diversity by vaccine adjuvants. Immunology 2010, 130, 16–22. [Google Scholar] [CrossRef]
- Lynch, H.E.; Stewart, S.M.; Kepler, T.B.; Sempowski, G.D.; Alam, S.M. Surface plasmon resonance measurements of plasma antibody avidity during primary and secondary responses to anthrax protective antigen. J. Immunol. Methods 2014, 404, 1–12. [Google Scholar] [CrossRef]
- Vajdy, M.; Lycke, N.Y. Cholera toxin adjuvant promotes long-term immunological memory in the gut mucosa to unrelated immunogens after oral immunization. Immunology 1992, 75, 488–492. [Google Scholar]
- Leitner, W.W. Vaccine adjuvants: Is the pipeline clogged? Immunotherapy 2012, 4, 565–567. [Google Scholar] [CrossRef]
- Khong, H.T.; Yang, J.C.; Topalian, S.L.; Sherry, R.M.; Mavroukakis, S.A.; White, D.E.; Rosenberg, S.A. Immunization of HLA-A*0201 and/or HLA-DPbeta1*04 patients with metastatic melanoma using epitopes from the NY-ESO-1 antigen. J. Immunother. 2004, 27, 472–477. [Google Scholar] [CrossRef]
- Brinckerhoff, L.H.; Kalashnikov, V.V.; Thompson, L.W.; Yamshchikov, G.V.; Pierce, R.A.; Galavotti, H.S.; Engelhard, V.H.; Slingluff, C.L., Jr. Terminal modifications inhibit proteolytic degradation of an immunogenic MART-1(27–35) peptide: Implications for peptide vaccines. Int. J. Cancer. 1999, 83, 326–334. [Google Scholar] [CrossRef]
- Querec, T.; Bennouna, S.; Alkan, S.; Laouar, Y.; Gorden, K.; Flavell, R.; Akira, S.; Ahmed, R.; Pulendran, B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef]
- Higgins, S.C.; Mills, K.H. TLR, NLR Agonists, and Other Immune Modulators as Infectious Disease Vaccine Adjuvants. Curr. Infect. Dis. Rep. 2010, 12, 4–12. [Google Scholar] [CrossRef]
- Kobayashi, H.; Horner, A.A.; Takabayashi, K.; Nguyen, M.D.; Huang, E.; Cinman, N.; Raz, E. Immunostimulatory DNA pre-priming: A novel approach for prolonged Th1-biased immunity. Cell. Immunol. 1999, 198, 69–75. [Google Scholar]
- Krieg, A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef]
- Lipford, G.B.; Sparwasser, T.; Bauer, M.; Zimmermann, S.; Koch, E.S.; Heeg, K.; Wagner, H. Immunostimulatory DNA: Sequence-dependent production of potentially harmful or useful cytokines. Eur. J. Immunol. 1997, 27, 3420–3426. [Google Scholar] [CrossRef]
- Mizel, S.B.; Bates, J.T. Flagellin as an adjuvant: Cellular mechanisms and potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [CrossRef]
- Nguyen, C.T.; Hong, S.H.; Sin, J.I.; Vu, H.V.; Jeong, K.; Cho, K.O.; Uematsu, S.; Akira, S.; Lee, S.E.; Rhee, J.H. Flagellin enhances tumor-specific CD8(+) T cell immune responses through TLR5 stimulation in a therapeutic cancer vaccine model. Vaccine 2013, 31, 3879–3887. [Google Scholar] [CrossRef]
- Braga, C.J.; Massis, L.M.; Sbrogio-Almeida, M.E.; Alencar, B.C.; Bargieri, D.Y.; Boscardin, S.B.; Rodrigues, M.M.; Ferreira, L.C. CD8+ T cell adjuvant effects of Salmonella FliCd flagellin in live vaccine vectors or as purified protein. Vaccine 2010, 28, 1373–1382. [Google Scholar] [CrossRef]
- Meyer, T.; Surber, C.; French, L.E.; Stockfleth, E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin. Investig. Drugs 2013, 22, 149–159. [Google Scholar] [CrossRef]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef]
- Dzopalic, T.; Dragicevic, A.; Vasilijic, S.; Vucevic, D.; Majstorovic, I.; Bozic, B.; Balint, B.; Colic, M. Loxoribine, a selective Toll-like receptor 7 agonist, induces maturation of human monocyte-derived dendritic cells and stimulates their Th-1- and Th-17-polarizing capability. Int. Immunopharmacol. 2010, 10, 1428–1433. [Google Scholar] [CrossRef]
- Crespo, M.I.; Zacca, E.R.; Nunez, N.G.; Ranocchia, R.P.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Moron, G. TLR7 triggering with polyuridylic acid promotes cross-presentation in CD8alpha+ conventional dendritic cells by enhancing antigen preservation and MHC class I antigen permanence on the dendritic cell surface. J. Immunol. 2013, 190, 948–960. [Google Scholar] [CrossRef]
- Ngoi, S.M.; Tovey, M.G.; Vella, A.T. Targeting poly(I:C) to the TLR3-independent pathway boosts effector CD8 T cell differentiation through IFN-alpha/beta. J. Immunol. 2008, 181, 7670–7680. [Google Scholar]
