
Development of a Potency Assay for Nous-209, a Multivalent Neoantigens-Based Genetic Cancer Vaccine
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Vaccine Vectors Production and Quantization
2.2. Vaccine Vectors Thermal Inactivation
2.3. Cell Culture
2.4. HeLa Cells Transduction
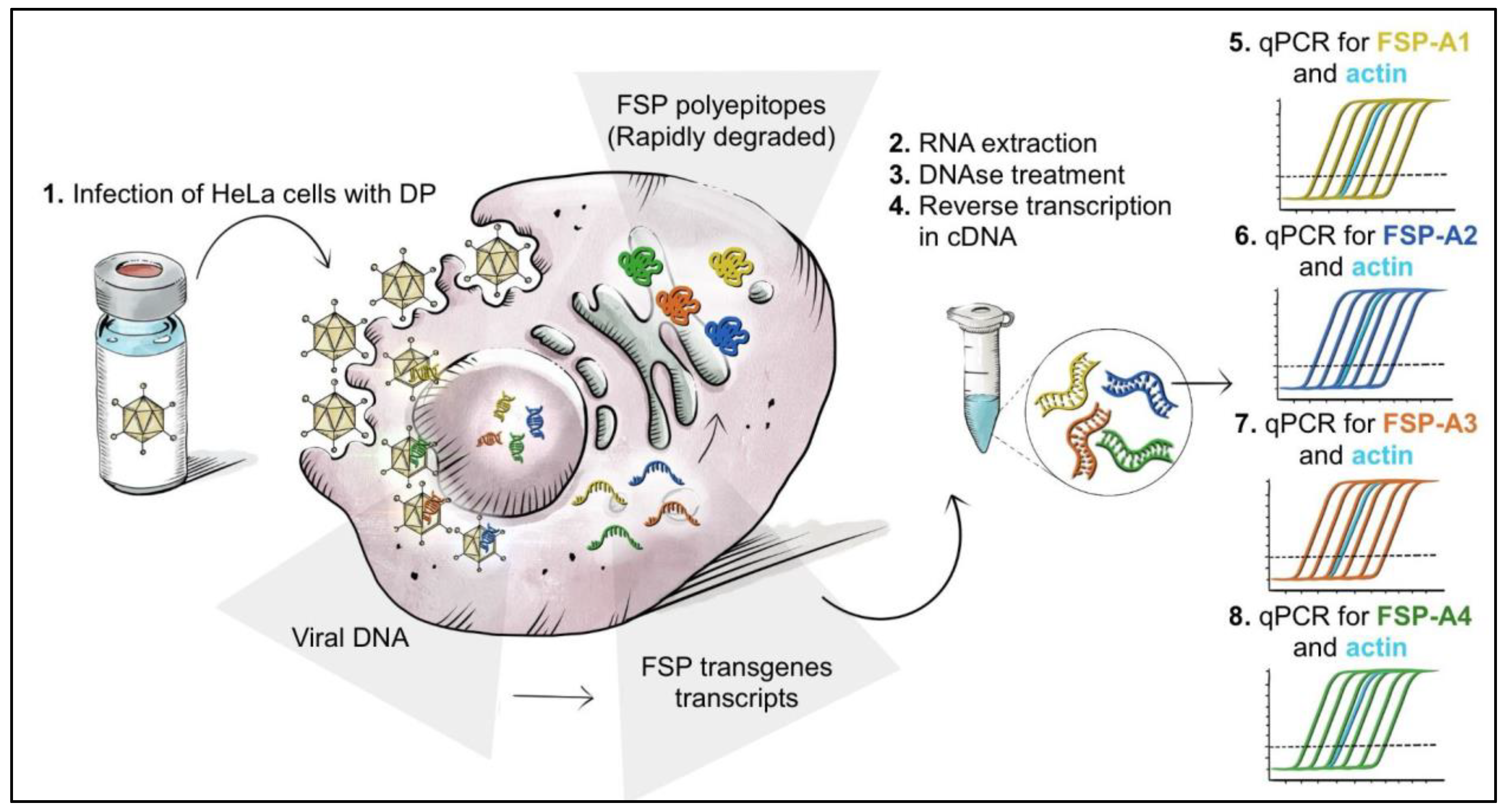
2.5. RNA Extraction and RT-Q-PCR
2.6. Infectivity Assay by Immunostaining
2.7. Mice and Vaccinations
