
CAR Beyond αβ T Cells: Unleashing NK Cells, Macrophages, and γδ T Lymphocytes Against Solid Tumors


Abstract

1. Introduction
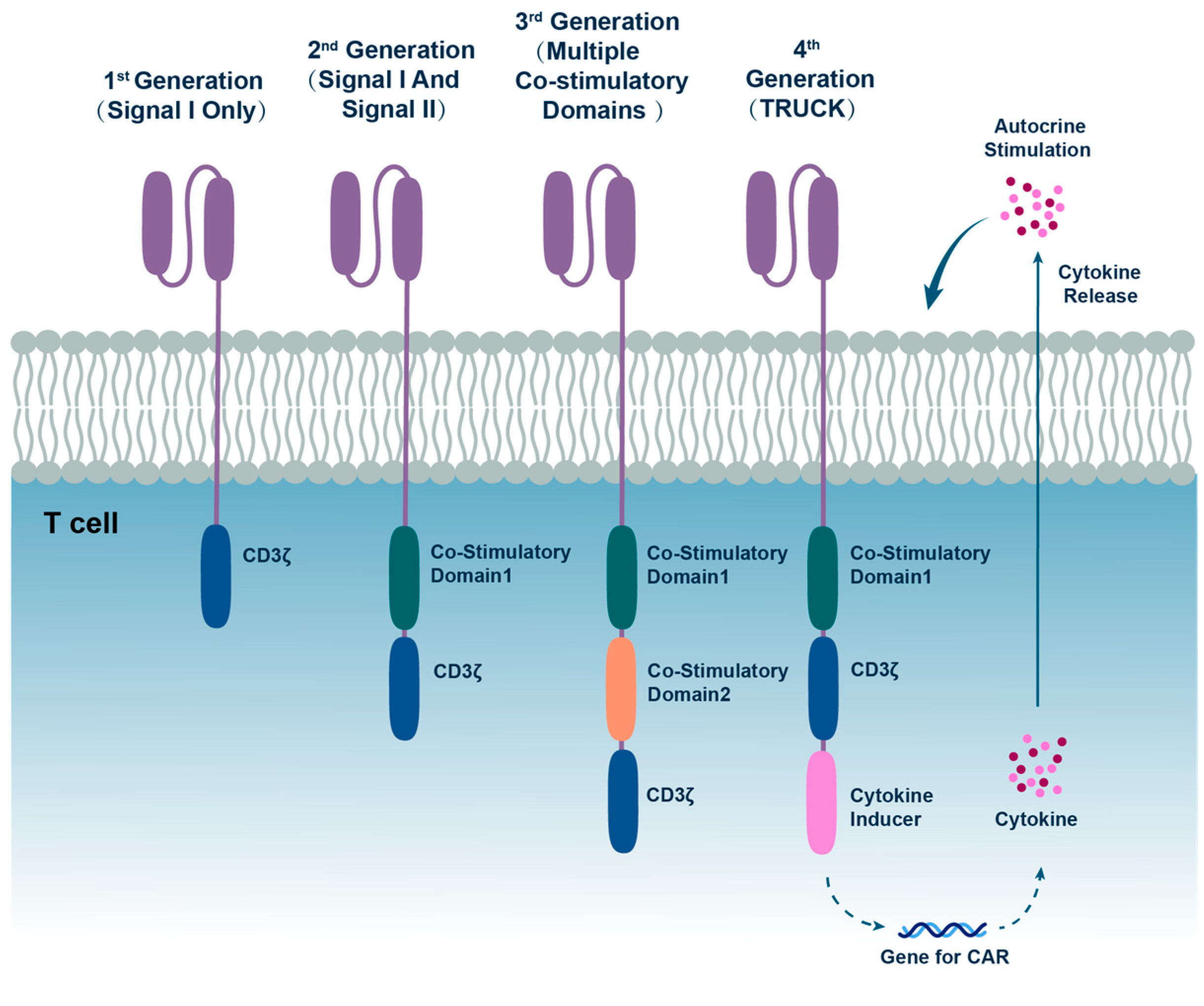
2. The Evolution of CAR Cell Therapy
3. Therapeutic Landscape of CAR-NK Therapy
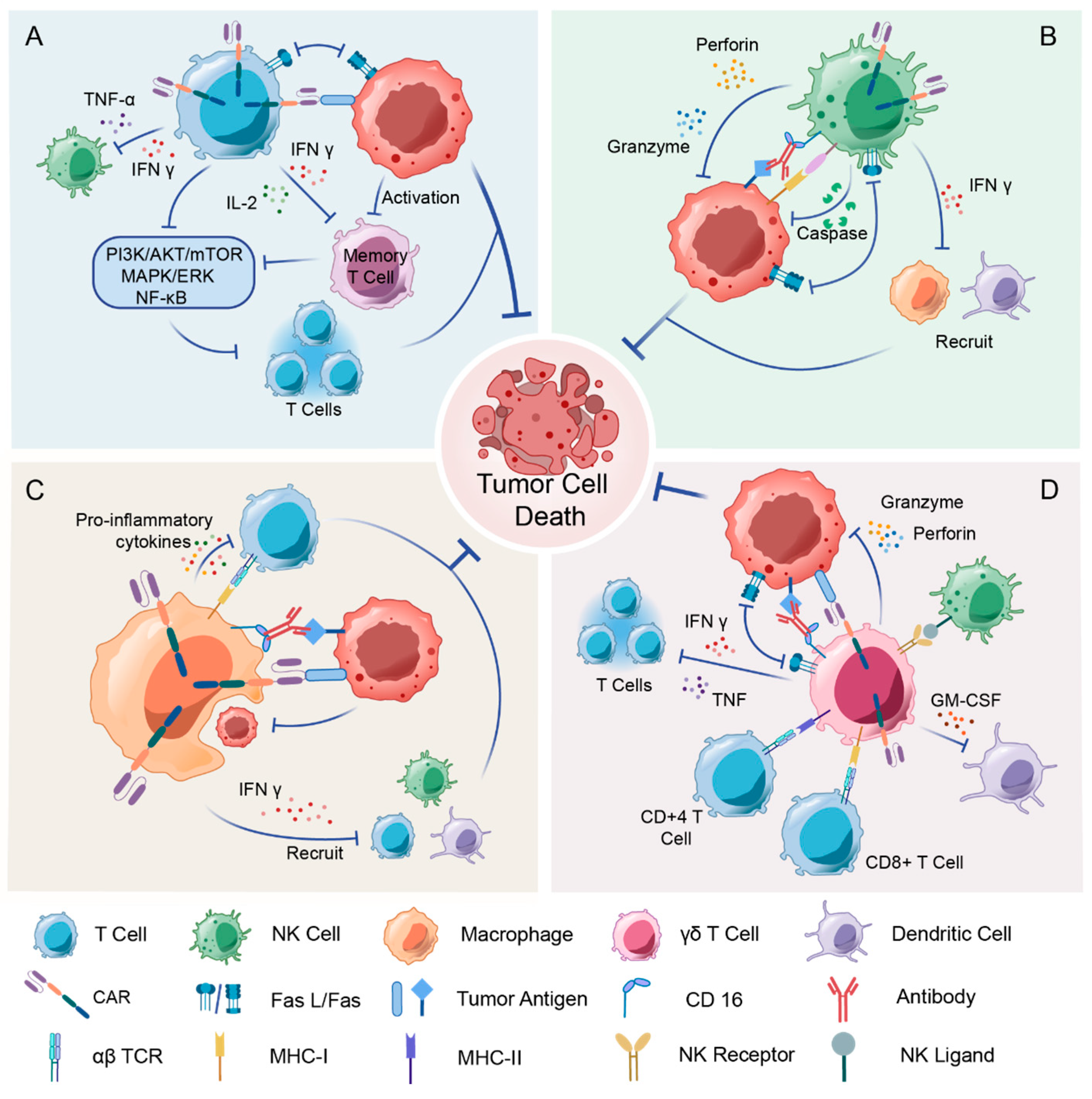
3.1. The Immune Function of NK Cell
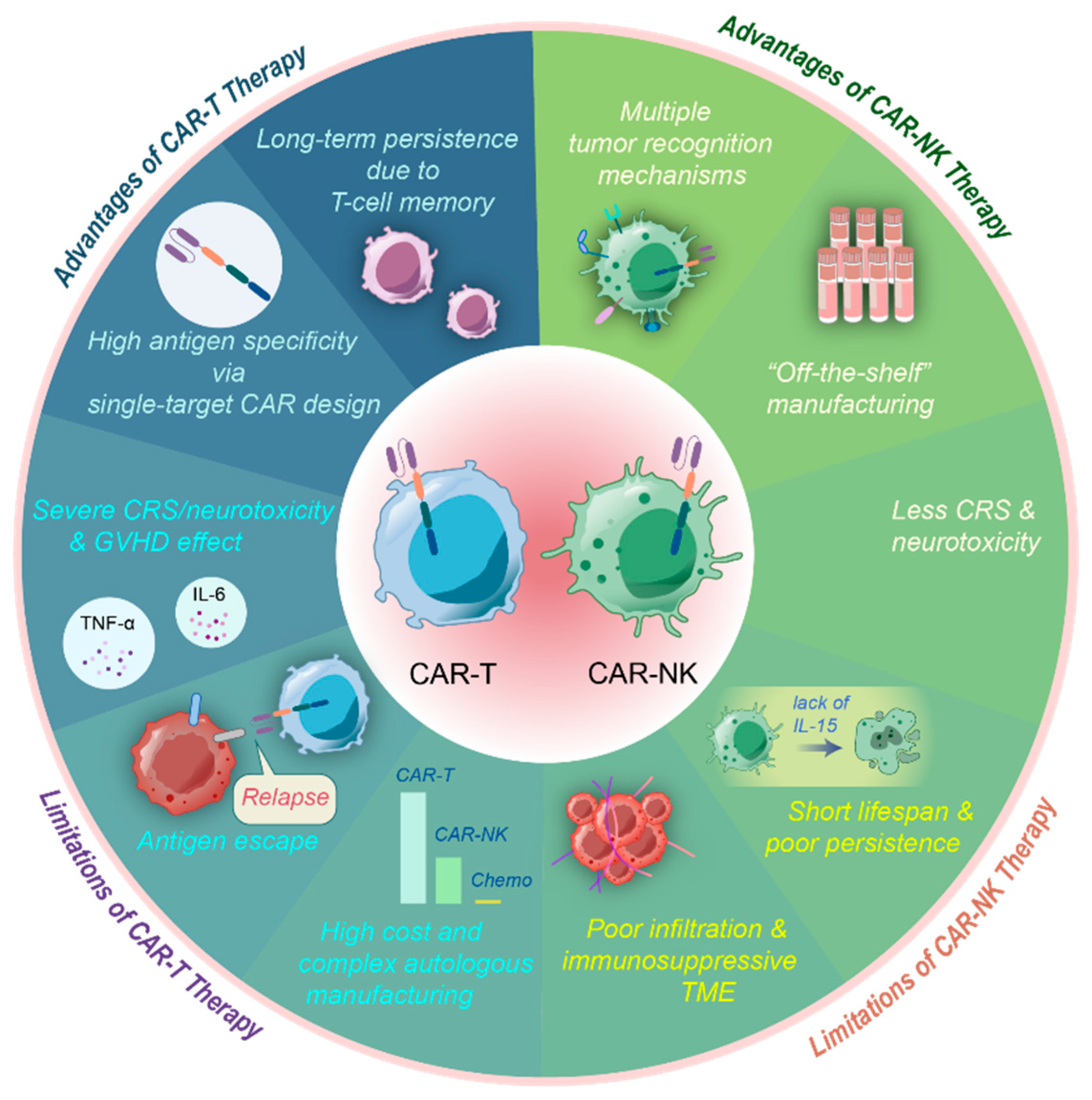
3.2. Advantages of CAR-NK Cells over CAR-T Cell Solid Tumors
3.3. Challenges and Therapies to Improve the Efficiency of CAR-NK
4. CAR-M Therapies as a Rising Horizon in Immunotherapy
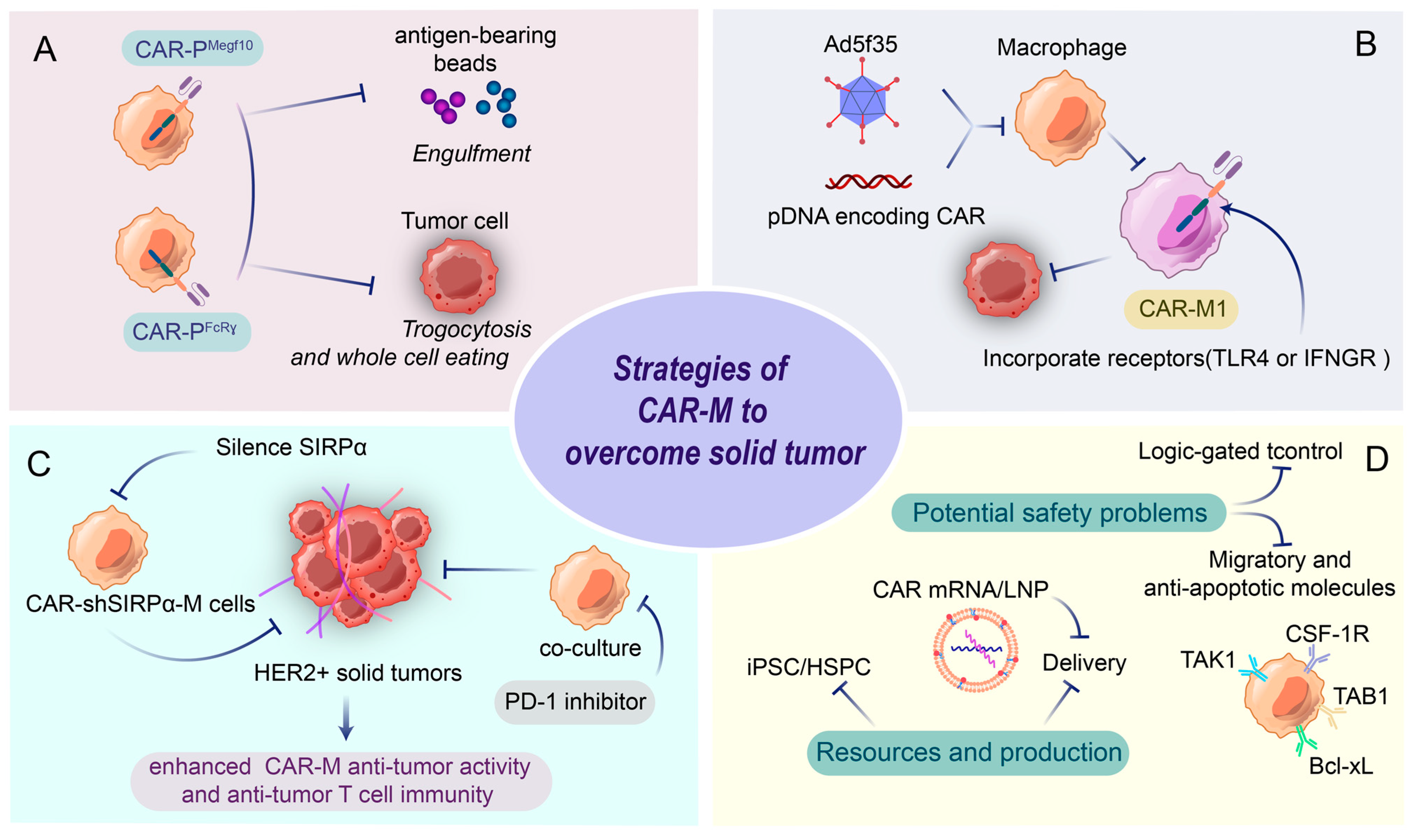
4.1. CAR-M in Solid Tumors
4.2. CAR Design for Macrophages: Boosting Phagocytosis and M1 Polarization
4.3. Challenges and Perspectives for CAR-M Therapy
5. CAR-γδ T: Appealing Immune Effector for Clinical Cancer Immunotherapy
5.1. The Biology of γδ T Cells
5.2. CAR-γδ T: Ideal Candidates for Cancer Immunotherapy
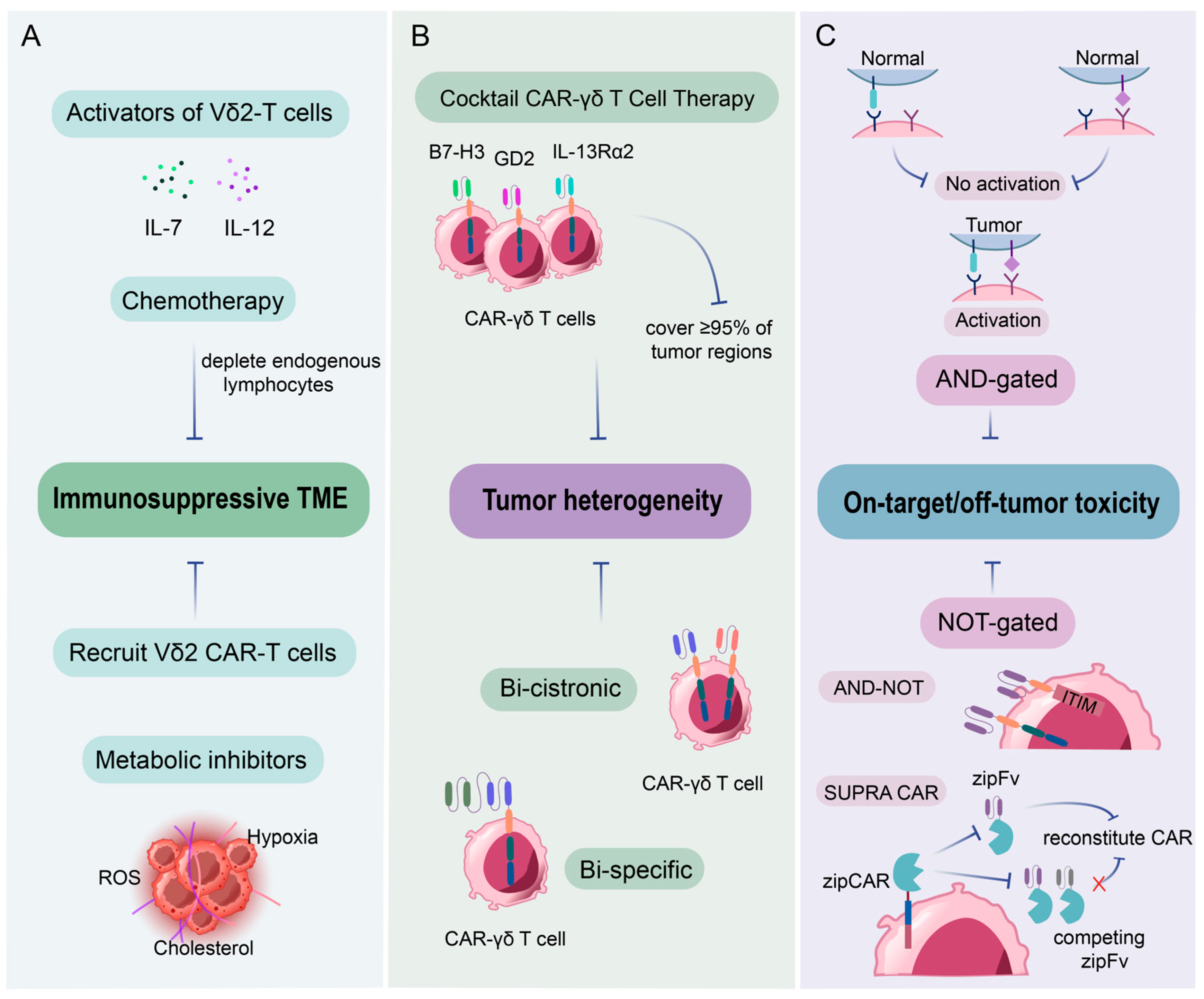
5.3. Challenges of CAR-γδ T Cells in Clinical Settings
6. Exploring Combination Therapies in CAR Cell Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Pan, S.; Wei, X.; Xu, X.; Wei, Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol. Ther. 2023, 31, 3146–3162. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar] [CrossRef]
- Leibson, P.J. Signal transduction during natural killer cell activation: Inside the mind of a killer. Immunity 1997, 6, 655–661. [Google Scholar] [CrossRef]
- Lanier, L.L. On guard--activating NK cell receptors. Nat. Immunol. 2001, 2, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Ravetch, J.V.; Lanier, L.L. Immune inhibitory receptors. Science 2000, 290, 84–89. [Google Scholar] [CrossRef]
- Stetson, D.B.; Mohrs, M.; Reinhardt, R.L.; Baron, J.L.; Wang, Z.E.; Gapin, L.; Kronenberg, M.; Locksley, R.M. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 2003, 198, 1069–1076. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Moretta, L.; Moretta, A. Cellular ligands of activating NK receptors. Trends Immunol. 2005, 26, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Kwon, D.; Moon, B.; Kim, D.H.; Kim, M.J.; Choi, J.; Kang, K.S. CD19 CAR-expressing iPSC-derived NK cells effectively enhance migration and cytotoxicity into glioblastoma by targeting to the pericytes in tumor microenvironment. Biomed. Pharmacother. 2024, 174, 116436. [Google Scholar] [CrossRef]
- Hu, Z.; Shen, R.; Campbell, A.; McMichael, E.; Yu, L.; Ramaswamy, B.; London, C.A.; Xu, T.; Carson, W.E., 3rd. Targeting Tissue Factor for Immunotherapy of Triple-Negative Breast Cancer Using a Second-Generation ICON. Cancer Immunol. Res. 2018, 6, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Yang, Y.; Li, W.; Tan, Y.; Guo, W.; Ao, L.; He, X.; Wu, X.; Xia, J.; Xu, X.; et al. Anti-αFR CAR-engineered NK-92 Cells Display Potent Cytotoxicity Against αFR-positive Ovarian Cancer. J. Immunother. 2019, 42, 284–296. [Google Scholar] [CrossRef]