- Wick, D.A.; Martin, S.D.; Nelson, B.H.; Webb, J.R. Profound CD8+ T cell immunity elicited by sequential daily immunization with exogenous antigen plus the TLR3 agonist poly(I:C). Vaccine 2011, 29, 984–993. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef]
- Sabbatini, P.; Tsuji, T.; Ferran, L.; Ritter, E.; Sedrak, C.; Tuballes, K.; Jungbluth, A.A.; Ritter, G.; Aghajanian, C.; Bell-McGuinn, K.; et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 2012, 18, 6497–6508. [Google Scholar] [CrossRef]
- Quinn, K.M.; Yamamoto, A.; Costa, A.; Darrah, P.A.; Lindsay, R.W.; Hegde, S.T.; Johnson, T.R.; Flynn, B.J.; Lore, K.; Seder, R.A. Coadministration of Polyinosinic:Polycytidylic Acid and Immunostimulatory Complexes Modifies Antigen Processing in Dendritic Cell Subsets and Enhances HIV Gag-Specific T Cell Immunity. J. Immunol. 2013, 191, 5085–5096. [Google Scholar] [CrossRef]
- Sanders, M.T.; Brown, L.E.; Deliyannis, G.; Pearse, M.J. ISCOM-based vaccines: The second decade. Immunol. Cell Biol. 2005, 83, 119–128. [Google Scholar] [CrossRef]
- Polakos, N.K.; Drane, D.; Cox, J.; Ng, P.; Selby, M.J.; Chien, D.; O’Hagan, D.T.; Houghton, M.; Paliard, X. Characterization of hepatitis C virus core-specific immune responses primed in rhesus macaques by a nonclassical ISCOM vaccine. J. Immunol. 2001, 166, 3589–3598. [Google Scholar]
- Arias, M.A.; van Roey, G.A.; Tregoning, J.S.; Moutaftsi, M.; Coler, R.N.; Windish, H.P.; Reed, S.G.; Carter, D.; Shattock, R.J. Glucopyranosyl Lipid Adjuvant (GLA), a Synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS One 2012, 7, e41144. [Google Scholar] [CrossRef]
- Hayashi, E.A.; Granato, A.; Paiva, L.S.; Bertho, A.L.; Bellio, M.; Nobrega, A. TLR4 promotes B cell maturation: Independence and cooperation with B lymphocyte-activating factor. J. Immunol. 2010, 184, 4662–4672. [Google Scholar] [CrossRef]
- Hwang, I.Y.; Park, C.; Harrison, K.; Kehrl, J.H. TLR4 signaling augments B lymphocyte migration and overcomes the restriction that limits access to germinal center dark zones. J. Exp. Med. 2009, 206, 2641–2657. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Vogel, L.A.; Zhang, W.; Loo, W.; Shnider, D.; Lind, E.F.; Ratliff, M.; Noelle, R.J.; Erickson, L.D. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J. Immunol. 2006, 177, 7723–7732. [Google Scholar]
- John, B.; Crispe, I.N. TLR-4 regulates CD8+ T cell trapping in the liver. J. Immunol. 2005, 175, 1643–1650. [Google Scholar]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef]
- Khodai, T.; Chappell, D.; Christy, C.; Cockle, P.; Eyles, J.; Hammond, D.; Gore, K.; McCluskie, M.J.; Evans, D.M.; Lang, S.; et al. Single and combination herpes simplex virus type 2 glycoprotein vaccines adjuvanted with CpG oligodeoxynucleotides or monophosphoryl lipid A exhibit differential immunity that is not correlated to protection in animal models. Clin. Vaccine Immunol. 2011, 18, 1702–1709. [Google Scholar] [CrossRef]
- Aachoui, Y.; Chowdhury, R.R.; Fitch, R.W.; Ghosh, S.K. Molecular signatures of phytol-derived immunostimulants in the context of chemokine-cytokine microenvironment and enhanced immune response. Cell. Immunol. 2011, 271, 227–238. [Google Scholar] [CrossRef]
- Aachoui, Y.; Schulte, M.L.; Fitch, R.W.; Ghosh, S.K. Synthetic adjuvants for vaccine formulations: Evaluation of new phytol derivatives in induction and persistence of specific immune response. Cell. Immunol. 2011, 271, 308–318. [Google Scholar] [CrossRef]
- Lim, S.Y.; Meyer, M.; Kjonaas, R.A.; Ghosh, S.K. Phytol-based novel adjuvants in vaccine formulation: 1. assessment of safety and efficacy during stimulation of humoral and cell-mediated immune responses. J. Immune Based Ther. Vaccines 2006, 4. [Google Scholar] [CrossRef]
- Bouvier, I.; Jusforgues-Saklani, H.; Lim, A.; Lemaitre, F.; Lemercier, B.; Auriau, C.; Nicola, M.A.; Leroy, S.; Law, H.K.; Bandeira, A.; et al. Immunization route dictates cross-priming efficiency and impacts the optimal timing of adjuvant delivery. Front. Immunol. 2011, 2. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Jiao, J.; Hu, H.M. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 2011, 6, 645–650. [Google Scholar] [CrossRef]
