

Safety, Tolerability, and Immunogenicity of a DNA Vaccine (pGX9501) Against SARS-CoV-2 in Healthy Volunteers: A Single-Center, Randomized, Double-Blind, Placebo-Controlled, and Dose-Ranging Phase I Trial
 ,
,
Abstract
1. Introduction
2. Methods
2.1. Study Design and Participants
2.2. Declaration
2.3. Randomization and Masking
2.4. Objectives and Endpoints
2.5. Procedures
2.5.1. Adverse Events Collection
2.5.2. Immunogenicity Analysis
2.5.3. Gut Microbiota Composition Testing Method
2.6. Statistical Analysis
3. Results
3.1. Study Population
3.2. Demographic and Baseline Characteristics
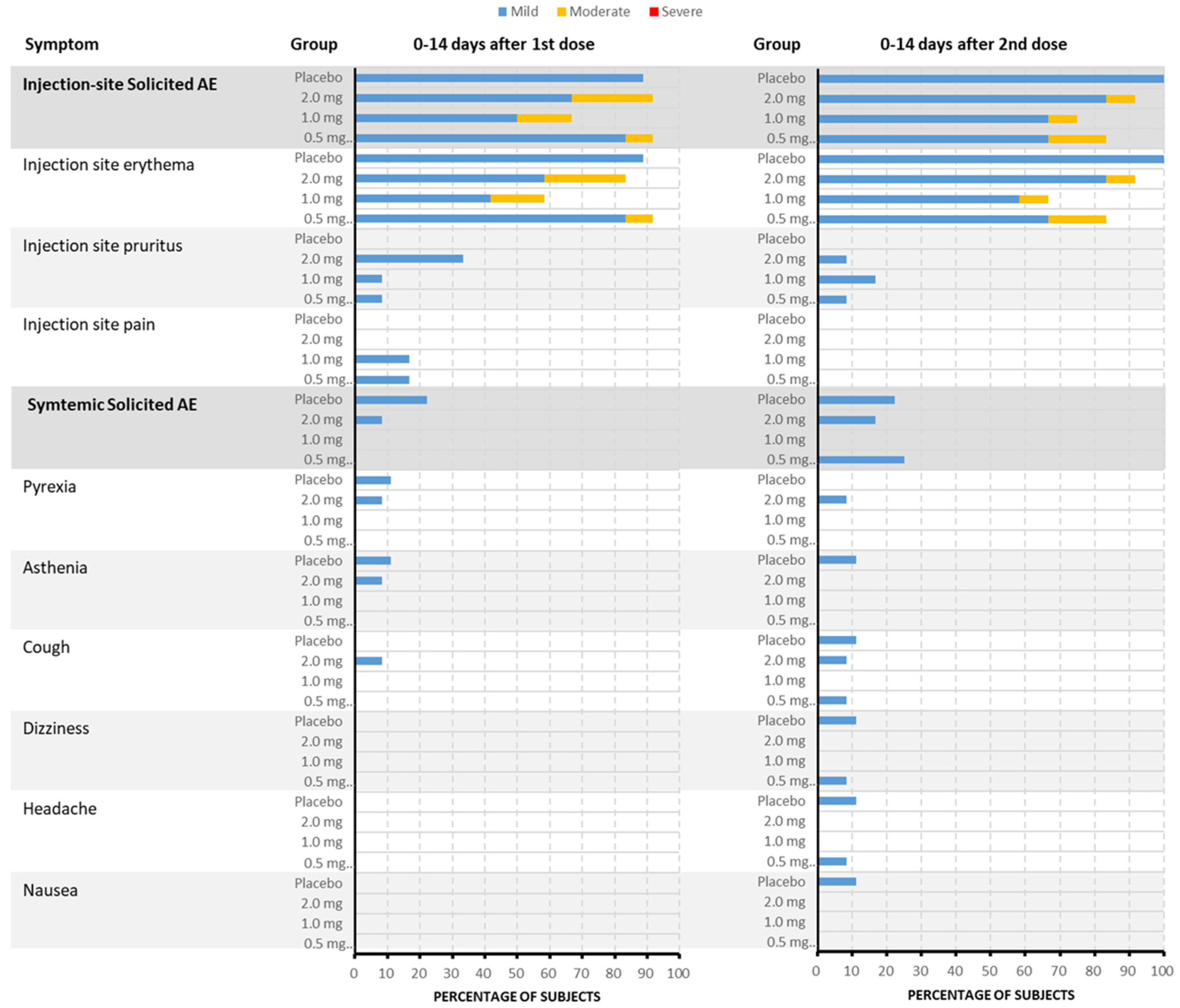
3.3. Safety and Tolerability
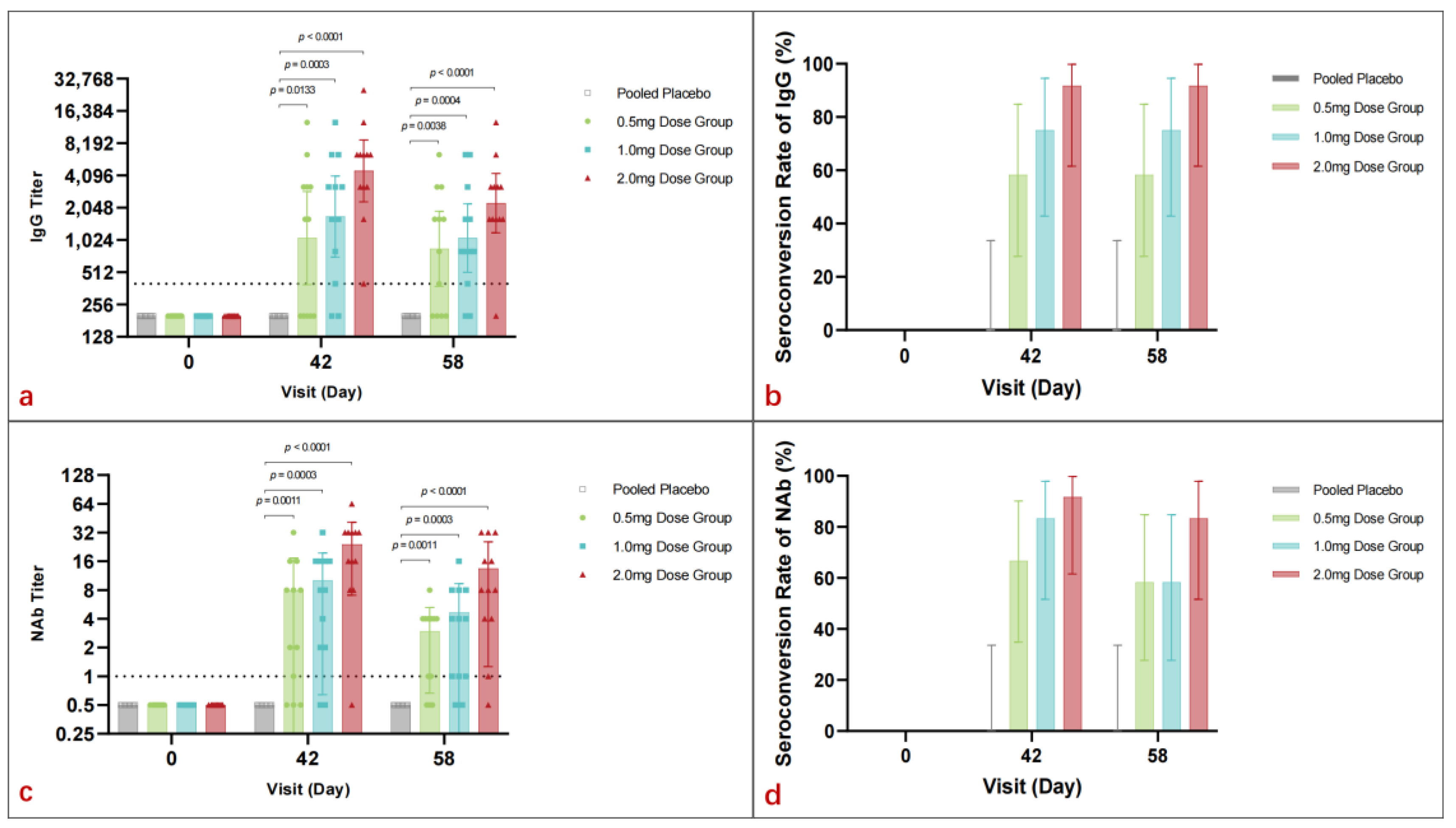
3.4. Immunogenicity
3.4.1. Spike-Binding Antibody
3.4.2. Neutralizing Antibody
3.4.3. IFN-γ Elispot Assay
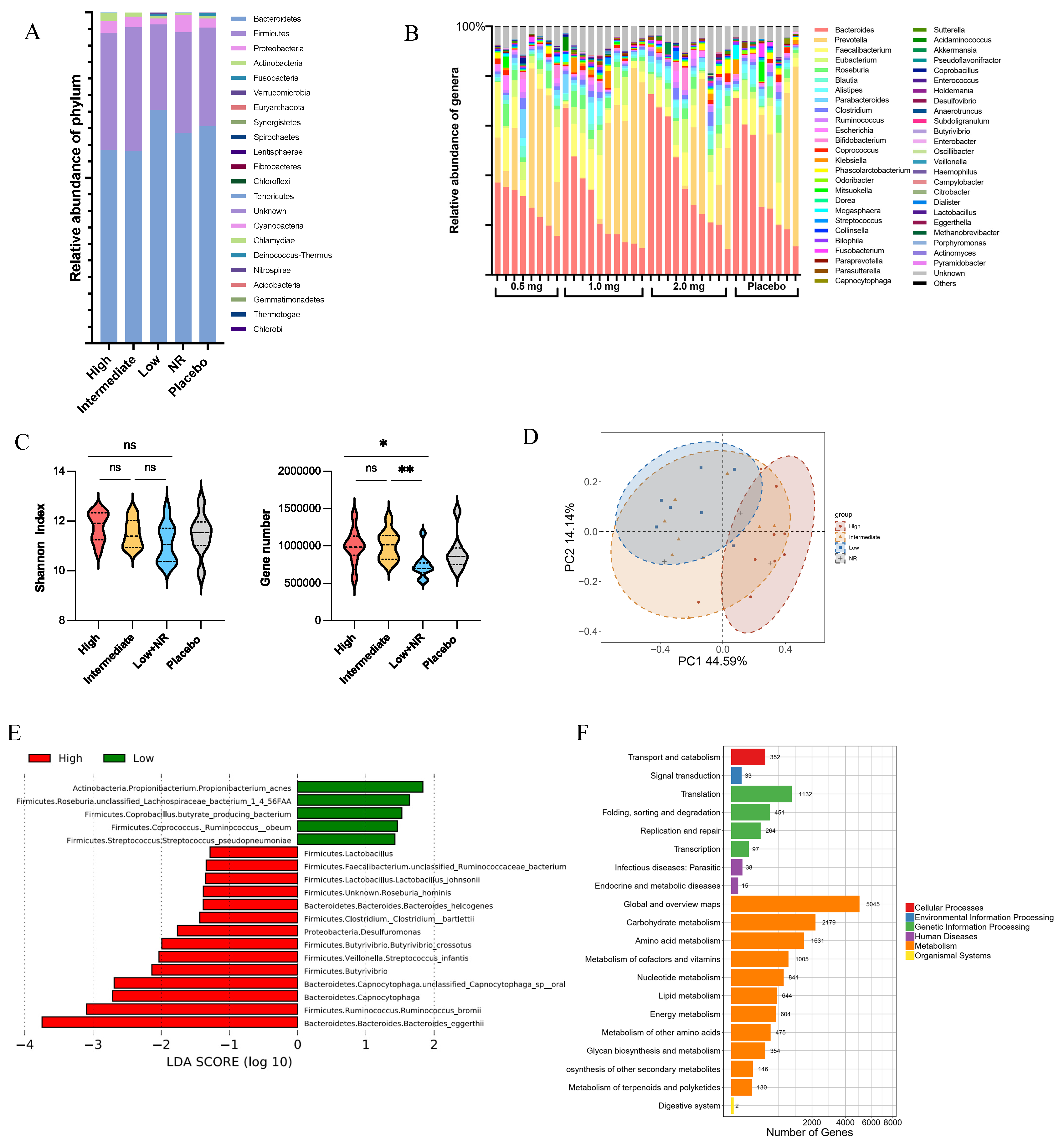