- Lee, Y.E.; Ju, A.; Choi, H.W.; Kim, J.C.; Kim, E.E.; Kim, T.S.; Kang, H.J.; Kim, S.Y.; Jang, J.Y.; Ku, J.L.; et al. Rationally designed redirection of natural killer cells anchoring a cytotoxic ligand for pancreatic cancer treatment. J. Control Release 2020, 326, 310–323. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Dagher, O.K.; Posey, A.D., Jr. Forks in the road for CAR T and CAR NK cell cancer therapies. Nat. Immunol. 2023, 24, 1994–2007. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e185. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Voshol, H.; Dullens, H.F.; Den Otter, W.; Vliegenthart, J.F. Human natural killer cells: A convenient purification procedure and the influence of cryopreservation on cytotoxic activity. J. Immunol. Methods 1993, 165, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Johnson, R.; Yu, G.; McKenna, D.H.; Hubel, A. Preservation of cell-based immunotherapies for clinical trials. Cytotherapy 2019, 21, 943–957. [Google Scholar] [CrossRef]
- Das, S.; Niemeyer, E.; Leung, Z.A.; Fritsch, T.; Matosevic, S. Human Natural Killer Cells Cryopreserved without DMSO Sustain Robust Effector Responses. Mol. Pharm. 2024, 21, 651–660. [Google Scholar] [CrossRef]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Russick, J.; Torset, C.; Hemery, E.; Cremer, I. NK cells in the tumor microenvironment: Prognostic and theranostic impact. Recent advances and trends. Semin. Immunol. 2020, 48, 101407. [Google Scholar] [CrossRef]
- Townsend, M.J.; Weinmann, A.S.; Matsuda, J.L.; Salomon, R.; Farnham, P.J.; Biron, C.A.; Gapin, L.; Glimcher, L.H. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 2004, 20, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Hess, L.U.; Martrus, G.; Ziegler, A.E.; Langeneckert, A.E.; Salzberger, W.; Goebels, H.; Sagebiel, A.F.; Hagen, S.H.; Poch, T.; Ravichandran, G.; et al. The Transcription Factor Promyelocytic Leukemia Zinc Finger Protein Is Associated With Expression of Liver-Homing Receptors on Human Blood CD56(bright) Natural Killer Cells. Hepatol. Commun. 2020, 4, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Ran, G.H.; Lin, Y.Q.; Tian, L.; Zhang, T.; Yan, D.M.; Yu, J.H.; Deng, Y.C. Natural killer cell homing and trafficking in tissues and tumors: From biology to application. Signal Transduct. Target. Ther. 2022, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Tay, J.C.K.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther. Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef]
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef]
- Somanchi, S.S.; Somanchi, A.; Cooper, L.J.; Lee, D.A. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood 2012, 119, 5164–5172. [Google Scholar] [CrossRef]