2.8. Ex Vivo Immune Analysis
2.9. Statistical Analyses
3. Results
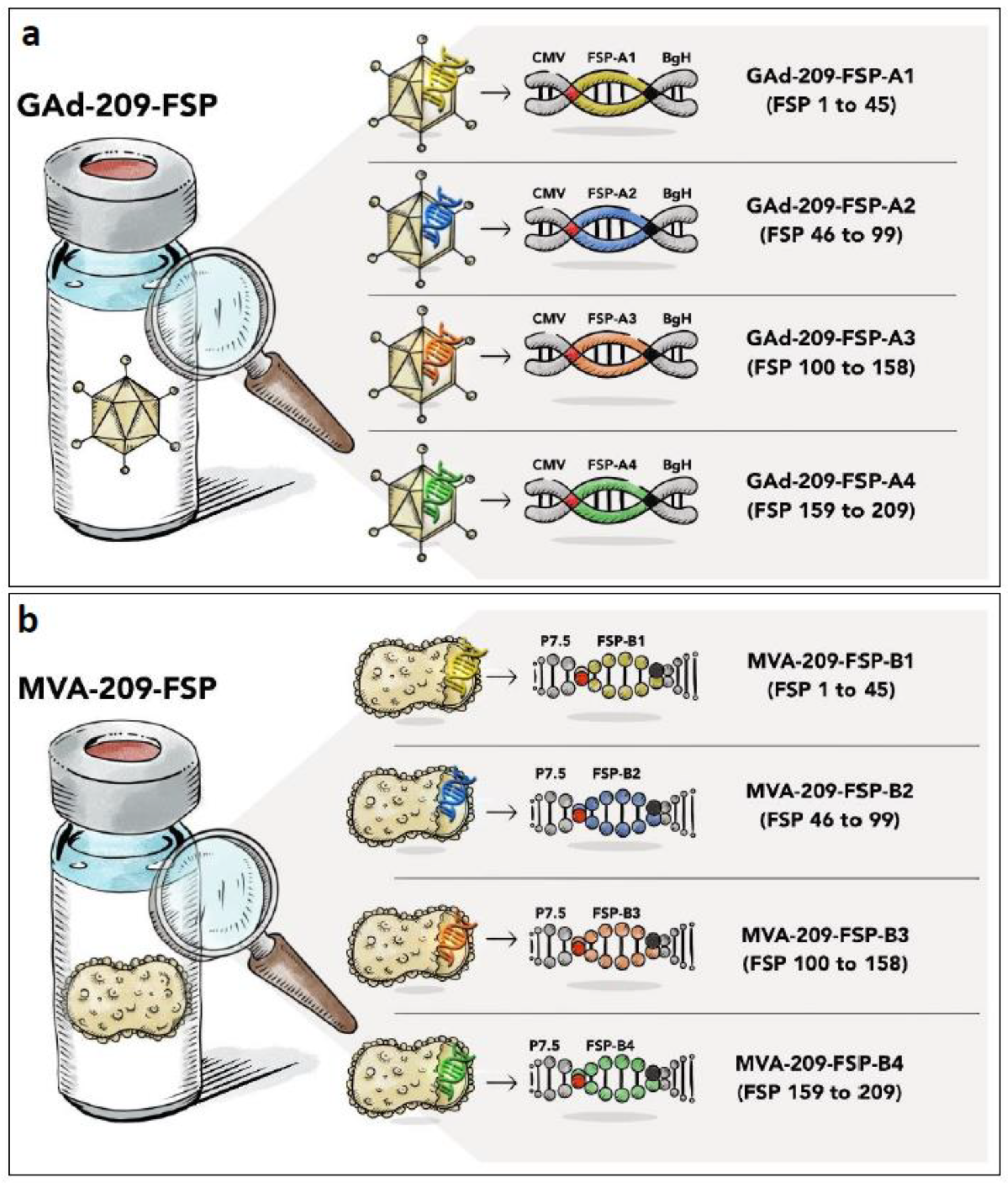
3.1. Nous-209 Vaccine Generation and Immunogenicity
3.2. Definition of Potency Testing Panel for GAd-209-FSP and MVA-209-FSP Vectors
