
The Platform Technology Approach to mRNA Product Development and Regulation


Abstract
1. Introduction
2. Streamlined Regulatory Pathways for mRNA Platform Technologies Are Possible
3. Regulators’ Experience and Expectations with Platform Technologies
Utilisation of Platform Technology in mRNA Regulatory Submissions and Review
4. mRNA-LNP Products in the Market or under Late-Stage Clinical Development
- Respiratory viruses—SARS-CoV-2, respiratory syncytial virus, seasonal influenza.
- Other infectious diseases—HIV, Lyme disease, cytomegalovirus.
- Cancers—malignant melanoma, uveal melanoma, lymphoma, solid tumours, pulmonary osteosarcoma, prostate cancer, head and neck cancers, gastric cancer, pancreatic cancer, ovarian cancer, biliary tract cancer.
- Rare or metabolic diseases—methylmalonic acidemia, ornithine transcarbamylase deficiency, phenylketonuria, propionic aciduria, primary ciliary dyskinesia.
5. Data Requirements for mRNA Product Development and Regulatory Submissions
- Module 1—Administrative information and prescribing information.
- Module 2—Overviews and summaries of Modules 3–5.
- Module 3—Quality (chemistry, manufacturing, and controls) reports for the drug substance (mRNA) and (finished) drug product.
- Module 4—Non-clinical reports (pharmacology, pharmacokinetics, and toxicology).
- Module 5—Clinical study reports (including biopharmaceutic studies, human pharmacokinetic and pharmacodynamic studies, clinical trial (efficacy and safety) studies, and post-marketing experience).
5.1. Quality (Chemistry, Manufacturing, and Controls (CMC))
5.2. Non-Clinical Study Reports
5.3. Application of the Platform Approach to Clinical Data
6. Use of Comparability and Bridging Studies
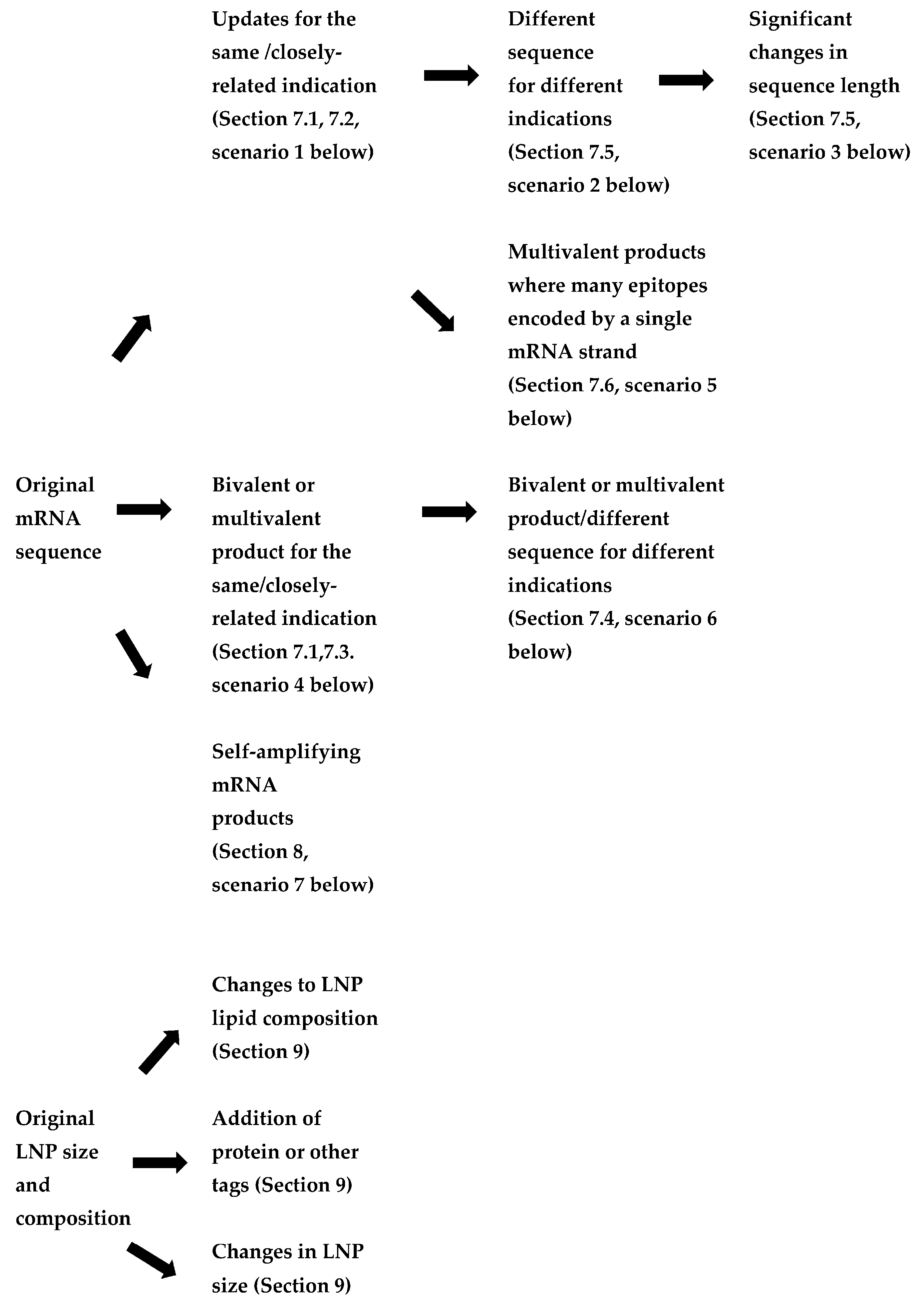
7. Applying the Platform Technology Approach to a Wider Range of mRNA Products
- Updates to mRNA sequence for the same or closely related indication (to enable improvement to a vaccine or therapeutic and/or for a vaccine against a viral variant). In these cases, changes to both the coding (open reading frame) and non-coding sequences could be made.