- Zhu, A.; Bai, Y.; Nan, Y.; Ju, D. Natural killer cell engagers: From bi-specific to tri-specific and tetra-specific engagers for enhanced cancer immunotherapy. Clin. Transl. Med. 2024, 14, e70046. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Arora, J.; Ayyappan, S.; Yin, C.; Smith, B.J.; Lemke-Miltner, C.D.; Wang, Z.; Farooq, U.; Weiner, G.J. T-cell help in the tumor microenvironment enhances rituximab-mediated NK-cell ADCC. Blood 2024, 143, 1816–1824. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Y.; He, Z.; Li, L.; Liu, S.; Jiang, M.; Zhao, B.; Deng, M.; Wang, W.; Mi, X.; et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Törnroos, H.; Hägerstrand, H.; Lindqvist, C. Culturing the Human Natural Killer Cell Line NK-92 in Interleukin-2 and Interleukin-15—Implications for Clinical Trials. Anticancer. Res. 2019, 39, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Guo, M.; Huang, M.; Liu, Y.; Qian, Y.; Liu, Q.; Cao, X. Neoleukin-2/15-armored CAR-NK cells sustain superior therapeutic efficacy in solid tumors via c-Myc/NRF1 activation. Signal Transduct. Target. Ther. 2025, 10, 78. [Google Scholar] [CrossRef]
- Glienke, W.; Esser, R.; Priesner, C.; Suerth, J.D.; Schambach, A.; Wels, W.S.; Grez, M.; Kloess, S.; Arseniev, L.; Koehl, U. Advantages and applications of CAR-expressing natural killer cells. Front. Pharmacol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Frankel, N.W.; Deng, H.; Yucel, G.; Gainer, M.; Leemans, N.; Lam, A.; Li, Y.; Hung, M.; Lee, D.; Lee, C.T.; et al. Precision off-the-shelf natural killer cell therapies for oncology with logic-gated gene circuits. Cell Rep. 2024, 43, 114145. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Reiss, K.A.; Angelos, M.G.; Dees, E.C.; Yuan, Y.; Ueno, N.T.; Pohlmann, P.R.; Johnson, M.L.; Chao, J.; Shestova, O.; Serody, J.S.; et al. CAR-macrophage therapy for HER2-overexpressing advanced solid tumors: A phase 1 trial. Nat. Med. 2025, 31, 1171–1182. [Google Scholar] [CrossRef]
- Shah, Z.; Tian, L.; Li, Z.; Jin, L.; Zhang, J.; Li, Z.; Barr, T.; Tang, H.; Feng, M.; Caligiuri, M.A.; et al. Human anti-PSCA CAR macrophages possess potent antitumor activity against pancreatic cancer. Cell Stem Cell 2024, 31, 803–817.e806. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Lei, A.; Yu, H.; Lu, S.; Lu, H.; Ding, X.; Tan, T.; Zhang, H.; Zhu, M.; Tian, L.; Wang, X.; et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat. Immunol. 2024, 25, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Lecoultre, M.; Dutoit, V.; Walker, P.R. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: A review. J. Immunother. Cancer 2020, 8, e001408. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Zhang, H.; Huo, Y.; Zheng, W.; Li, P.; Li, H.; Zhang, L.; Sa, L.; He, Y.; Zhao, Z.; Shi, C.; et al. Silencing of SIRPα enhances the antitumor efficacy of CAR-M in solid tumors. Cell Mol. Immunol. 2024, 21, 1335–1349. [Google Scholar] [CrossRef]
- Pierini, S.; Gabbasov, R.; Oliveira-Nunes, M.C.; Qureshi, R.; Worth, A.; Huang, S.; Nagar, K.; Griffin, C.; Lian, L.; Yashiro-Ohtani, Y.; et al. Chimeric antigen receptor macrophages (CAR-M) sensitize HER2+ solid tumors to PD1 blockade in pre-clinical models. Nat. Commun. 2025, 16, 706. [Google Scholar] [CrossRef]