3.3. Setup of a Potency Assay by Quantitative Measure of Transgene mRNA Expression via RT-Q-PCR
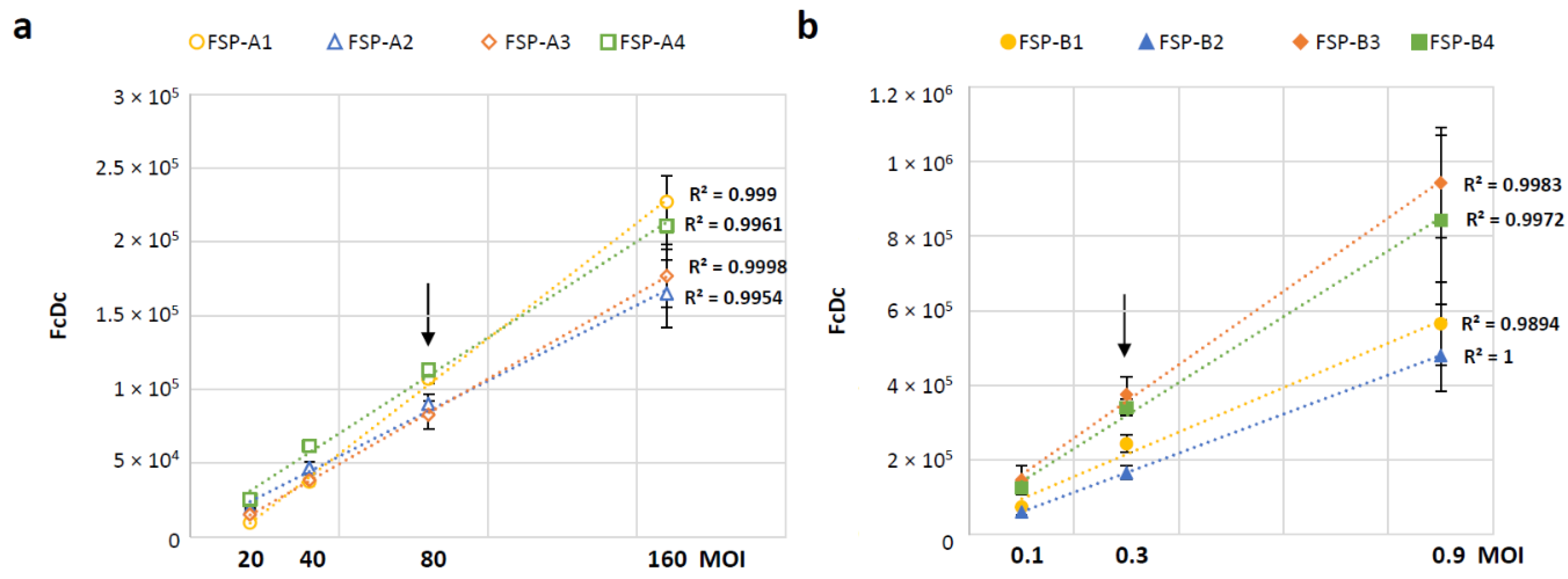
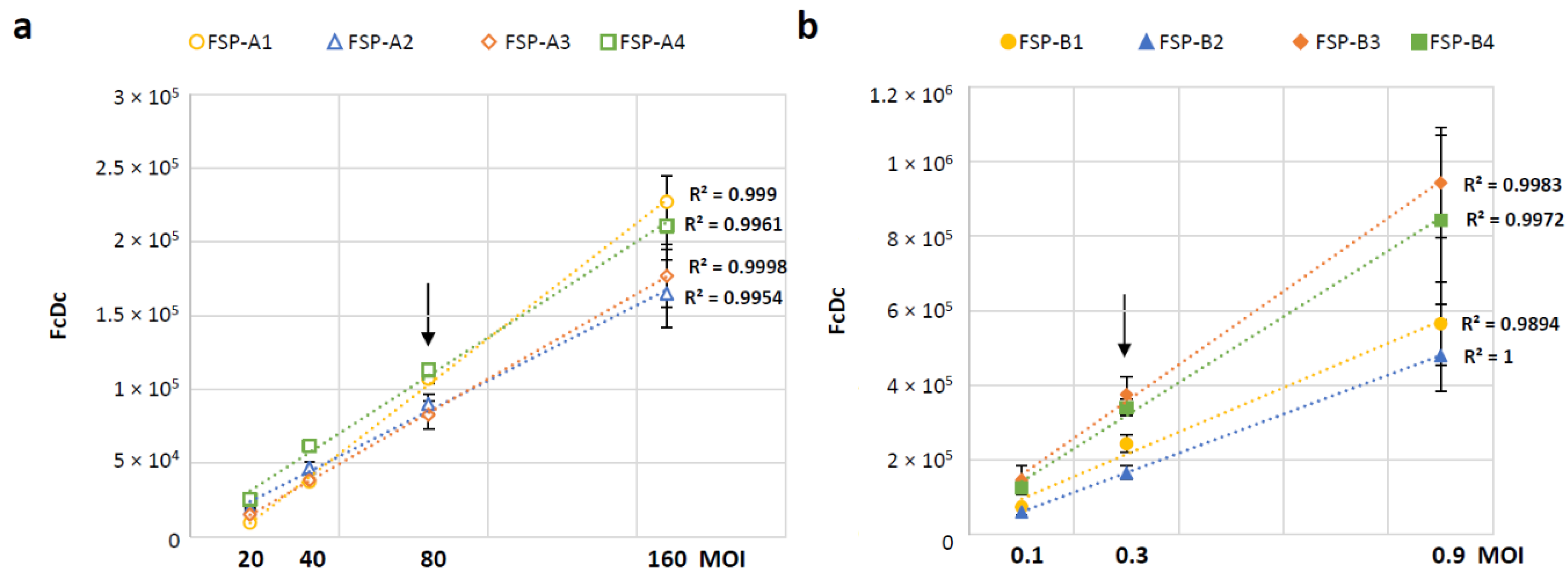
3.4. Selection of Time Points and MOI