- The use of a different mRNA sequence for a vaccine or therapeutic treating different indications within the same family of products (e.g., respiratory viruses, metabolic diseases).
- Significant changes in mRNA sequence length, but targeting the same disease (for example, a shorter mRNA encoding a subunit or epitope rather than the full protein), which affect its stability and delivery.
- Monovalent products (single mRNA sequence) vs. bivalent or multivalent products, where the encoding mRNAs are on separate strands. These may either be for variants or seasonal updates to an ancestral vaccine or products targeting diseases which require multiple proteins to be expressed.
- Products where multiple short antigenic peptides are encoded in a single sequence where each set of selected epitopes are specific to an individual (e.g., individualised neoantigen therapies).
- Therapeutic products addressing different targets of the same metabolic pathway (e.g., a group of rare diseases caused by different faulty enzymes of the same cellular pathway).
- Self-amplifying mRNA products, where the replicon mRNA included both mRNAs for enzymes involved in amplification as well as the coding sequence for the target antigen.
7.1. Experience with COVID Vaccines
7.2. Updates to mRNA Sequence for the Same Indication
- Information on the sequence accuracy for the new DNA template and mRNA product. However, the same manufacturing process, controls, and analytical methods are applied for the new cell bank generating the new plasmid containing the varied sequence.
- Demonstration that the levels of expression of the new mRNA are comparable.
- Demonstration that the process and product-related impurities are comparable.
- If changes affect the coding regions, identity testing and analysis of expression levels of the altered protein is also required.
7.3. Bivalent Products for the Same Indication (e.g., Certain COVID-19 Vaccines)
7.4. Other Bivalent or Multivalent Products
- Two or more existing mRNA sequences against multiple variants or in combination products (e.g., COVID-19 and influenza mRNAs).
- A combination of existing and new mRNA sequences.
- Two or more new mRNA sequences, e.g., for a vaccine, where use of a range of antigens is considered important to mount a broad immune response [28], or for a therapeutic where both subunits of an enzyme must be expressed to regain function.
7.5. Different mRNA Sequences for Different Indications Using the Same LNP and Route of Administration
- Vaccines against infectious diseases when there is wide genetic or antigenic diversity with an infection—e.g., HIV mRNA candidate vaccines with a single sequence co-expressing two antigens [20], mRNA seasonal influenza vaccines [37], or where multiple antigens may be required, e.g., for norovirus vaccines. For some antigens, such as cytomegalovirus, the candidate mRNA vaccine is reflective of the complex viral structure [20].
- Where expression of multiple enzyme subunits is required to regain function in a treated patient, e.g., propionic acidemia [59].
- Oncology mRNA vaccines encoding several tumour-associated antigens or that express a mix of cytokines that mediate tumour regression [61].
- When the vaccine needs to cover more than one species of a pathogen (e.g., Borrelia in Lyme disease).
7.6. Multivalent Products Where Many Epitopes Are Encoded by a Single Strand of mRNA
8. Considerations for Self-Amplifying mRNA Products
9. Lipid Nanoparticle Variations
10. Particular Considerations for mRNA Therapeutics
11. Implications for Personalised Medicine and Rare Disease mRNA Products
- The manufacturing stream for a given individualised neoantigen product could be split from a single DNA template starting material throughout two parallel manufacturing processes to produce a pair of batches, which can then be compared at the range of process validation steps.
- Design a set of sequences that encompasses the extremes of potential patient-specific sequences and assess the reliability of expected versus actual product characteristics.
- Evaluate manufacturing updates at the process level, including updates to genome sequencing and bioinformatics.