- Duan, Z.; Li, Z.; Wang, Z.; Chen, C.; Luo, Y. Chimeric antigen receptor macrophages activated through TLR4 or IFN-γ receptors suppress breast cancer growth by targeting VEGFR2. Cancer Immunol. Immunother. 2023, 72, 3243–3257. [Google Scholar] [CrossRef]
- Shi, Y.; Li, X.; Dong, Y.; Yuan, H.; Wang, Y.; Yang, R. Exploring the potential of CAR-macrophage therapy. Life Sci. 2025, 361, 123300. [Google Scholar] [CrossRef]
- Dong, X.; Fan, J.; Xie, W.; Wu, X.; Wei, J.; He, Z.; Wang, W.; Wang, X.; Shen, P.; Bei, Y. Efficacy evaluation of chimeric antigen receptor-modified human peritoneal macrophages in the treatment of gastric cancer. Br. J. Cancer 2023, 129, 551–562. [Google Scholar] [CrossRef]
- Ye, Z.; Chen, J.; Zhao, X.; Li, Y.; Harmon, J.; Huang, C.; Chen, J.; Xu, Q. In Vitro Engineering Chimeric Antigen Receptor Macrophages and T Cells by Lipid Nanoparticle-Mediated mRNA Delivery. ACS Biomater. Sci. Eng. 2022, 8, 722–733. [Google Scholar] [CrossRef]
- Li, N.; Geng, S.; Dong, Z.Z.; Jin, Y.; Ying, H.; Li, H.W.; Shi, L. A new era of cancer immunotherapy: Combining revolutionary technologies for enhanced CAR-M therapy. Mol. Cancer 2024, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Wong, N.M.; Tague, E.; Ngo, J.T.; Khalil, A.S.; Wong, W.W. High-performance multiplex drug-gated CAR circuits. Cancer Cell 2022, 40, 1294–1305.e4. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D. γδ T-cells: Cross-talk between innate and adaptive immunity. Cell Mol. Life Sci. 2011, 68, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Kozbor, D.; Trinchieri, G.; Monos, D.S.; Isobe, M.; Russo, G.; Haney, J.A.; Zmijewski, C.; Croce, C.M. Human TCR-gamma+/delta+, CD8+ T lymphocytes recognize tetanus toxoid in an MHC-restricted fashion. J. Exp. Med. 1989, 169, 1847–1851. [Google Scholar] [CrossRef]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef]
- Costa, G.P.; Mensurado, S.; Silva-Santos, B. Therapeutic avenues for γδ T cells in cancer. J. Immunother. Cancer 2023, 11, e007955. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Cortés-Selva, D.; Dasgupta, B.; Singh, S.; Grewal, I.S. Innate and Innate-Like Cells: The Future of Chimeric Antigen Receptor (CAR) Cell Therapy. Trends Pharmacol. Sci. 2021, 42, 45–59. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Girardi, M.; Oppenheim, D.E.; Steele, C.R.; Lewis, J.M.; Glusac, E.; Filler, R.; Hobby, P.; Sutton, B.; Tigelaar, R.E.; Hayday, A.C. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001, 294, 605–609. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, B.L.; Gehrs, B.C.; Nan, L.; Lopez, R.D. Ex vivo expanded human Vgamma9Vdelta2+ gammadelta-T cells mediate innate antitumor activity against human prostate cancer cells in vitro. J. Urol. 2005, 173, 1552–1556. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Eltoum, I.E.; Guo, B.; Beck, B.H.; Cloud, G.A.; Lopez, R.D. Protective immunosurveillance and therapeutic antitumor activity of gammadelta T cells demonstrated in a mouse model of prostate cancer. J. Immunol. 2008, 180, 6044–6053. [Google Scholar] [CrossRef]