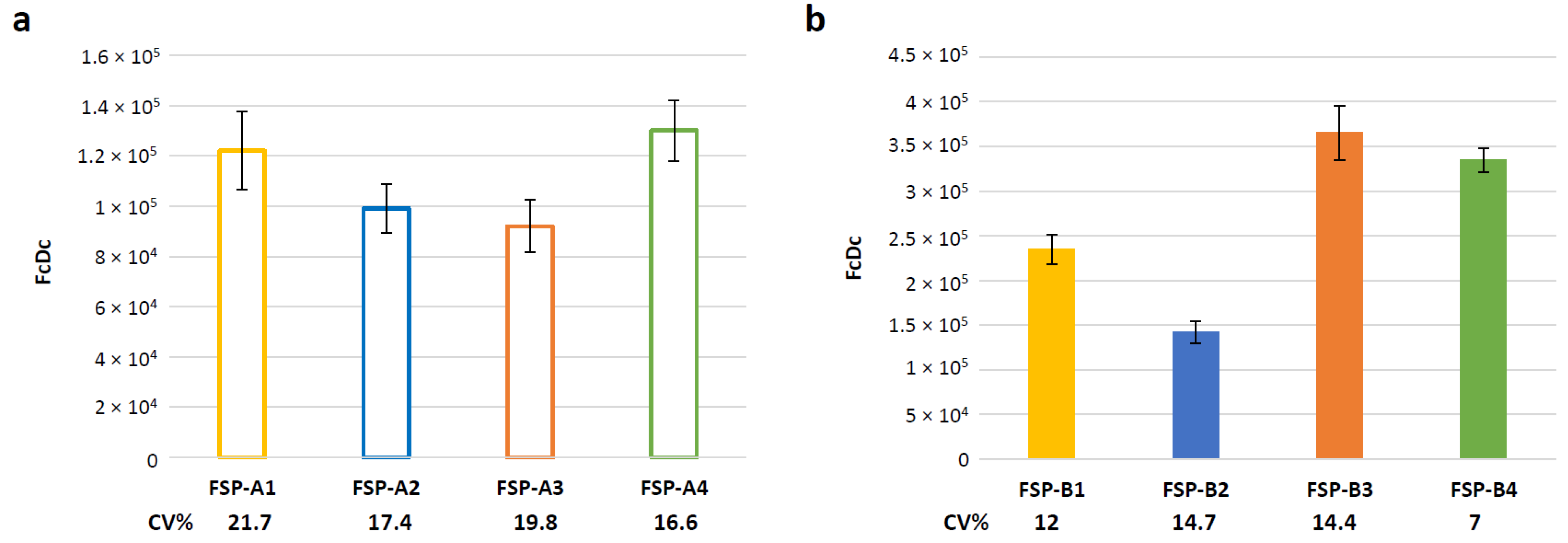
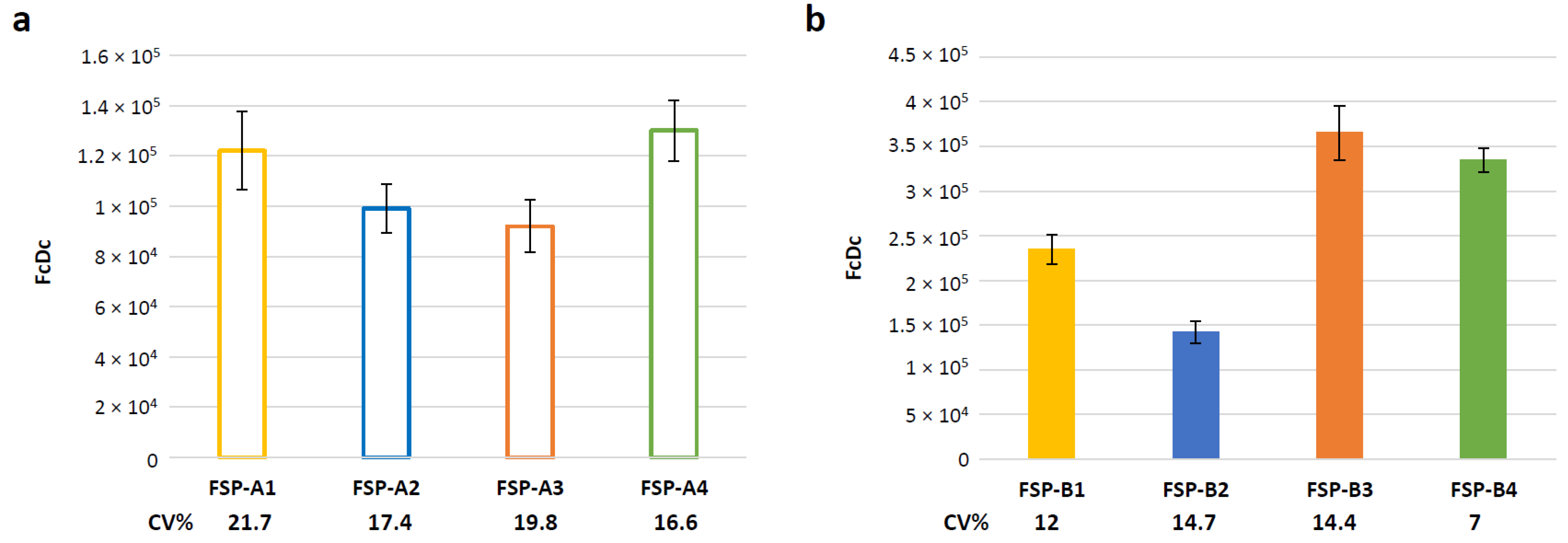
3.5. Evaluation of Assay Variability
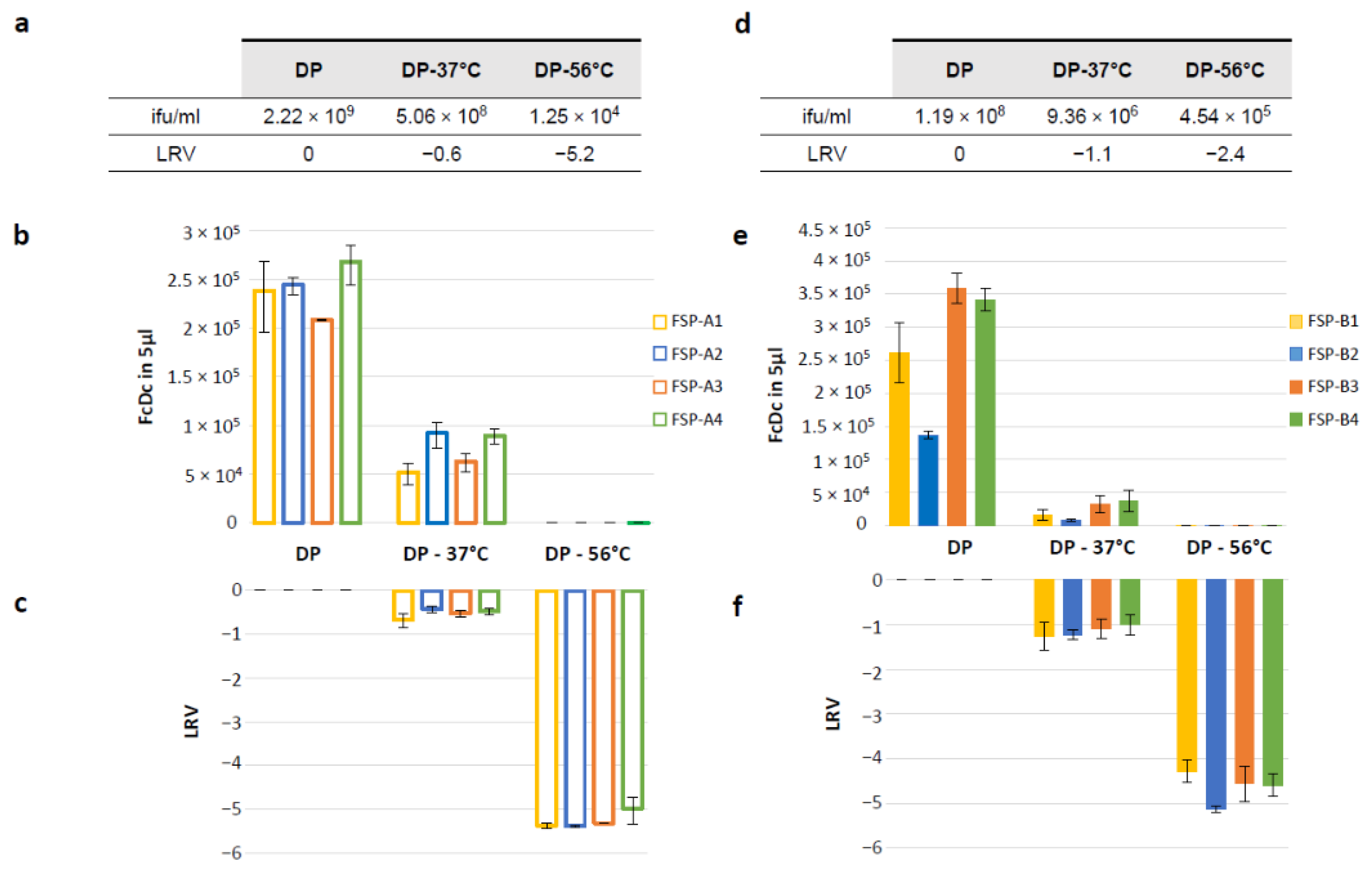
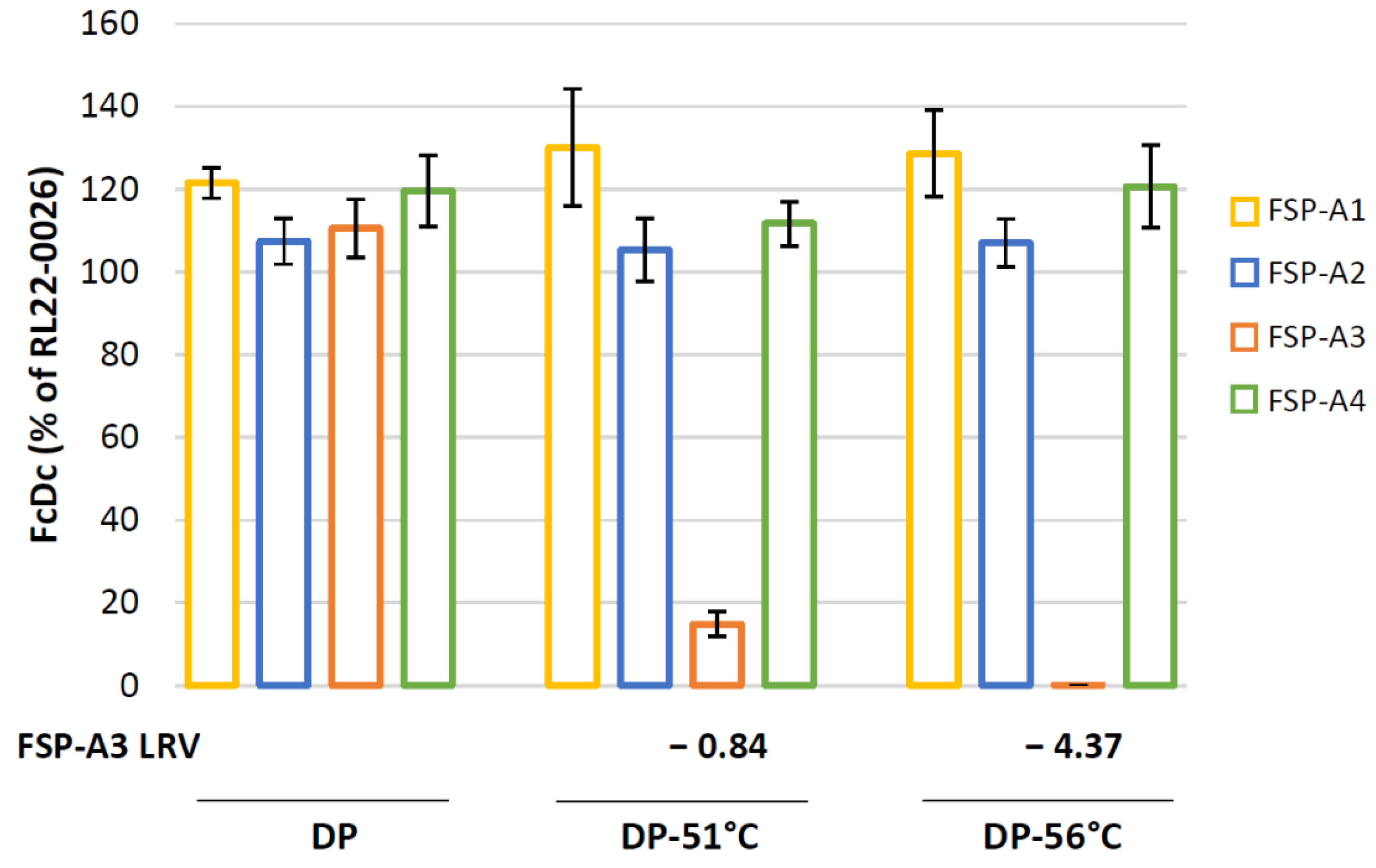