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect Dis. 2022, 22, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; Snare, M.; Liao, H.; Chiou, S.; Marmura, T.; et al. Developments of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl. Res. 2021, 242, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.; Kaufmann, M.; Glozman, V.; Chakrabarti, A. Disease X: Accelerating the development of medical countermeasures for the next pandemic. Lancet Infect Dis. 2020, 20, e108–e115. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Z.; Xie, C.; Xia, X. Advances in mRNA vaccines. Int. Rev. Cell Mol. Biol. 2022, 372, 295–316. [Google Scholar] [PubMed]
- Metkar, M.; Pepin, C.S.; Moore, M.J. Tailor made: The art of therapeutic mRNA design. Nat. Rev. Drug Discov. 2024, 24, 67–83. [Google Scholar] [CrossRef]
- Zhang, G.; Tang, T.; Chen, Y.; Huang, X.; Liang, T. mRNA vaccines in disease prevention and treatment. Signal Transduct. Targeted Ther. 2023, 8, 365–394. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA vaccine era—Mechanisms, drug platform and clinical prospection. Int. J. Mol. Sci. 2020, 21, 6582–6617. [Google Scholar] [CrossRef]
- Sparrow, E.; Hasso-Agopsowicz, M.; Kaslow, D.C.; Singh, K.; Rao, R.; Chibi, M.; Makubalo, L.E.; Reeder, J.C.; Kang, G.; Karron, R.A.; et al. Leveraging mRNA technology to accelerate development of vaccines for some emerging and neglected tropical diseases through local vaccine production. Front. Trop. Dis. 2022, 3, 844039. [Google Scholar] [CrossRef]
- European Medicines Agency. Concept Paper on the Development of a Guideline on the 5 Quality Aspects of mRNA Vaccines. Available online: www.ema.europa.eu/en/documents/scientific-guideline/concept-paper-development-guideline-quality-aspects-mrna-vaccines_en.pdf (accessed on 26 March 2024).
- European Medicines Agency. EMA and ECDC Statement on Updating COVID-19 Vaccines to Target New SARS-CoV-2 Virus Variants. Available online: www.ema.europa.eu/en/news/ema-and-ecdc-statement-updating-covid-19-vaccines-target-new-sars-cov-2-virus-variants (accessed on 26 March 2024).
- World Health Organization. Evaluation of the Quality, Safety and Efficacy of Messenger RNA Vaccines for the Prevention of Infectious Diseases: Regulatory Considerations. World Health Organization Expert Committee on Biological Standardization 74th Report, 2022, Annex 3. Available online: www.who.int/publications/m/item/evaluation-of-the-quality-safety-and-efficacy-of-messenger-rna-vaccines-for-the-prevention-of-infectious-diseases-regulatory-considerations (accessed on 1 May 2024).
- Madabushi, R.; Seo, P.; Zhao, L.; Tegenge, M.; Zhu, H. Review: Role of model-informed drug development approaches in the lifecycle of drug development and regulatory decision-making. Pharm. Res. 2022, 39, 1669–1680. [Google Scholar] [CrossRef]
- Schrieber, S.J.; Putnam, W.S.; Chiu Yuen Chow, E.; Cieslak, J.; Zhuang, Y.; Martin, S.W.; Hanson, P.; Maggio, F.; Rivera Rosada, L.A. Comparability considerations and challenges for expedited development programs for biological products. Drugs R D 2020, 20, 301–306. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Comparability of Biotechnology-Derived Medicinal Products after a Change in the Manufacturing Process Non-Clinical and Clinical Issues. Available online: www.ema.europa.eu/en/documents/scientific-guideline/guideline-comparability-biotechnology-derived-medicinal-products-after-change-manufacturing-process-non-clinical-and-clinical-issues_en.pdf (accessed on 4 May 2024).
- Castellanos, M.M.; Gressard, H.; Li, X.; Magagnoli, C.; Moriconi, A.; Strangers, D.; Strodiot, L.; Tello Soto, M.; Zwierzyna, M.; Campa, C. CMC Strategies and advanced technologies for vaccine development to boost acceleration and pandemic preparedness. Vaccines 2023, 11, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Guidance for Industry: Comparability Protocols—Chemistry, Manufacturing, and Controls Information. Available online: www.fda.gov/files/drugs/published/Comparability-Protocols----Chemistry--Manufacturing--and-Controls-Information.pdf (accessed on 26 March 2024).
- European Medicines Agency. Questions and Answers on Comparability Considerations for Advanced Therapy Medicinal Products (ATMP)—Scientific Guideline. Available online: www.ema.europa.eu/en/questions-answers-comparability-considerations-advanced-therapy-medicinal-products-atmp (accessed on 26 March 2024).