- Kabelitz, D.; Wesch, D.; He, W. Perspectives of gammadelta T cells in tumor immunology. Cancer Res. 2007, 67, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, H.Y.; Peng, W.; Kiniwa, Y.; Seo, K.H.; Wang, R.F. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 2007, 27, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- van der Veken, L.T.; Coccoris, M.; Swart, E.; Falkenburg, J.H.; Schumacher, T.N.; Heemskerk, M.H. Alpha beta T cell receptor transfer to gamma delta T cells generates functional effector cells without mixed TCR dimers in vivo. J. Immunol. 2009, 182, 164–170. [Google Scholar] [CrossRef]
- van der Veken, L.T.; Hagedoorn, R.S.; van Loenen, M.M.; Willemze, R.; Falkenburg, J.H.; Heemskerk, M.H. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006, 66, 3331–3337. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Mirzaei, H.; Lee, S.Y.; Hadjati, J.; Till, B.G. Prospects for chimeric antigen receptor (CAR) γδ T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–423. [Google Scholar] [CrossRef]
- Almeida, A.R.; Correia, D.V.; Fernandes-Platzgummer, A.; da Silva, C.L.; da Silva, M.G.; Anjos, D.R.; Silva-Santos, B. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin. Cancer Res. 2016, 22, 5795–5804. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Ribot, J.C.; Ribeiro, S.T.; Correia, D.V.; Sousa, A.E.; Silva-Santos, B. Human γδ thymocytes are functionally immature and differentiate into cytotoxic type 1 effector T cells upon IL-2/IL-15 signaling. J. Immunol. 2014, 192, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yin, S.; Zhang, J.; Hu, Y.; Huang, B.; Cui, L.; Kang, N.; He, W. A new effect of IL-4 on human γδ T cells: Promoting regulatory Vδ1 T cells via IL-10 production and inhibiting function of Vδ2 T cells. Cell Mol. Immunol. 2016, 13, 217–228. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Abramowski, P.; Wisidagamage Don, N.D.; Flutter, B.; Capsomidis, A.; Cheung, G.W.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wang, W.; Hawes, I.; Han, J.; Yao, Z.; Bertaina, A. Advancements in γδT cell engineering: Paving the way for enhanced cancer immunotherapy. Front. Immunol. 2024, 15, 1360237. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Farhood, B.; Eleojo Musa, A.; Taeb, S.; Rezaeyan, A.; Najafi, M. Abscopal effect in radioimmunotherapy. Int. Immunopharmacol. 2020, 85, 106663. [Google Scholar] [CrossRef]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef]
- Chang, Y.; Chang, M.; Bao, X.; Dong, C. Advancements in adoptive CAR immune cell immunotherapy synergistically combined with multimodal approaches for tumor treatment. Bioact. Mater. 2024, 42, 379–403. [Google Scholar] [CrossRef]
- Wang, A.X.; Ong, X.J.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front. Immunol. 2023, 14, 1140541. [Google Scholar] [CrossRef]
- Noordam, L.; Kaijen, M.E.H.; Bezemer, K.; Cornelissen, R.; Maat, L.; Hoogsteden, H.C.; Aerts, J.; Hendriks, R.W.; Hegmans, J.; Vroman, H. Low-dose cyclophosphamide depletes circulating naïve and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology 2018, 7, e1474318. [Google Scholar] [CrossRef]