- Scott, E.A.; Stano, A.; Gillard, M.; Maio-Liu, A.C.; Swartz, M.A.; Hubbell, J.A. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials 2012, 33, 6211–6219. [Google Scholar] [CrossRef]
- Xiang, S.D.; Wilson, K.; Day, S.; Fuchsberger, M.; Plebanski, M. Methods of effective conjugation of antigens to nanoparticles as non-inflammatory vaccine carriers. Methods 2013, 60, 232–241. [Google Scholar] [CrossRef]
- Duewell, P.; Kisser, U.; Heckelsmiller, K.; Hoves, S.; Stoitzner, P.; Koernig, S.; Morelli, A.B.; Clausen, B.E.; Dauer, M.; Eigler, A.; et al. ISCOMATRIX adjuvant combines immune activation with antigen delivery to dendritic cells in vivo leading to effective cross-priming of CD8+ T cells. J. Immunol. 2011, 187, 55–63. [Google Scholar] [CrossRef]
- Wilson, N.S.; Yang, B.; Morelli, A.B.; Koernig, S.; Yang, A.; Loeser, S.; Airey, D.; Provan, L.; Hass, P.; Braley, H.; et al. ISCOMATRIX vaccines mediate CD8+ T-cell cross-priming by a MyD88-dependent signaling pathway. Immunol. Cell. Biol. 2012, 90, 540–552. [Google Scholar] [CrossRef]
- Kastenmuller, K.; Wille-Reece, U.; Lindsay, R.W.; Trager, L.R.; Darrah, P.A.; Flynn, B.J.; Becker, M.R.; Udey, M.C.; Clausen, B.E.; Igyarto, B.Z.; et al. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J. Clin. Investig. 2011, 121, 1782–1796. [Google Scholar] [CrossRef]
- Jelinek, I.; Leonard, J.N.; Price, G.E.; Brown, K.N.; Meyer-Manlapat, A.; Goldsmith, P.K.; Wang, Y.; Venzon, D.; Epstein, S.L.; Segal, D.M. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J. Immunol. 2011, 186, 2422–2429. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef]
- Wilson, N.S.; Behrens, G.M.; Lundie, R.J.; Smith, C.M.; Waithman, J.; Young, L.; Forehan, S.P.; Mount, A.; Steptoe, R.J.; Shortman, K.D.; et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 2006, 7, 165–172. [Google Scholar]
- Rhee, E.G.; Kelley, R.P.; Agarwal, I.; Lynch, D.M.; La Porte, A.; Simmons, N.L.; Clark, S.L.; Barouch, D.H. TLR4 ligands augment antigen-specific CD8+ T lymphocyte responses elicited by a viral vaccine vector. J. Virol. 2010, 84, 10413–10419. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Aldhamen, Y.A.; Depas, W.; Seregin, S.S.; Liu, C.J.; Schuldt, N.; Quach, D.; Quiroga, D.; Godbehere, S.; Zlatkin, I.; et al. A new adenovirus based vaccine vector expressing an Eimeria tenella derived TLR agonist improves cellular immune responses to an antigenic target. PLoS One 2010, 5, e9579. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Overwijk, W.W. Peptide-based anticancer vaccines: The making and unmaking of a T-cell graveyard. Oncoimmunology 2013, 2, e24743. [Google Scholar] [CrossRef]
- Salerno, E.P.; Shea, S.M.; Olson, W.C.; Petroni, G.R.; Smolkin, M.E.; McSkimming, C.; Chianese-Bullock, K.A.; Slingluff, C.L., Jr. Activation, dysfunction and retention of T cells in vaccine sites after injection of incomplete Freund’s adjuvant, with or without peptide. Cancer Immunol. Immunother. 2013, 62, 1149–1159. [Google Scholar] [CrossRef]
- Song, X.T.; Evel-Kabler, K.; Rollins, L.; Aldrich, M.; Gao, F.; Huang, X.F.; Chen, S.Y. An alternative and effective HIV vaccination approach based on inhibition of antigen presentation attenuators in dendritic cells. PLoS Med. 2006, 3, e11. [Google Scholar] [CrossRef]
- Seubert, A.; Calabro, S.; Santini, L.; Galli, B.; Genovese, A.; Valentini, S.; Aprea, S.; Colaprico, A.; D’Oro, U.; Giuliani, M.M.; et al. Adjuvanticity of the oil-in-water emulsion MF59 is independent of Nlrp3 inflammasome but requires the adaptor protein MyD88. Proc. Natl. Acad. Sci. USA 2011, 108, 11169–11174. [Google Scholar] [CrossRef]
- Shenderov, K.; Barber, D.L.; Mayer-Barber, K.D.; Gurcha, S.S.; Jankovic, D.; Feng, C.G.; Oland, S.; Hieny, S.; Caspar, P.; Yamasaki, S.; et al. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J. Immunol. 2013, 190, 5722–5730. [Google Scholar] [CrossRef]
- Gandhapudi, S.K.; Chilton, P.M.; Mitchell, T.C. TRIF is required for TLR4 mediated adjuvant effects on T cell clonal expansion. PLoS One 2013, 8, e56855. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Kool, M.; Fierens, K.; Lambrecht, B.N. Alum adjuvant: Some of the tricks of the oldest adjuvant. J. Med. Microbiol. 2012, 61, 927–934. [Google Scholar] [CrossRef]
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef]