- Perrotta, C.; Fenizia, C.; Carnovale, C.; Pozzi, M.; Trabattoni, D.; Cervia, D.; Clementi, E. Updated considerations for the immunopharmacological aspects of the “talented mRNA vaccines”. Vaccines 2023, 11, 1481–1509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Narayan, E.; Liu, Q.; Tsybovsky, Y.; Boswell, K.L.; Ding, S.; Hu, Z.; Follmann, D.; Lin, Y.; Miao, H.; et al. A multiclade env-gag VLP mRNA vaccine elicits tier-2 HIV-1-neutalising antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat. Med. 2021, 27, 2234–2245. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Karthigeyan, K.P.; Herbek, S.; Valencia, S.M.; Jenks, J.A.; Webster, H.; Miller, I.G.; Connors, M.; Pollara, J.; Andy, C.; et al. Human Cytomegalovirus mRNA-1647 vaccine candidate elicits potent and broad neutralization and higher antibody-dependent cellular cytotoxicity responses than the gB/MF59 vaccine. J. Infect Dis. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, Q.; Durnell, L.A.; et al. Intramuscular delivery of replicon RNA encoding ZIKV-117 human monoclonal antibody protects against zika virus infection. Mol. Ther. Methods Clin. Dev. 2020, 18, 402–414. [Google Scholar] [CrossRef]
- Clarke, L.A.; Amaral, M.D. What can RNA-based therapy do for monogenetic diseases? Pharmaceutics 2023, 15, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.E.; Erasmus, J.H.; Reese, B.; Pucor, T.; Archer, J.; Kandahar, A.; Hsu, F.-C.; Nicholes, K.; Reed, S.; Baldwin, S.; et al. An RNA-based vaccine platform for use against Mycobacterium tuberculosis. Vaccines 2023, 11, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; DiPiazza, A.T.; Falcone, S.; Ruckwardt, T.J.; Morabito, K.M.; Abiona, O.M.; Chang, L.A.; Caringal, R.T.; Presnyak, V.; Narayan, E.; et al. Chimeric Fusion (F) and attachment (G) glycoprotein antigen delivery by mRNA as a candidate Nipah vaccine. Front. Immunol. 2021, 12, 772864. [Google Scholar] [CrossRef]
- Pine, M.; Arona, G.; Hart, T.M.; Bettini, E.; Gaudette, B.T.; Muramatsu, H.; Tombacz, I.; Kanbayashi, T.; Tam, Y.K.; Brisson, D.; et al. Development of an mRNA-lipid nanoparticle vaccine against Lyme disease. Mol. Therap. 2023, 31, 2702–2714. [Google Scholar] [CrossRef]
- Sajid, A.; Matias, J.; Arora, G.; Kurokawa, C.; De Pont, K.; Tang, X.; Lynn, G.; Wu, M.-J.; Pal, U.; Strank, N.O.; et al. mRNA vaccination induces tick resistance and prevents transmission of the Lyme disease agent. Sci. Transl. Med. 2021, 13, eabj9827. [Google Scholar] [CrossRef]
- Nitika, W.J.; Hui, A.-M. The development of mRNA vaccines for infectious diseases: Recent updates. Infect. Drug Resist. 2021, 14, 5271–5285. [Google Scholar] [CrossRef]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Sanyal, G.; Sarnefalt, A.; Kumar, A. Considerations for bioanalytical characterisation and batch release of COVID-19 vaccines. Npj Vaccines 2021, 6, 53–62. [Google Scholar] [CrossRef]
- Challener, C.A. Analysis of mRNA Therapeutics and Vaccines; Pharmaceutical Technology: Piscataway, NJ, USA, 2023; pp. 16–21. [Google Scholar]
- Cheng, F.; Wang, Y.; Bai, Y.; Liang, Z.; Mao, Q.; Liu, D.; Wu, X.; Xu, M. Research advances on the stability of mRNA vaccines. Viruses 2023, 15, 668–683. [Google Scholar] [CrossRef]
- United States Pharmacopeia. Analytical Procedures for mRNA Vaccine Quality. Draft Guidelines: 2nd Edition, 2023. Available online: www.usp.org/sites/default/files/usp/document/our-work/biologics/documents/vaccine-mrna-guidelines-2.pdf (accessed on 26 March 2024).
- Li, H.H.; Xu, J.; He, L.; Denny, L.I.; Rustandi, R.R.; Dornadula, G.; Fiorito, B.; Zhang, Z.-Q. Development and qualification of cell-based relative potency assay for a human respiratory syncytial virus mRNA vaccine. J. Pharm. Biomed. Anal. 2023, 234, 115523. [Google Scholar] [CrossRef]
- Sanyal, G. Development of functionally relevant potency assays for monovalent and multivalent vaccines delivered by evolving technologies. Npj Vaccines 2022, 7, 50–59. [Google Scholar] [CrossRef]
- Demongeot, J.; Fougere, C. mRNA COVID-19 Vaccines—Facts and hypotheses on fragmentation and encapsulation. Vaccines 2023, 11, 40–65. [Google Scholar] [CrossRef]
- Chivulka, S.; Plitnik, T.; Tibbits, T.; Karve, S.; Dias, A.; Zhang, D.; Goldman, R.; Gopani, H.; Khanmohammed, A.; Sarode, A.; et al. Development of multivalent mRNA vaccine candidates for seasonal or pandemic influenza. Npj Vaccines 2021, 6, 153–167. [Google Scholar]
- United States Food and Drug Administration. Q5C Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products. Available online: www.fda.gov/regulatory-information/search-fda-guidance-documents/q5c-quality-biotechnological-products-stability-testing-biotechnologicalbiological-products (accessed on 4 May 2024).
- Covarrubias, C.E.; Rivera, T.A.; Soto, C.A.; Deeks, T.; Kalergis, A.M. Current GMP standards for the production of vaccines and antibodies: An overview. Front. Public Health 2022, 10, 1021905. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef]
- European Medicines Agency. Clinical Evaluation on New Vaccines—Scientific Guideline. Available online: www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical/evaluation-vaccines_en.pdf (accessed on 26 March 2024).