- Hou, Z.; Jiang, Y.; Fu, Y.; Ruan, M.; Meng, D.; Li, Y.; Zhao, D.; Yang, J.; Long, Z.; Ge, J. Cyclophosphamide Abrogates Immune Effector Cell-Associated Neurotoxicity Syndrome Associated With CAR-T Cell Therapy. Am. J. Hematol. 2025, 100, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, T.; Jiang, G.; Zeng, Q.; Li, Z.; Huang, X. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol. Cancer 2023, 22, 131. [Google Scholar] [CrossRef]
- Wei, J.; Luo, C.; Wang, Y.; Guo, Y.; Dai, H.; Tong, C.; Ti, D.; Wu, Z.; Han, W. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J. Immunother. Cancer 2019, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, e99573. [Google Scholar] [CrossRef]
- Porter, C.E.; Rosewell Shaw, A.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Zarezadeh Mehrabadi, A.; Roozbahani, F.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Esmaeili Gouvarchin Ghaleh, H. Overview of the pre-clinical and clinical studies about the use of CAR-T cell therapy of cancer combined with oncolytic viruses. World J. Surg. Oncol. 2022, 20, 16. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Gardell, J.L.; Matsumoto, L.R.; Chinn, H.; DeGolier, K.R.; Kreuser, S.A.; Prieskorn, B.; Balcaitis, S.; Davis, A.; Ellenbogen, R.G.; Crane, C.A. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J. Immunother. Cancer 2020, 8, e001202. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, J.; Liang, Z.; Dai, K.; Gan, J.; Wang, Q.; Xu, Y.; Chen, Y.H.; Wan, X. CAR-Macrophages and CAR-T Cells Synergistically Kill Tumor Cells In Vitro. Cells 2022, 11, 3692. [Google Scholar] [CrossRef] [PubMed]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H.; Rostami, S.; Jalil, A.T.; Al-Gazally, M.E.; Alsaikhan, F.; Rizaev, J.A.; Mohammad, T.A.M.; et al. CAR-T cell combination therapy: The next revolution in cancer treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Bhat, H.; Adusumilli, P.S.; Hou, Z. Combinational CAR T-cell therapy for solid tumors: Requisites, rationales, and trials. Pharmacol. Ther. 2025, 266, 108763. [Google Scholar] [CrossRef]
- Hovhannisyan, L.; Riether, C.; Aebersold, D.M.; Medová, M.; Zimmer, Y. CAR T cell-based immunotherapy and radiation therapy: Potential, promises and risks. Mol. Cancer 2023, 22, 82. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Key Events | Structure Features | Meanings |
|---|---|---|---|
| 1988 | TILs used to treat metastatic cancer | Enhanced tumor-specific immune response against metastatic cancer | |
| 1993 | A chimeric gene developed to provide effector lymphocytes with antibody-type recognition [4] | CAR consists of the extracellular domain, the transmembrane domain, and the intracellular signal transduction domain (CD3ζ chain) | 1st generation CAR-T prototype described |
| 1998 | CD3/CD28 beads induce ex vivo expansion of human T cell | A co-stimulation domain (CD 28) added to CAR | 2nd generation CAR-T |