- Hutchison, S.; Benson, R.A.; Gibson, V.B.; Pollock, A.H.; Garside, P.; Brewer, J.M. Antigen depot is not required for alum adjuvanticity. FASEB J. 2012, 26, 1272–1279. [Google Scholar] [CrossRef]
- Zuidscherwoude, M.; de Winde, C.M.; Cambi, A.; van Spriel, A.B. Microdomains in the membrane landscape shape antigen-presenting cell function. J. Leukoc. Biol. 2014, 95, 251–263. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Li, H.; Willingham, S.B.; Ting, J.P.; Re, F. Cutting edge: Inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J. Immunol. 2008, 181, 17–21. [Google Scholar]
- Kool, M.; Petrilli, V.; de Smedt, T.; Rolaz, A.; Hammad, H.; van Nimwegen, M.; Bergen, I.M.; Castillo, R.; Lambrecht, B.N.; Tschopp, J. Cutting edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 2008, 181, 3755–3759. [Google Scholar]
- Vaine, C.A.; Patel, M.K.; Zhu, J.; Lee, E.; Finberg, R.W.; Hayward, R.C.; Kurt-Jones, E.A. Tuning innate immune activation by surface texturing of polymer microparticles: The role of shape in inflammasome activation. J. Immunol. 2013, 190, 3525–3532. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, X.; Leitner, W.W.; Aldridge, M.E.; Zheng, J.; Yu, Z.; Restifo, N.P.; Weiss, R.; Scheiblhofer, S.; Xie, C.; et al. Polymeric structure and host Toll-like receptor 4 dictate immunogenicity of NY-ESO-1 antigen in vivo. J. Biol. Chem. 2011, 286, 37077–37084. [Google Scholar] [CrossRef]
- Timmermans, K.; Plantinga, T.S.; Kox, M.; Vaneker, M.; Scheffer, G.J.; Adema, G.J.; Joosten, L.A.; Netea, M.G. Blueprints of signaling interactions between pattern recognition receptors: Implications for the design of vaccine adjuvants. Clin. Vaccine Immunol. 2013, 20, 427–432. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Quaak, S.G.; Beijnen, J.H.; Hennink, W.E.; Storm, G.; Schumacher, T.N.; Haanen, J.B.; Nuijen, B. Lipopolysaccharide contamination in intradermal DNA vaccination: Toxic impurity or adjuvant? Int. J. Pharm. 2010, 390, 32–36. [Google Scholar] [CrossRef]
- Kool, M.; Soullie, T.; van Nimwegen, M.; Willart, M.A.; Muskens, F.; Jung, S.; Hoogsteden, H.C.; Hammad, H.; Lambrecht, B.N. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 2008, 205, 869–882. [Google Scholar] [CrossRef]
- Ferguson, T.A.; Choi, J.; Green, D.R. Armed response: How dying cells influence T-cell functions. Immunol. Rev. 2011, 241, 77–88. [Google Scholar] [CrossRef]
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef]
- Schon, M.P.; Wienrich, B.G.; Drewniok, C.; Bong, A.B.; Eberle, J.; Geilen, C.C.; Gollnick, H.; Schon, M. Death receptor-independent apoptosis in malignant melanoma induced by the small-molecule immune response modifier imiquimod. J. Investig. Dermatol. 2004, 122, 1266–1276. [Google Scholar] [CrossRef]
- Leitner, W.W.; Hwang, L.N.; Bergmann-Leitner, E.S.; Finkelstein, S.E.; Frank, S.; Restifo, N.P. Apoptosis is essential for the increased efficacy of alphaviral replicase-based DNA vaccines. Vaccine 2004, 22, 1537–1544. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Leitner, W.W. Danger, death and DNA vaccines. Microbes Infect. 2004, 6, 319–327. [Google Scholar] [CrossRef]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptors’ two-edged sword: When immunity meets apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef]
- Schon, M.; Bong, A.B.; Drewniok, C.; Herz, J.; Geilen, C.C.; Reifenberger, J.; Benninghoff, B.; Slade, H.B.; Gollnick, H.; Schon, M.P. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J. Natl. Cancer Inst. 2003, 95, 1138–1149. [Google Scholar] [CrossRef]
- Wegmann, F.; Gartlan, K.H.; Harandi, A.M.; Brinckmann, S.A.; Coccia, M.; Hillson, W.R.; Kok, W.L.; Cole, S.; Ho, L.P.; Lambe, T.; et al. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat. Biotechnol. 2012, 30, 883–888. [Google Scholar] [CrossRef]
- Yang, Y.W.; Wu, C.A.; Morrow, W.J. The apoptotic and necrotic effects of tomatine adjuvant. Vaccine 2004, 22, 2316–2327. [Google Scholar] [CrossRef]