- Vervaeke, P.; Borgos, S.E.; Sanders, N.N.; Combes, F. Regulatory guidelines and preclinical tools to study the biodistribution of RNA therapeutics. Adv. Drug Deliv. Res. 2022, 184, 114236. [Google Scholar] [CrossRef]
- World Health Organization. Clinical Evaluation of Vaccines. Available online: www.who.int/teams/health-product-policy-and-standards/standards-and-specifications/vaccine-standardization/clinical-evaluation-of-vaccines (accessed on 26 March 2024).
- Kim, D.; Robertson, J.S.; Excler, J.-L.; Condt, R.C.; Fast, P.E.; Gurwith, M.; Pavlakis, G.; Monath, T.P.; Smith, J.; Wood, D.; et al. The Brighton Collaboration standardized template for collection of key information for benefit-risk assessment of nucleic acid (RNA and DNA) vaccines. Vaccine 2020, 38, 5556–5561. [Google Scholar] [CrossRef]
- Baldwin, J.; Piplani, S.; Sakala, I.G.; Honda-Okubo, Y.; Li, L.; Petrovsky, N. Rapid development of analytical methods for evaluating pandemic vaccine: A COVID-19 perspective. Bioanalysis 2021, 13, 1805–1826. [Google Scholar] [CrossRef]
- Weir, J.P.; Gruber, M.F. An overview of the regulation of influenza vaccines in the United States. Influenza Other Resp. Vir. 2016, 10, 354–360. [Google Scholar] [CrossRef]
- Fritzell, B. Bridging studies. Dev. Biol. Stand. 1998, 95, 181–188. [Google Scholar]
- Finch, C.L.; Dowling, W.E.; King, T.H.; Martinez, C.; Nguyen, B.V.; Roozerdaal, R.; Rustomjee, R.; Skiadopoulous, M.H.; Vert-Wong, E.; Yellowlees, A.; et al. Bridging human and animal data in pursuit of vaccine licensure. Vaccines 2022, 10, 1384. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Guidance for Industry: Development and Licensure of Vaccines to Prevent COVID-19. Available online: www.fda.gov/media/139638/download (accessed on 26 March 2024).
- Van Tilbeurgh, M.; Lemdani, K.; Beignon, A.-S.; Chapon, C.; Tchitchek, N.; Cheraitia, L.; Lopez, E.M.; Pascal, Q.; Le Grand, R.; Maisonnasse, P.; et al. Predictive markers of immunogenicity and efficacy for human vaccines. Vaccines 2012, 9, 579–614. [Google Scholar] [CrossRef]
- Granados-Riveron, J.; Aquini-Jarquin, G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed. Pharmacother. 2021, 142, 111–953. [Google Scholar] [CrossRef]
- Scheaffer, S.M.; Lee, D.; Whitener, B.; Ying, B.; Wu, K.; Liang, C.-Y.; Jani, H.; Martin, P.; Amato, N.J.; Avena, L.E.; et al. Bivalent SARS-CoV-2 mRNA vaccines increase breadth of neutralization and protect against the BA.5 Omicron variant in mice. Nat. Med. 2023, 29, 247–257. [Google Scholar] [CrossRef]
- European Medicines Agency. Spikevax (Previously COVID-19 Vaccines (Moderna)). Available online: www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf (accessed on 26 March 2024).
- Sanjuan, R.; Domigo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Schlake, T.; Thran, M.; Fiedler, K.; Heidenreich, R.; Petsch, B.; Fotin-Mleczek, M. mRNA: A novel avenue to antibody therapy? Mol. Therap. 2019, 27, 773–784. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, P.B.; Donis, R.O.; Koup, R.A.; Fong, Y.; Plotkin, S.A.; Fallman, D. A COVID-19 milestone attained—A correlate of protection for vaccines. N. Engl. J. Med. 2022, 387, 2203–2206. [Google Scholar] [CrossRef]
- Kackos, C.M.; De Beauchamp, J.; Davitt, C.J.H.; Lonzaric, J.; Sealy, R.E.; Hurwitz, J.L.; Samsa, M.M.; Webby, R.J. Seasonal quadrivalent mRNA vaccine prevents and mitigates influenza infection. Npj Vaccines 2023, 8, 157–168. [Google Scholar] [CrossRef]
- Attarwala, H.; Lumley, M.; Liang, M.; Ivaturi, V.; Senn, J. Translational pharmacokinetic/pharmacodynamic model for mRNA-3927, an investigational therapeutic for the treatment of propionic acidemia. Nucleic Acids Therap. 2023, 33, 141–147. [Google Scholar] [CrossRef]
- Deal, C.E.; Carfi, A.; Plante, O.J. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines 2021, 9, 108–123. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, O.; Svane, I.M. Clinical advances and ongoing trials of mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef]
- Poveda, C.; Biter, A.B.; Bottazzi, M.E.; Strych, U. Establishing preferred product characterization for the evaluation of RNA vaccine antigens. Vaccines 2019, 7, 131–144. [Google Scholar] [CrossRef]
- Umscheid, C.A.; Margolis, D.J.; Grossman, C.E. Key concepts of clinical trials—A narrative review. Postgrad. Med. 2011, 123, 194–204. [Google Scholar] [CrossRef]
- Schober, G.B.; Story, S.; Arya, D.P. A careful look at lipid nanoparticle characterization: Analysis of benchmark formulations for encapsulation of RNA cargo size gradient. Nat. Sci. Rep. 2024, 14, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomized, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Altmann, G. Japan approves first self-amplifying mRNA COVID-19 vaccines. Precision Vaccinations News, 28 November 2023. Available online: https://precisionvaccinations.com/2023/11/28/ (accessed on 26 March 2024).