3.5. Gut Microbiota Composition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tebas, P.; Kraynyak, K.A.; Patel, A.; Maslow, J.N.; Morrow, M.P.; Sylvester, A.J.; Knoblock, D.; Gillespie, E.; Amante, D.; Racine, T.; et al. Intradermal SynCon® Ebola GP DNA Vaccine Is Temperature Stable and Safely Demonstrates Cellular and Humoral Immunogenicity Advantages in Healthy Volunteers. J. Infect. Dis. 2019, 220, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.-D.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect. Dis. 2019, 19, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Maslow, J.N. Vaccine development for emerging virulent infectious diseases. Vaccine 2017, 35, 5437–5443. [Google Scholar] [CrossRef] [PubMed]
- Gooch, K.E.; Smith, T.R.F.; Salguero, F.J.; Fotheringham, S.A.; Watson, R.J.; Dennis, M.J.; Handley, A.; Humphries, H.E.; Longet, S.; Tipton, T.; et al. One or two dose regimen of the SARS-CoV-2 synthetic DNA vaccine INO-4800 protects against respiratory tract disease burden in nonhuman primate challenge model. Vaccine 2021, 39, 4885–4894. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Yang, S.; Boyera, J.D.; Reuschel, E.L.; Patel, A.; Christensen-Quick, A.; Andrade, V.M.; Morrow, M.P.; Kraynyak, K.; Agnes, J.; et al. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: A preliminary report of an open-label, Phase 1 clinical trial. eClinicalMedicine 2021, 31, 100689. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.R.F.; Schultheis, K.; Morrow, M.P.; Kraynyak, K.A.; McCoy, J.R.; Yim, K.C.; Muthumani, K.; Humeau, L.; Weiner, D.B.; Sardesai, N.Y.; et al. Development of an intradermal DNA vaccine delivery strategy to achieve single-dose immunity against respiratory syncytial virus. Vaccine 2017, 35, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. 27 November 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf (accessed on 25 May 2025).