- Marichal, T.; Ohata, K.; Bedoret, D.; Mesnil, C.; Sabatel, C.; Kobiyama, K.; Lekeux, P.; Coban, C.; Akira, S.; Ishii, K.J.; et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat. Med. 2011, 17, 996–1002. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Lima, H., Jr.; Jacobson, L.S.; Goldberg, M.F.; Chandran, K.; Diaz-Griffero, F.; Lisanti, M.P.; Brojatsch, J. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle 2013, 12, 1868–1878. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Complete lysosomal disruption: A route to necrosis, not to the inflammasome. Cell Cycle 2013, 12. [Google Scholar] [CrossRef]
- Kono, H.; Orlowski, G.M.; Patel, Z.; Rock, K.L. The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J. Immunol. 2012, 189, 3734–3740. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Seubert, A.; Monaci, E.; Pizza, M.; O’Hagan, D.T.; Wack, A. The adjuvants aluminum hydroxide and MF59 induce monocyte and granulocyte chemoattractants and enhance monocyte differentiation toward dendritic cells. J. Immunol. 2008, 180, 5402–5412. [Google Scholar]
- Vinuesa, C.G.; Chang, P.P. Innate B cell helpers reveal novel types of antibody responses. Nat. Immunol. 2013, 14, 119–126. [Google Scholar] [CrossRef]
- Schmieg, J.; Yang, G.; Franck, R.W.; Tsuji, M. A multifactorial mechanism in the superior antimalarial activity of alpha-C-GalCer. J. Biomed. Biotechnol. 2010. [Google Scholar] [CrossRef]
- Schmieg, J.; Gonzalez-Aseguinolaza, G.; Tsuji, M. The role of natural killer T cells and other T cell subsets against infection by the pre-erythrocytic stages of malaria parasites. Microbes Infect. 2003, 5, 499–506. [Google Scholar] [CrossRef]
- Padte, N.N.; Li, X.; Tsuji, M.; Vasan, S. Clinical development of a novel CD1d-binding NKT cell ligand as a vaccine adjuvant. Clin. Immunol. 2011, 140, 142–151. [Google Scholar] [CrossRef]
- Guillonneau, C.; Mintern, J.D.; Hubert, F.X.; Hurt, A.C.; Besra, G.S.; Porcelli, S.; Barr, I.G.; Doherty, P.C.; Godfrey, D.I.; Turner, S.J. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 3330–3335. [Google Scholar]
- Kamijuku, H.; Nagata, Y.; Jiang, X.; Ichinohe, T.; Tashiro, T.; Mori, K.; Taniguchi, M.; Hase, K.; Ohno, H.; Shimaoka, T.; et al. Mechanism of NKT cell activation by intranasal coadministration of alpha-galactosylceramide, which can induce cross-protection against influenza viruses. Mucosal Immunol. 2008, 1, 208–218. [Google Scholar] [CrossRef]
- Gonzalez-Aseguinolaza, G.; van Kaer, L.; Bergmann, C.C.; Wilson, J.M.; Schmieg, J.; Kronenberg, M.; Nakayama, T.; Taniguchi, M.; Koezuka, Y.; Tsuji, M. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J. Exp. Med. 2002, 195, 617–624. [Google Scholar] [CrossRef]
- Devera, T.S.; Shah, H.B.; Lang, G.A.; Lang, M.L. Glycolipid-activated NKT cells support the induction of persistent plasma cell responses and antibody titers. Eur. J. Immunol. 2008, 38, 1001–1011. [Google Scholar] [CrossRef]
- Bai, L.; Deng, S.; Reboulet, R.; Mathew, R.; Teyton, L.; Savage, P.B.; Bendelac, A. Natural killer T (NKT)-B-cell interactions promote prolonged antibody responses and long-term memory to pneumococcal capsular polysaccharides. Proc. Natl. Acad. Sci. USA 2013, 110, 16097–16102. [Google Scholar]
- Chang, D.H.; Osman, K.; Connolly, J.; Kukreja, A.; Krasovsky, J.; Pack, M.; Hutchinson, A.; Geller, M.; Liu, N.; Annable, R.; et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 2005, 201, 1503–1517. [Google Scholar] [CrossRef]
- McKee, A.S.; Munks, M.W.; MacLeod, M.K.; Fleenor, C.J.; van Rooijen, N.; Kappler, J.W.; Marrack, P. Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J. Immunol. 2009, 183, 4403–4414. [Google Scholar] [CrossRef]
- Paton, W.D. Compound 48/80: A potent histamine liberator. Br. J. Pharmacol. Chemother. 1951, 6, 499–508. [Google Scholar] [CrossRef]
- McLachlan, J.B.; Shelburne, C.P.; Hart, J.P.; Pizzo, S.V.; Goyal, R.; Brooking-Dixon, R.; Staats, H.F.; Abraham, S.N. Mast cell activators: A new class of highly effective vaccine adjuvants. Nat. Med. 2008, 14, 536–541. [Google Scholar] [CrossRef]
- Esposito, S.; Montinaro, V.; Groppali, E.; Tenconi, R.; Semino, M.; Principi, N. Live attenuated intranasal influenza vaccine. Hum. Vaccin. Immunother. 2012, 8, 76–80. [Google Scholar] [CrossRef]
- Harkema, J.R.; Carey, S.A.; Wagner, J.G. The nose revisited: A brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol. Pathol. 2006, 34, 252–269. [Google Scholar] [CrossRef]