- Oda, Y.; Kumagai, Y.; Kanai, M.; Iwama, Y.; Okura, I.; Minamida, T.; Yagi, Y.; Kurosawa, T.; Greener, B.; Zhang, Y.; et al. Immunogenicity and safety of a booster dose of a self-amplifying mRNA vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: A double blind, multicentre, randomized, controlled, phase 3 non-inferiority trials. Lancet Infect Dis. 2024, 4, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Briones, M.C.; Silva-Pilipich, N.; Herrardor-Canete, G.; Vanrell, L.; Smerdou, C. A new generation of vaccines based on alphavirus self-amplifying RNA. Curr. Opin. Virol. 2020, 44, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; Ip, S.; Geall, A.J. An update on self-amplifying mRNA vaccine development. Vaccines 2021, 9, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Aliahmad, P.; Miyake-Stoner, S.J.; Geall, A.J.; Wang, N.S. Next generation self-replicating RNA vectors for vaccines and immunotherapies. Cancer Gene Therap. 2022, 30, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thompson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotech. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Self-copying RNA vaccine wins first full approval: What’s next? Nature 2023, 624, 236–237. [Google Scholar] [CrossRef]
- Comes, J.D.G.; Pijman, G.P.; Hick, T.A.H. Rise of the RNA machines—Self-amplification in mRNA vaccine design. Trends Biotech. 2023, 41, 1417–1429. [Google Scholar] [CrossRef]
- Sittplangkoon, C.; Alameh, M.-G.; Weissman, D.; Lin, P.J.C.; Tam, Y.K.; Prompetchara, E.; Palaga, T. mRNA vaccines with unmodified uridine induces robust type 1 interferon-dependent anti-tumor immunity in a melanoma model. Front. Immunol. 2022, 13, 983000. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Therap. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2023, 6, 1078–1094. [Google Scholar] [CrossRef]
- Albertsen, C.H. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef]
- Liu, T.; Tian, Y.; Zheng, A.; Cui, C. Design strategies for and stability of mRNA-lipid nanoparticle COVID-19 vaccines. Polymers 2022, 6, 4195. [Google Scholar] [CrossRef]
- Herrera-Barrera, M.; Ryals, R.C.; Gautam, M.; Jozic, A.; Landry, M.; Korzum, T.; Gupta, M.; Acosta, C.; Stoddard, J.; Reynaga, R.; et al. Peptide-guided lipid nano particles deliver mRNA to the neural retina of rodents and non-human primates. Sci. Adv. 2023, 9, eadd4623. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.L.; Persano, F.; Persano, S. Advances in lipid nanoparticles for mRNA-based cancer immunotherapy. Front. Chem. 2020, 8, 589959. [Google Scholar] [CrossRef]
- Gote, V.; Bolla, P.K.; Kommineni, N.; Butreddy, A.; Nukala, P.K.; Pallakurthi, S.S.; Khan, W. A comprehensive review of mRNA vaccines. Int. J. Mol. Sci. 2023, 24, 2700–2735. [Google Scholar] [CrossRef]
- Mendonca, M.C.P.; Kont, A.; Kowalski, P.S.; O’Driscoll, C.M. Design of lipid-based nanoparticles for delivery of therapeutic nucleic acids. Drug Discov. Today 2023, 28, 103505. [Google Scholar] [CrossRef]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Damase, T.R.; Sukhovershin, R.; Boda, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The limitless future of RNA therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef]
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the promise of mRNA therapeutics. Nat. Biotech 2022, 40, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.; Li, Y.; Tenchov, R.; Sasso, J.M.; Zhang, D.; Li, D.; Zou, L.; Wang, X.; Zhou, Q. Messenger RNA-based therapeutics and vaccines: What’s beyond COVID-19? ACS Pharmacol. Translat. Sci. 2023, 6, 943–969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-S.; Kumari, M.; Chen, G.-H.; Hong, M.H.; Yuan, J.P.-Y.; Tsai, J.-L.; Wu, H.-C. mRNA-based vaccines and therapeutics: An in-depth survey of current and upcoming clinical applications. J Biomed. Sci. 2023, 30, 84–118. [Google Scholar] [CrossRef] [PubMed]
- Koeberl, D.; Schulze, A.; Sondheimer, N.; Lipschultz, G.S.; Geberhiwot, T.; Li, R.; Saini, R.; Luo, J.; Sikirica, V.; Jin, L.; et al. Interim analysis of a first in human phase 1/2 mRNA trial for propionic aciduria. Nature 2024, 628, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Sanchez, A.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating cystic fibrosis with mRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Atochina-Vasserman, E.N.; Weissman, D. mRNA-based therapeutics: Looking beyond COVID-19 vaccines. Lancet 2024, 403, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges and prospects. Sig. Transduct. Target Ther. 2023, 8, 450–462. [Google Scholar] [CrossRef]