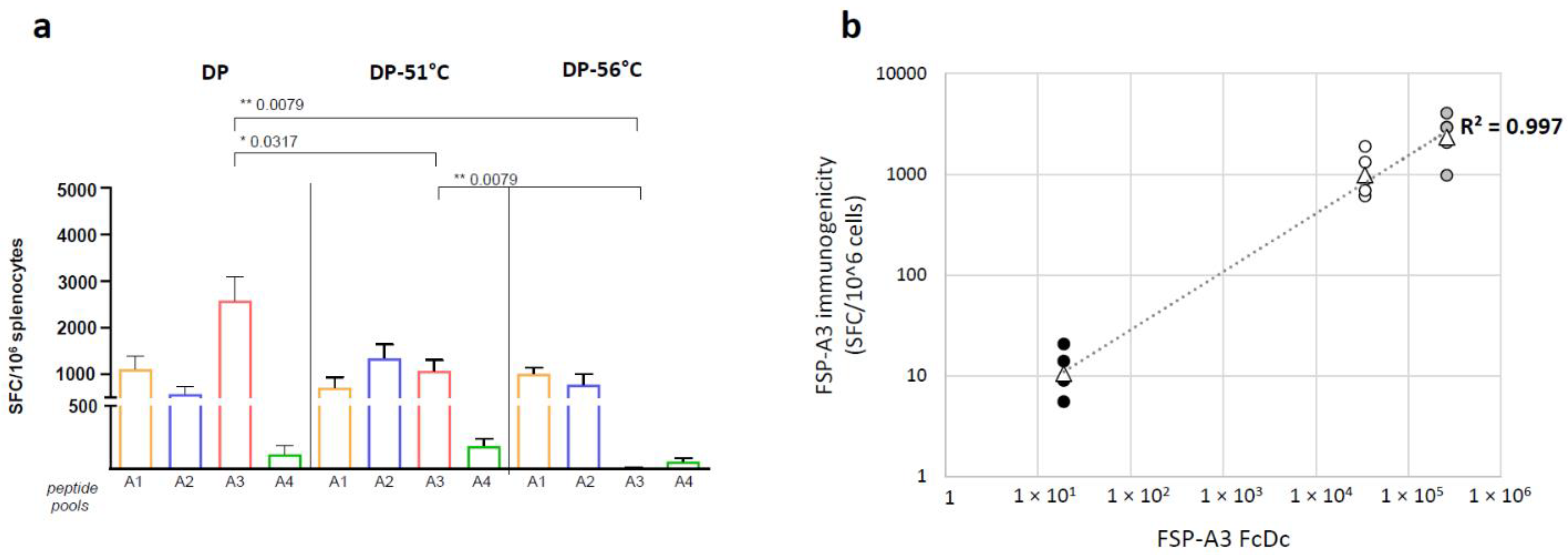
3.6. The RT-Q-PCR Potency Assay Can Detect Loss of Potency Correlating with In Vivo Immunogenicity Data
- DP: mix of GAd-209-FSP-A1 DS; GAd-209-FSP-A2 DS; GAd-209-FSP-A4 DS; GAd-209-FSP-A3 DS.