- National Medical Products Administration. Classification Standards for Adverse Events in Clinical Trials of Preventive Vaccines. Announcement No. 102. 31 December 2019. Available online: http://www.nmpa.gov.cn/ (accessed on 25 May 2025).
- Xie, H.; Guo, R.; Zhong, H.; Feng, Q.; Lan, Z.; Qin, B.; Ward, K.J.; Jackson, M.A.; Xia, Y.; Chen, X.; et al. Shotgun Metagenomics of 250 Adult Twins Reveals Genetic and Environmental Impacts on the Gut Microbiome. Cell Syst. 2016, 3, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Avila-Perez, G.; Nogales, A.; Park, J.G.; Vasquez, D.M.; Dean, D.A.; Barravecchia, M.; Perez, D.R.; Almazán, F.; Martínez-Sobrido, L. In vivo rescue of recombinant Zika virus from an infectious cDNA clone and its implications in vaccine development. Sci. Rep. 2020, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Richie, T.L.; Baraceros, M.F.; Rahardjo, N.; Gay, T.; Banania, J.-G.; Charoenvit, Y.; Epstein, J.E.; Luke, T.; Freilich, D.A.; et al. Boosting of DNA vaccine-elicited gamma interferon responses in humans by exposure to malaria parasites. Infect. Immun. 2005, 73, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Manoj, S.; Griebel, P.J.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Modulation of immune responses to bovine herpesvirus-1 in cattle by immunization with a DNA vaccine encoding glycoprotein D as a fusion protein with bovine CD154. Immunology 2004, 112, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Momin, T.; Kansagra, K.; Patel, H.; Sharma, S.; Sharma, B.; Patel, J.; Mittal, R.; Sanmukhani, J.; Maithal, K.; Dey, A.; et al. Safety and Immunogenicity of a DNA SARS-CoV-2 vaccine (ZyCoV-D): Results of an open-label, non-randomized phase I part of phase I/II clinical study by intradermal route in healthy subjects in India. eClinicalMedicine 2021, 38, 101020. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, safety, and immunogenicity of the DNA SARS-CoV-2 vaccine (ZyCoV-D): The interim efficacy results of a phase 3, randomised, double-blind, placebo-controlled study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.E.; Louder, M.K.; Holman, L.A.; Gordon, I.J.; Enama, M.E.; Larkin, B.D.; Andrews, C.A.; Vogel, L.; Koup, R.A.; Roederer, M.; et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a Phase I clinical trial. Vaccine 2008, 26, 6338–6343. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Roberts, C.C.; Muthumani, K.; Reuschel, E.L.; Kudchodkar, S.B.; Zaidi, F.I.; White, S.; Khan, A.S.; Racine, T.; Choi, H.; et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine. N. Engl. J. Med. 2021, 385, e35. [Google Scholar] [CrossRef] [PubMed]
- Kalams, S.A.; Parker, S.D.; Elizaga, M.; Metch, B.; Edupuganti, S.; Hural, J.; De Rosa, S.; Carter, D.K.; Rybczyk, K.; Frank, I.; et al. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J. Infect. Dis. 2013, 208, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Mammen Jr, M.P.; Tebas, P.; Agnes, J.; Giffear, M.; Kraynyak, K.A.; Blackwood, E.; Amante, D.; Reuschel, E.L.; Purwar, M.; Christensen-Quick, A.; et al. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: A preliminary report of a randomized, blinded, placebo-controlled, Phase 2 clinical trial in adults at high risk of viral exposure. medRxiv 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0.5 mg (N = 12) | 1.0 mg (N = 12) | 2.0 mg (N = 12) | Placebo (N = 9) | Total (N = 45) | |