- Perrinjaquet, M.; Schlegel, C.R. Personalised neoantigen cancer vaccines: An analysis of the clinical and commercial potential of ongoing development programs. Drug Discov. Today 2023, 28, 103773. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic aciduria. Nat. Commun. 2020, 11, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, M.; Dwivedi, G.; Lavertu, M.; Alameh, M.-G. Vaccine technologies and platforms for infectious diseases: Current progress, challenges and opportunities. Vaccines 2021, 9, 1490–1520. [Google Scholar] [CrossRef]

{kind=link}
| Sponsor | Commercial or Submitted for Regulatory Review | Phase 3 | Phase 2 | Phase 1/2 | Phase 1 |
|---|---|---|---|---|---|
| Moderna (https://trials.modernatx.com/search-results/) (accessed 1 May 2024) | Various SARS-CoV-2 vaccines mRNA1345—Respiratory Syncytial Virus | mRNA1647—Cytomegalovirus (women) mRNA1010—Seasonal influenza mRNA1083—Seasonal influenza—COVID combination mRNA4157—Melanoma, personalised individual neoantigen therapy (with pembrolizumab) | mRNA1647—Cytomegalovirus (extension trials) mRNA1893—Zika virus | mRNA1769—Smallpox/ monkeypox mRNA1608—Genital herpes mRNA1975/1982—Lyme disease mRNA3210—Phenylketonuria mRNA1468—Shingles | mRNA3745—Glycogen storage disease mRNA2752—Lymphoma, Triple negative breast cancer mRNA0184—Chronic heart failure mRNA 1195—Epstein–Barr virus mRNA1365—RSV plus human metapneumovirus mRNA 4157—Personalised neoantigen therapy mRNA1403—Norovirus mRNA1653—Metapneumovirus and parainfluenza mRNA1944—Chikungunya mRNA6231—Autoimmune diseases |
| BioNTech (in some case with partners, e.g., Pfizer, Genetech and Genmab) (www.biontech.com/int/en/home/pipeline-and-products/pipeline.html) (accessed 1 May 2024) | A range of COVID-19 vaccines (with Pfizer) | BNT161—Seasonal influenza BNT162b2+ BNT161 COVID-19- Influenza combination | BNT111—Advanced, R/R melanoma BNT113—Metastatic/R/R HPV16+ head and neck cancer, metastatic NSCLC BNT116—Metastatic non-small cell lung cancer BNT122 (autogene cevumeran)—Advanced melanoma, advanced colorectal cancer, Adjuvant pancreatic ductal adenocarcinoma BNT162b5/6/7—COVID-19 (ancestral + BA 2) | BNT142—Solid tumours BNT151—Solid tumours BNT165—Malaria BNT166—Mpox BNT167—Shingles | BNT112—Metastatic/localised prostate cancer BNT116—Advanced/ metastatic NSCLC BNT122 (autogene cevumeran)—Solid tumours BNT152+153—Solid tumours BNT163—HSV BNT164—Tuberculosis BNT162b2 BNT162b4—COVID-19 |
| Sanofi (www.sanofi.com/en/our-science/our-pipeline) (accessed 1 May 2024) | SP 0256—RSV and RSV/hMPV bivalent older adults | SP 0273—QIV Influenza SAR441000—Cytokine mRNA for Solid tumours | |||
| GSK (www.gsk.com/en-gb/innovation/pipeline/) (accessed 1 May 2024) | GSK4382276—Seasonal influenza GSK4396687—SARS-CoV 2 | ||||
| CureVac (www.curevac.com/en/pipeline/) (accessed 1 May 2024) | COVID-19 (with GSK) | Non-small cell lung cancer | Avian and seasonal Influenza (with GSK) Rabies Glioblastoma Solid tumours | ||
| Arcturus Therapeutics (https://arcturusrx.com/mrna-medicines-pipeline/) (accessed 1 May 2024) | ARCT-154 COVID-19 sa RNA vaccine (with CSL) | ARCT-2301 Bivalent COVID-19 Ancestral/Omicron BA.4/5 (with CSL) ARCT-2303) Monovalent: COVID XBB.1.5 (with CSL) | Ornithine Transcarbamylase Deficiency | Cystic Fibrosis ARCT-2138—Seasonal influenza Quadrivalent—(with CSL) | |
| Daichi Sankyo (www.daiichisankyo.com/rd/pipeline/) (accessed 1 May 2024) | DS-5670 Monovalent COVID booster (ancestral strain) | ||||
| Suzhou Abogen (https://abogenbio.com/en/about) (accessed 1 May 2024) | ABO1020 Monovalent: COVID BA4/5 |
| Nature of the mRNA Change | Example of Product | mRNA Characteristics | Impact on Quality Data Requirements | Impact On Preclinical Data Requirements | Impact on Clinical Data Requirements |
|---|---|---|---|---|---|
| Updates to original sequence for the same or closely-related indication | XBB 1.5 COVID vaccine | Single sequence, similar length as reference mRNA sequence; same LNP Changes to either or both the coding and non-coding regions could be made | Re-use: most CMC approaches Bridge: Sequence specific analysis, expression and potency assays | Re-use: Toxicology, biodistribution Bridge: Comparison of immune responses to reference product New: Possible single species toxicology studies. | Re-use: If same indication or for variant (some regulators may want bridging data) New: If indication is different |