| 2004 | 4-1BB signaling capacity provoked potent cytotoxicity against ALL [5] | 4-1BB used as the co-stimulation domain | 2nd generation CAR-T |
| 2008 | GD2- CAR-T cells showed antitumor activity and safety in neuroblastoma [6] | CAR is directed to the diasialoganglioside GD2 | Virus-specific CTLs can be modified to function as tumor-directed effector cells |
| 2010 | CD28 endodomain showed remarking enhanced expansion and persistence in lymphoma patients [7] | Combining two co-stimulatory molecules (CD28 ICOS, 4-1BB, OX40, and CD27) | 3rd generation CAR-T arise |
| 2011 | The remission of leukemia patients is ongoing after CAR-T treatment (2nd generation CAR-T) [8] | Promise of 2nd generation CAR-T in leukemia treatment. | |
| 2012 | Decade-long clinical trials proved the safety and function of 1st generation CAR-T for HIV [9]; engineered CAR-T cells deliver inducible IL-12 to combat cancer | Engineer CAR-T with IL-15/IL-12/IL-18 and a suicide gene | Paved the way for CAR-T therapy in HIV treatment; 4th generation CAR-T arise |
| 2017 | FDA approval CD19 CAR-T therapy for leukemia | Marked a major breakthrough in cancer treatment, especially offering new hope for patients with relapsed or refractory leukemia | |
| 2021 | FDA approval of: abecma for multiple myeloma; CAR-T for autoimmune disease (SLE) | The achievement of CAR-T in multiple myeloma treatment and the expansion of CAR-T therapy into new disease areas | |
| 2023 | Mutated c-KIT added to CAR-T | Focus on solid tumors |
| CAR-T | CAR-NK | CAR-M | CAR-γδ T | |
|---|---|---|---|---|
| Mechanisms of cell killing | CAR-dependent T-mediated cell killing; cytokine release; antigen presentation; TME remodeling | CAR-dependent NK -mediated cell killing; innate cytotoxicity; cytokine release; ADCC | CAR-dependent phagocytosis, cytotoxicity, pro-inflammatory secretion, antigen presentation, TME remodeling | CAR-dependent cell killing, indirect antitumor contribution, antigen presentation, direct cytotoxicity, cytokine release, ADCC |
| Cellular sources | Autologous, MHC-matched allogeneic, T cell lines | Autologous, non-MHC-matched allogeneic, NK cell lines | Autologous (iPSCs and cell lines are used in preclinical studies) | Autologous or allogeneic γδ T cell lines (peripheral blood, iPSC-derived, tissue-resident γδ T cells) |
| In vitro expansion | Effectively expanded in vivo using optimized culture conditions and cytokines | Efficiently expanded in vitro with specific cytokines | Limited ability to expand, but alternative sources (iPSC and cell lines) can be used | Readily expandable in vitro (especially Vγ9Vδ2) |
| Production | Time-consuming and costly | “Off-the-shelf” products | Time-consuming, but with potential for “off-the-shelf”, low-cost, and standardized products | Potentially “off-the-shelf” products |