- Ichinohe, T.; Tamura, S.I.; Kawaguchi, A.; Ninomiya, A.; Imai, M.; Itamura, S.; Odagiri, T.; Tashiro, M.; Takahashi, H.; Sawa, H.; et al. Cross-protection against H5N1 influenza virus infection is afforded by intranasal inoculation with seasonal trivalent inactivated influenza vaccine. J. Infect. Dis. 2007, 196, 1313–1320. [Google Scholar] [CrossRef]
- Martinez-Gil, L.; Goff, P.H.; Hai, R.; Garcia-Sastre, A.; Shaw, M.L.; Palese, P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J. Virol. 2013, 87, 1290–1300. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef]
- Fujihashi, K.; Koga, T.; van Ginkel, F.W.; Hagiwara, Y.; McGhee, J.R. A dilemma for mucosal vaccination: Efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine 2002, 20, 2431–2438. [Google Scholar] [CrossRef]
- Das, S.C.; Hatta, M.; Wilker, P.R.; Myc, A.; Hamouda, T.; Neumann, G.; Baker, J.R.; Kawaoka, Y. Nanoemulsion W(80)5EC improves immune responses upon intranasal delivery of an inactivated pandemic H1N1 influenza vaccine. Vaccine 2012, 30, 6871–6877. [Google Scholar] [CrossRef]
- Bielinska, A.U.; Gerber, M.; Blanco, L.P.; Makidon, P.E.; Janczak, K.W.; Beer, M.; Swanson, B.; Baker, J.R., Jr. Induction of Th17 cellular immunity with a novel nanoemulsion adjuvant. Crit. Rev. Immunol. 2010, 30, 189–199. [Google Scholar] [CrossRef]
- Miller, M.S.; Gardner, T.J.; Krammer, F.; Aguado, L.C.; Tortorella, D.; Basler, C.F.; Palese, P. Neutralizing antibodies against previously encountered influenza virus strains increase over time: A longitudinal analysis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Ballou, W.R. The development of the RTS,S malaria vaccine candidate: Challenges and lessons. Parasite Immunol. 2009, 31, 492–500. [Google Scholar] [CrossRef]
- Hutnick, N.A.; Myles, D.J.; Bian, C.B.; Muthumani, K.; Weiner, D.B. Selected approaches for increasing HIV DNA vaccine immunogenicity in vivo. Curr. Opin. Virol. 2011, 1, 233–240. [Google Scholar] [CrossRef]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef]
- Leitner, W.W.; Seguin, M.C.; Ballou, W.R.; Seitz, J.P.; Schultz, A.M.; Sheehy, M.J.; Lyon, J.A. Immune responses induced by intramuscular or gene gun injection of protective deoxyribonucleic acid vaccines that express the circumsporozoite protein from Plasmodium berghei malaria parasites. J. Immunol. 1997, 159, 6112–6119. [Google Scholar]
- Kester, K.E.; Cummings, J.F.; Ockenhouse, C.F.; Nielsen, R.; Hall, B.T.; Gordon, D.M.; Schwenk, R.J.; Krzych, U.; Holland, C.A.; Richmond, G.; et al. Phase 2a trial of 0, 1, and 3 month and 0, 7, and 28 day immunization schedules of malaria vaccine RTS,S/AS02 in malaria-naive adults at the Walter Reed Army Institute of Research. Vaccine 2008, 26, 2191–2202. [Google Scholar] [CrossRef]
- Stoute, J.A.; Slaoui, M.; Heppner, D.G.; Momin, P.; Kester, K.E.; Desmons, P.; Wellde, B.T.; Garcon, N.; Krzych, U.; Marchand, M. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. N. Engl. J. Med. 1997, 336, 86–91. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From vaccines to memory and back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. [Google Scholar]
- Leitner, W.W.; Hwang, L.N.; deVeer, M.J.; Zhou, A.; Silverman, R.H.; Williams, B.R.; Dubensky, T.W.; Ying, H.; Restifo, N.P. Alphavirus-based DNA vaccine breaks immunological tolerance by activating innate antiviral pathways. Nat. Med. 2003, 9, 33–39. [Google Scholar]
- Hawkins, W.G.; Gold, J.S.; Blachere, N.E.; Bowne, W.B.; Hoos, A.; Lewis, J.J.; Houghton, A.N. Xenogeneic DNA immunization in melanoma models for minimal residual disease. J. Surg. Res. 2002, 102, 137–143. [Google Scholar] [CrossRef]
- Crotty, S.; Johnston, R.J.; Schoenberger, S.P. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 2010, 11, 114–120. [Google Scholar] [CrossRef]
- Eng, N.F.; Bhardwaj, N.; Mulligan, R.; Diaz-Mitoma, F. The potential of 1018 ISS adjuvant in hepatitis B vaccines: HEPLISAV review. Hum. Vaccin Immunother. 2013, 9, 1661–1672. [Google Scholar] [CrossRef]
- FDA approves first adjuvanted vaccine for prevention of H5N1 avian influenza. Available online: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm376444.htm (accessed on 20 March 2014).