|---|---|---|---|---|---|
| Age (Years) | |||||
| n | 12 | 12 | 12 | 9 | 45 |
| Mean (SD) | 29.8 (11.30) | 34.8 (9.92) | 27.8 (6.90) | 29.2 (9.31) | 30.5 (9.57) |
| Median | 26.0 | 33.0 | 26.0 | 24.0 | 27.0 |
| Min | 21 | 20 | 21 | 22 | 20 |
| Max | 54 | 52 | 40 | 48 | 54 |
| Age Group | |||||
| n | 12 | 12 | 12 | 9 | 45 |
| ≤40 Years | 10 (83.3%) | 9 (75.0%) | 12 (100.0%) | 7 (77.8%) | 38 (84.4%) |
| >40 Years | 2 (16.7%) | 3 (25.0%) | 0 | 2 (22.2%) | 7 (15.6%) |
| Sex | |||||
| n | 12 | 12 | 12 | 9 | 45 |
| Male | 8 (66.7%) | 10 (83.3%) | 9 (75.0%) | 4 (44.4%) | 31 (68.9%) |
| Female | 4 (33.3%) | 2 (16.7%) | 3 (25.0%) | 5 (55.6%) | 14 (31.1%) |
| Race | |||||
| n | 12 | 12 | 12 | 9 | 45 |
| Asian | 12 (100.0%) | 12 (100.0%) | 12 (100.0%) | 9 (100.0%) | 45 (100.0%) |
| Black | 0 | 0 | 0 | 0 | 0 |
| White | 0 | 0 | 0 | 0 | 0 |
| Other | 0 | 0 | 0 | 0 | 0 |
| Chinese Nationality | |||||
| n | 12 | 12 | 12 | 9 | 45 |
| Han | 12 (100.0%) | 11 (91.7%) | 10 (83.3%) | 9 (100.0%) | 42 (93.3%) |
| Other | 0 | 1 (8.3%) | 2 (16.7%) | 0 | 3 (6.7%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhou, Y.; Cheng, X.; Qiu, C.; Wang, S.; Xia, Y.; Huai, X.; Xiu, Z.; Wang, J.; He, Y.; et al. Safety, Tolerability, and Immunogenicity of a DNA Vaccine (pGX9501) Against SARS-CoV-2 in Healthy Volunteers: A Single-Center, Randomized, Double-Blind, Placebo-Controlled, and Dose-Ranging Phase I Trial. Vaccines 2025, 13, 573. https://doi.org/10.3390/vaccines13060573
Yang H, Zhou Y, Cheng X, Qiu C, Wang S, Xia Y, Huai X, Xiu Z, Wang J, He Y, et al. Safety, Tolerability, and Immunogenicity of a DNA Vaccine (pGX9501) Against SARS-CoV-2 in Healthy Volunteers: A Single-Center, Randomized, Double-Blind, Placebo-Controlled, and Dose-Ranging Phase I Trial. Vaccines. 2025; 13(6):573. https://doi.org/10.3390/vaccines13060573
Chicago/Turabian StyleYang, Haijing, Yang Zhou, Xin Cheng, Chao Qiu, Shuo Wang, Yu Xia, Xuefen Huai, Zhenning Xiu, Jiarong Wang, Yue He, and et al. 2025. "Safety, Tolerability, and Immunogenicity of a DNA Vaccine (pGX9501) Against SARS-CoV-2 in Healthy Volunteers: A Single-Center, Randomized, Double-Blind, Placebo-Controlled, and Dose-Ranging Phase I Trial" Vaccines 13, no. 6: 573. https://doi.org/10.3390/vaccines13060573
APA StyleYang, H., Zhou, Y., Cheng, X., Qiu, C., Wang, S., Xia, Y., Huai, X., Xiu, Z., Wang, J., He, Y., Cao, G., Wei, Q., Wang, J., Ai, J., Zhang, H., Zhang, Y., Zhang, J., Zhang, W., & Wang, B. (2025). Safety, Tolerability, and Immunogenicity of a DNA Vaccine (pGX9501) Against SARS-CoV-2 in Healthy Volunteers: A Single-Center, Randomized, Double-Blind, Placebo-Controlled, and Dose-Ranging Phase I Trial. Vaccines, 13(6), 573. https://doi.org/10.3390/vaccines13060573