- DP-51 °C: mix of GAd-209-FSP-A1 DS; GAd-209-FSP-A2 DS; GAd-209-FSP-A4 DS; GAd-209-FSP-A3 DS incubated 3′ at 51 °C.
- DP-56 °C: mix of GAd-209-FSP-A1 DS; GAd-209-FSP-A2 DS; GAd-209-FSP-A4 DS; GAd-209-FSP-A3 DS incubated 10′ at 56 °C.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanyal, G. Development of Functionally Relevant Potency Assays for Monovalent and Multivalent Vaccines Delivered by Evolving Technologies. NPJ Vaccines 2022, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Potency Tests for Cellular and Gene Therapy Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/potency-tests-cellular-and-gene-therapy-products (accessed on 9 February 2024).
- Capelle, M.A.H.; Babich, L.; van Deventer-Troost, J.P.E.; Salerno, D.; Krijgsman, K.; Dirmeier, U.; Raaby, B.; Adriaansen, J. Stability and Suitability for Storage and Distribution of Ad26.ZEBOV/MVA-BN®-Filo Heterologous Prime-Boost Ebola Vaccine. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2018, 129, 215–221. [Google Scholar] [CrossRef]
- Zabdeno|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zabdeno (accessed on 9 February 2024).
- Mvabea|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/mvabea (accessed on 9 February 2024).
- Jcovden (Previously COVID-19 Vaccine Janssen)|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/jcovden-previously-covid-19-vaccine-janssen (accessed on 9 February 2024).
- Vaxzevria (Previously COVID-19 Vaccine AstraZeneca)|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vaxzevria-previously-covid-19-vaccine-astrazeneca (accessed on 9 February 2024).
- Human Cell-Based Medicinal Products—Scientific Guideline|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-cell-based-medicinal-products-scientific-guideline (accessed on 9 February 2024).
- Quality, Non-Clinical and Clinical Aspects of Medicinal Products Containing Genetically Modified Cells—Scientific Guideline|European Medicines Agency. Available online: https://www.ema.europa.eu/en/quality-non-clinical-and-clinical-aspects-medicinal-products-containing-genetically-modified-cells-scientific-guideline (accessed on 9 February 2024).
- Quality, Preclinical and Clinical Aspects of Gene Therapy Medicinal Products—Scientific Guideline|European Medicines Agency. Available online: https://www.ema.europa.eu/en/quality-preclinical-and-clinical-aspects-gene-therapy-medicinal-products-scientific-guideline (accessed on 9 February 2024).
- FDA Delays a Biotech’s Cancer Cell Therapy Once Again. Available online: https://www.biopharmadive.com/news/iovance-fda-delay-lifileucel-ceo-resign/600447/ (accessed on 9 February 2024).
- Salmikangas, P.; Carlsson, B.; Klumb, C.; Reimer, T.; Thirstrup, S. Potency Testing of Cell and Gene Therapy Products. Front. Med. 2023, 10, 1190016. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Brasu, N.; De Intinis, C.; Leoni, G.; Russo, V.; Langone, F.; Baev, D.; Micarelli, E.; Petiti, L.; Picelli, S.; et al. Adenoviral-Based Vaccine Promotes Neoantigen-Specific CD8+ T Cell Stemness and Tumor Rejection. Sci. Transl. Med. 2022, 14, eabo7604. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Rojas, L.A.; Sethna, Z.; Soares, K.; Derhovanessian, E.; Mueller, F.; Yadav, M.; Basturk, O.; Gonen, M.; Wei, A.C.; et al. Phase I Trial of Adjuvant Autogene Cevumeran, an Individualized mRNA Neoantigen Vaccine, for Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2022, 40, 2516. [Google Scholar] [CrossRef]
- Palmer, C.D.; Rappaport, A.R.; Davis, M.J.; Hart, M.G.; Scallan, C.D.; Hong, S.-J.; Gitlin, L.; Kraemer, L.D.; Kounlavouth, S.; Yang, A.; et al. Individualized, Heterologous Chimpanzee Adenovirus and Self-Amplifying mRNA Neoantigen Vaccine for Advanced Metastatic Solid Tumors: Phase 1 Trial Interim Results. Nat. Med. 2022, 28, 1619–1629. [Google Scholar] [CrossRef]