- Tsai, T.F.; Crucitti, A.; Nacci, P.; Nicolay, U.; Della Cioppa, G.; Ferguson, J.; Clemens, R. Explorations of clinical trials and pharmacovigilance databases of MF59(R)-adjuvanted influenza vaccines for associated cases of narcolepsy. Scand. J. Infect. Dis. 2011, 43, 702–706. [Google Scholar]
- Singh, A.K.; Mahlios, J.; Mignot, E. Genetic association, seasonal infections and autoimmune basis of narcolepsy. J. Autoimmun. 2013, 43, 26–31. [Google Scholar] [CrossRef]
- De la Herran-Arita, A.K.; Kornum, B.R.; Mahlios, J.; Jiang, W.; Lin, L.; Hou, T.; Macaubas, C.; Einen, M.; Plazzi, G.; Crowe, C.; et al. CD4+ T Cell Autoimmunity to Hypocretin/Orexin and Cross-Reactivity to a 2009 H1N1 Influenza A Epitope in Narcolepsy. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Arav-Boger, R.; Wojcik, G.L.; Duggal, P.; Ingersoll, R.G.; Beaty, T.; Pass, R.F.; Yolken, R.H. Polymorphisms in Toll-like receptor genes influence antibody responses to cytomegalovirus glycoprotein B vaccine. BMC Res. Notes. 2012, 5. [Google Scholar] [CrossRef]
- Cho, P.; Gelinas, L.; Corbett, N.P.; Tebbutt, S.J.; Turvey, S.E.; Fortuno, E.S., 3rd; Kollmann, T.R. Association of common single-nucleotide polymorphisms in innate immune genes with differences in TLR-induced cytokine production in neonates. Genes Immun. 2013, 14, 199–211. [Google Scholar] [CrossRef]
- Pothlichet, J.; Quintana-Murci, L. The genetics of innate immunity sensors and human disease. Int. Rev. Immunol. 2013, 32, 157–208. [Google Scholar] [CrossRef]
- Clifford, H.D.; Hayden, C.M.; Khoo, S.K.; Naniche, D.; Mandomando, I.M.; Zhang, G.; Richmond, P.; Le Souef, P.N. Polymorphisms in key innate immune genes and their effects on measles vaccine responses and vaccine failure in children from Mozambique. Vaccine 2012, 30, 6180–6185. [Google Scholar] [CrossRef]
- Ranatunga, D.; Hedrich, C.M.; Wang, F.; McVicar, D.W.; Nowak, N.; Joshi, T.; Feigenbaum, L.; Grant, L.R.; Stager, S.; Bream, J.H. A human IL10 BAC transgene reveals tissue-specific control of IL-10 expression and alters disease outcome. Proc. Natl. Acad. Sci. USA 2009, 106, 17123–17128. [Google Scholar] [CrossRef]
- Lambert, N.D.; Ovsyannikova, I.G.; Pankratz, V.S.; Jacobson, R.M.; Poland, G.A. Understanding the immune response to seasonal influenza vaccination in older adults: A systems biology approach. Expert Rev. Vaccines 2012, 11, 985–994. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Levy, O.; Montgomery, R.R.; Goriely, S. Innate immune function by Toll-like receptors: Distinct responses in newborns and the elderly. Immunity 2012, 37, 771–783. [Google Scholar] [CrossRef]
- Weber, D.J.; Rutala, W.A.; Samsa, G.P.; Santimaw, J.E.; Lemon, S.M. Obesity as a predictor of poor antibody response to hepatitis B plasma vaccine. JAMA 1985, 254, 3187–3189. [Google Scholar] [CrossRef]
- Eliakim, A.; Schwindt, C.; Zaldivar, F.; Casali, P.; Cooper, D.M. Reduced tetanus antibody titers in overweight children. Autoimmunity 2006, 39, 137–141. [Google Scholar] [CrossRef]
- Mastelic, B.; Kamath, A.T.; Fontannaz, P.; Tougne, C.; Rochat, A.F.; Belnoue, E.; Combescure, C.; Auderset, F.; Lambert, P.H.; Tacchini-Cottier, F.; et al. Environmental and T cell-intrinsic factors limit the expansion of neonatal follicular T helper cells but may be circumvented by specific adjuvants. J. Immunol. 2012, 189, 5764–5772. [Google Scholar] [CrossRef]
- McCarron, M.; Reen, D.J. Activated human neonatal CD8+ T cells are subject to immunomodulation by direct TLR2 or TLR5 stimulation. J. Immunol. 2009, 182, 55–62. [Google Scholar]
- Clemens, J.; Jodar, L. Introducing new vaccines into developing countries: Obstacles, opportunities and complexities. Nat. Med. 2005, 11, S12–S15. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Duncan, E.H.; Mease, R.M.; Angov, E. Impact of pre-existing MSP1(42)-allele specific immunity on potency of an erythrocytic Plasmodium falciparum vaccine. Malar. J. 2012, 11. [Google Scholar] [CrossRef]