| Bivalent or multivalent product for the same or closely-related indication | Bivalent COVID vaccine, influenza vaccines | Several sequences, homologous to ancestral sequence; same LNP | Re-use: CMC for original product Bridge: Manufacturing and quality for new sequence Sequence specific analysis, expression/identity, and potency assays that can distinguish products of each sequence | Re-use: Preclinical data for original product Bridge: Adapt pharmacokinetics, biodistribution, and toxicology analysis for original product New: Assessment of contribution of each sequence to the immune response | Re-use: Clinical data for original products Bridge: Some regulators may require data on a correlate of immunity or protection |
| Different mRNA sequence for a vaccine or therapeutic treating different indications | Respiratory Syncytial Virus (RSV) | Single sequence, similar length to reference mRNA sequence; same LNP | Bridge: CMC for original product if LNP is same; mRNA manufacturing process similar New: Sequence specific analysis, expression/identity and potency assays | Bridge: Aspects of pharmacology, toxicology and biodistribution data common to reference product New: More data will be required for some diseases or if tissue target is different from reference product | Bridge: potentially use biomarker if product acts on similar pathways to existing mRNA product New: New clinical trial data required to support the new indication |
| Bivalent or multivalent product, different sequence for different indications | Cytomegalovirus Lyme disease | Multiple sequences, similar length to reference mRNA sequence; Same LNP | Re-use: If mRNAs have been in previous products New: Encapsulation efficiency (if co-formulated), identities and quantities of expressed mRNA and proteins | Bridging: Single species study for biodistribution and toxicology New: New non-clinical efficacy data | New: New clinical trial data required even for existing mRNAs |
| Significant changes in mRNA sequence length (including potentially to the non-coding region) | Norovirus Tumour-associated antigens Monoclonal antibodies | May potentially change size and nature of LNP used for delivery | Bridge: Manufacturing, analytical and stability studies | Bridge: Single species study for biodistribution and toxicology New: New non-clinical efficacy (and possibly safety) data | New: New clinical trial data required |
| Multivalent products where many epitopes encoded by single strand of mRNA | Individualised neoantigen therapies | Sequences encoding epitopes included in a single mRNA strand and co-translated | New: Require quality and manufacturing approaches that recognise bounds of product manufacture | New: Full toxicology, biodistribution and pharmacology data required for representative products, but not all products | New: New clinical trial data required for representative products, but not all products |
| Self-amplifying mRNA products | COVID-19 vaccine Infectious diseases Oncology | Encoding replicon RNA as well as encoding protein of interest A platform within a platform | New: Origin of replicon genes, whether encoded on same or different mRNA strand to protein of interest, manufacturing and quality assessments | Bridge: Genotoxicity and pregnancy data (if available for another sa-mRNA product) New: Full toxicology, biodistribution and pharmacology data | New: New clinical trial data required |
| LNP variations | Oncology Therapeutics | Changes in LNP size, lipid composition or addition of protein or other tags | Bridge: If LNP composition similar and only size is changed, or slight changes to composition New: If new lipids or proteins are included | Bridge: If LNP composition similar and only size is changed, or slight changes to composition New: Full toxicology, biodistribution and pharmacology data otherwise required | New: New clinical trial data usually required |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skerritt, J.H.; Tucek-Szabo, C.; Sutton, B.; Nolan, T. The Platform Technology Approach to mRNA Product Development and Regulation. Vaccines 2024, 12, 528. https://doi.org/10.3390/vaccines12050528
Skerritt JH, Tucek-Szabo C, Sutton B, Nolan T. The Platform Technology Approach to mRNA Product Development and Regulation. Vaccines. 2024; 12(5):528. https://doi.org/10.3390/vaccines12050528
Chicago/Turabian StyleSkerritt, John H., Carolyn Tucek-Szabo, Brett Sutton, and Terry Nolan. 2024. "The Platform Technology Approach to mRNA Product Development and Regulation" Vaccines 12, no. 5: 528. https://doi.org/10.3390/vaccines12050528
APA StyleSkerritt, J. H., Tucek-Szabo, C., Sutton, B., & Nolan, T. (2024). The Platform Technology Approach to mRNA Product Development and Regulation. Vaccines, 12(5), 528. https://doi.org/10.3390/vaccines12050528