- Morse, M.A.; Gwin, W.R.; Mitchell, D.A. Vaccine Therapies for Cancer: Then and Now. Target. Oncol. 2021, 16, 121–152. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA Vaccines: Current Preclinical and Clinical Developments and Future Perspectives. J. Exp. Clin. Cancer Res. CR 2019, 38, 146. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Mahajan, R.; Feher, B.; Jones, B.; Jones, D.; Marjerison, L.; Sam, M.; Hartikka, J.; Wloch, M.; Lalor, P.; Kaslow, D.; et al. A TaqMan Reverse Transcription Polymerase Chain Reaction (RT-PCR) in Vitro Potency Assay for Plasmid-Based Vaccine Products. Mol. Biotechnol. 2008, 40, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res. 2020, 80, 3972–3982. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A Human Vaccine Strategy Based on Chimpanzee Adenoviral and MVA Vectors That Primes, Boosts, and Sustains Functional HCV-Specific T Cell Memory. Sci. Transl. Med. 2014, 6, 261ra153. [Google Scholar] [CrossRef] [PubMed]
- Bewig, B.; Schmidt, W.E. Accelerated Titering of Adenoviruses. BioTechniques 2000, 28, 870–873. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Leoni, G.; Cotugno, G.; Troise, F.; Langone, F.; Fichera, I.; De Lucia, M.; Avalle, L.; Vitale, R.; Leuzzi, A.; et al. Adenoviral Vaccine Targeting Multiple Neoantigens as Strategy to Eradicate Large Tumors Combined with Checkpoint Blockade. Nat. Commun. 2019, 10, 2688. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S. Vaccinia Virus Transcription. J. Gen. Virol. 2003, 84, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Rafajko, R.R.; Young, J.C. Thermal and ph stability of adenovirus types 12, 14, and 18. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1964, 116, 683–685. [Google Scholar] [CrossRef]
- Kaplan, C. The Heat Inactivation of Vaccinia Virus. J. Gen. Microbiol. 1958, 18, 58–63. [Google Scholar] [CrossRef]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular Strategies of Protein Quality Control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Wang, X.; Kim, S.W.; Herndon, J.M.; Becker-Hapak, M.K.; Carreno, B.M.; Myers, N.B.; Sturmoski, M.A.; McLellan, M.D.; et al. Optimized Polyepitope Neoantigen DNA Vaccines Elicit Neoantigen-Specific Immune Responses in Preclinical Models and in Clinical Translation. Genome Med. 2021, 13, 56. [Google Scholar] [CrossRef]
- Moss, B. Poxvirus Cell Entry: How Many Proteins Does It Take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C. Adenoviruses: Update on Structure and Function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bechter, O.; D’Alise, A.M.; Leoni, G.; Cotugno, G.; Siani, L.; Vitale, R.; Ruzza, V.; Garzia, I.; Antonucci, L.; Micarelli, E.; et al. NOUS-PEV, a Personalized Cancer Immunotherapy Targeting Neoantigens, Induces Long Lasting, Tumor Infiltrating Memory T Cells. Cancer Res. 2023, 83, LB196. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infectivity Assay * | RT-Q-PCR ** | Immunogenicity In Vivo ** | ||||
|---|---|---|---|---|---|---|
| Sample with | ifu/mL | % | FcDc | % | SFC/106 Cells | % |
| Fully potent GAd-209-FSP-A3 | 2.10 × 109 | 100% | 2.41 × 105 | 100% | 2.33 × 103 | 100% |
| Partially inactivated GAd-209-FSP-A3 | 4.14 × 108 | 19.70% | 3.71 × 104 | 15.40% | 9.81 × 102 | 42% |
| Fully inactivated GAd-209-FSP-A3 | 8.40 × 105 | 0.04% | 8.54 × 100 | 0.003% | 1.05 × 101 | 0.45% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartolomeo, R.; Troise, F.; Allocca, S.; Sdruscia, G.; Vitale, R.; Bignone, V.; Petrone, A.M.; Romano, G.; D’Alise, A.M.; Ruzza, V.; et al. Development of a Potency Assay for Nous-209, a Multivalent Neoantigens-Based Genetic Cancer Vaccine. Vaccines 2024, 12, 325. https://doi.org/10.3390/vaccines12030325
Bartolomeo R, Troise F, Allocca S, Sdruscia G, Vitale R, Bignone V, Petrone AM, Romano G, D’Alise AM, Ruzza V, et al. Development of a Potency Assay for Nous-209, a Multivalent Neoantigens-Based Genetic Cancer Vaccine. Vaccines. 2024; 12(3):325. https://doi.org/10.3390/vaccines12030325
Chicago/Turabian StyleBartolomeo, Rosa, Fulvia Troise, Simona Allocca, Giulia Sdruscia, Rosa Vitale, Veronica Bignone, Anna Maria Petrone, Giuseppina Romano, Anna Morena D’Alise, Valentino Ruzza, and et al. 2024. "Development of a Potency Assay for Nous-209, a Multivalent Neoantigens-Based Genetic Cancer Vaccine" Vaccines 12, no. 3: 325. https://doi.org/10.3390/vaccines12030325
APA StyleBartolomeo, R., Troise, F., Allocca, S., Sdruscia, G., Vitale, R., Bignone, V., Petrone, A. M., Romano, G., D’Alise, A. M., Ruzza, V., Garzia, I., Leoni, G., Merone, R., Lanzaro, F., Colloca, S., Siani, L., Scarselli, E., & Cotugno, G. (2024). Development of a Potency Assay for Nous-209, a Multivalent Neoantigens-Based Genetic Cancer Vaccine. Vaccines, 12(3), 325. https://doi.org/10.3390/vaccines12030325