| Antigen recognition | Specific antigen recognition via CAR | Specific antigen recognition via CAR, with additional innate cytotoxicity | Specific antigen recognition via CAR, with phagocytic and antigen-presenting capabilities | Specific antigen recognition via CAR, with additional innate TCR-mediated recognition |
| Infiltration in solid tumors | Poor | Poor | Abundant | Moderate |
| Persistence | Long-term | Short-term | Moderate to long-term (depends on the immune environment) | Lack of clear persistence data |
| Toxicities | Common and serious CRS/neurotoxicity; GVHD; on-target/off-tumor toxicity | Less common and serious CRS/neurotoxicity; do not cause GVHD | Lack of clear clinical data | No formal study comparing the toxicities so far; serious CRS has not been reported in preclinical studies so far |
| Clinical status | Proven efficacy in hematologic malignancies; six CAR-T therapies approved by the FDA | Limited clinical trials and no approved therapy. Three trials have been completed; one trial has been published | Still at an early stage; a first-in-human (phase 1) multicenter clinical trial has been published | No approved therapy; several early-phase trials are ongoing |
| Advantages | Prolonged durability; proved strong efficiency in hematologic malignancies; high antigen specificity | Abundant cell sources; providing “Off-the-shelf” products; low toxicity; multiple cell killing mechanisms | Abundant infiltration in solid tumor; various alternative sources; M1 macrophages in TME have pro-inflammatory and anti-tumor effects; phagocytosis of TAMs is key for tumor metastasis closely related to TME | Multiple killing mechanisms; broad-spectrum cancer cell recognition; ease of expansion; specialized antigens presenting |
| Disadvantages | Poor tumor trafficking and infiltration; lack of antigen heterogeneity; limited persistence in the immunosuppressive TME; serious toxicities; high cost of manufacturing | Very poor tumor infiltration; low CAR transduction efficiency; short-term persistence; limited ex vivo expansion | Difficult to transduce with virus; the phenotypes of macrophages change dynamically in TME | Low clearance rate of tumor cells in vivo; immunosuppressive TME has a significant impact on persistence and cytotoxic activity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xian, Y.; Wen, L. CAR Beyond αβ T Cells: Unleashing NK Cells, Macrophages, and γδ T Lymphocytes Against Solid Tumors. Vaccines 2025, 13, 654. https://doi.org/10.3390/vaccines13060654
Xian Y, Wen L. CAR Beyond αβ T Cells: Unleashing NK Cells, Macrophages, and γδ T Lymphocytes Against Solid Tumors. Vaccines. 2025; 13(6):654. https://doi.org/10.3390/vaccines13060654
Chicago/Turabian StyleXian, Yunjia, and Lu Wen. 2025. "CAR Beyond αβ T Cells: Unleashing NK Cells, Macrophages, and γδ T Lymphocytes Against Solid Tumors" Vaccines 13, no. 6: 654. https://doi.org/10.3390/vaccines13060654
APA StyleXian, Y., & Wen, L. (2025). CAR Beyond αβ T Cells: Unleashing NK Cells, Macrophages, and γδ T Lymphocytes Against Solid Tumors. Vaccines, 13(6), 654. https://doi.org/10.3390/vaccines13060654