- Halsey, N.A.; Boulos, R.; Mode, F.; Andre, J.; Bowman, L.; Yaeger, R.G.; Toureau, S.; Rohde, J.; Boulos, C. Response to measles vaccine in Haitian infants 6 to 12 months old. Influence of maternal antibodies, malnutrition, and concurrent illnesses. N. Engl. J. Med. 1985, 313, 544–549. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. FDA licensure of bivalent human papillomavirus vaccine (HPV2, Cervarix) for use in females and updated HPV vaccination recommendations from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 626–629. [Google Scholar]
- Condie, R.M.; Zak, S.J.; Good, R.A. Effect of meningococcal endotoxin on the immune response. Proc. Soc. Exp. Biol. Med. 1955, 90, 355–360. [Google Scholar] [CrossRef]
- McConkey, S.J.; Reece, W.H.; Moorthy, V.S.; Webster, D.; Dunachie, S.; Butcher, G.; Vuola, J.M.; Blanchard, T.J.; Gothard, P.; Watkins, K.; et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 2003, 9, 729–735. [Google Scholar] [CrossRef]
- Koup, R.A.; Roederer, M.; Lamoreaux, L.; Fischer, J.; Novik, L.; Nason, M.C.; Larkin, B.D.; Enama, M.E.; Ledgerwood, J.E.; Bailer, R.T.; et al. Priming immunization with DNA augments immunogenicity of recombinant adenoviral vectors for both HIV-1 specific antibody and T-cell responses. PLoS One 2010, 5, e9015. [Google Scholar] [CrossRef]
- Leroux-Roels, I.; Roman, F.; Forgus, S.; Maes, C.; de Boever, F.; Drame, M.; Gillard, P.; van der Most, R.; van Mechelen, M.; Hanon, E.; et al. Priming with AS03 A-adjuvanted H5N1 influenza vaccine improves the kinetics, magnitude and durability of the immune response after a heterologous booster vaccination: An open non-randomised extension of a double-blind randomised primary study. Vaccine 2010, 28, 849–857. [Google Scholar] [CrossRef]
- Zhu, Q.; Egelston, C.; Gagnon, S.; Sui, Y.; Belyakov, I.M.; Klinman, D.M.; Berzofsky, J.A. Using 3 TLR ligands as a combination adjuvant induces qualitative changes in T cell responses needed for antiviral protection in mice. J. Clin. Investig. 2010, 120, 607–616. [Google Scholar] [CrossRef]
- Kwissa, M.; Amara, R.R.; Robinson, H.L.; Moss, B.; Alkan, S.; Jabbar, A.; Villinger, F.; Pulendran, B. Adjuvanting a DNA vaccine with a TLR9 ligand plus Flt3 ligand results in enhanced cellular immunity against the simian immunodeficiency virus. J. Exp. Med. 2007, 204, 2733–2746. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Kaslow, D.C. Adjuvant-dependent immune response to malarial transmission-blocking vaccine candidate antigens. J. Exp. Med. 1992, 176, 1483–1487. [Google Scholar] [CrossRef]
- Ireton, R.C.; Gale, M., Jr. RIG-I like receptors in antiviral immunity and therapeutic applications. Viruses 2011, 3, 906–919. [Google Scholar] [CrossRef]
- Mercado-Lopez, X.; Cotter, C.R.; Kim, W.K.; Sun, Y.; Munoz, L.; Tapia, K.; Lopez, C.B. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 2013, 31, 5713–5721. [Google Scholar] [CrossRef]
- Yount, J.S.; Kraus, T.A.; Horvath, C.M.; Moran, T.M.; Lopez, C.B. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J. Immunol. 2006, 177, 4503–4513. [Google Scholar]
- Desel, C.; Werninghaus, K.; Ritter, M.; Jozefowski, K.; Wenzel, J.; Russkamp, N.; Schleicher, U.; Christensen, D.; Wirtz, S.; Kirschning, C.; et al. The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One 2013, 8, e53531. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bergmann-Leitner, E.S.; Leitner, W.W. Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators. Vaccines 2014, 2, 252-296. https://doi.org/10.3390/vaccines2020252
Bergmann-Leitner ES, Leitner WW. Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators. Vaccines. 2014; 2(2):252-296. https://doi.org/10.3390/vaccines2020252
Chicago/Turabian StyleBergmann-Leitner, Elke S., and Wolfgang W. Leitner. 2014. "Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators" Vaccines 2, no. 2: 252-296. https://doi.org/10.3390/vaccines2020252
APA StyleBergmann-Leitner, E. S., & Leitner, W. W. (2014). Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators. Vaccines, 2(2), 252-296. https://doi.org/10.3390/vaccines2020252