
mRNA-Based Vaccines Are Highly Immunogenic and Confer Protection in the Gnotobiotic Pig Model of Human Rotavirus Diarrhea

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Rotaviruses for Challenge and Immunoassays
2.2. mRNA and Protein Vaccines
2.3. Vaccine Inoculation, Virus Challenge, and Sample Collection of Gn Pigs
2.4. Assessment of Diarrhea and Detection of Fecal Virus Shedding by Antigen ELISA and CCIF
2.5. Detection of Interferon-α Induction after Prime Immunization
2.6. Detection of P2-VP8*-Specific Serum and Intestinal IgA and IgG Antibody by ELISA
2.7. Flow Cytometry for Detection of IFN-γ-Producing CD3+CD4+ and CD3+CD8+ T Cells
2.8. Detection of P2-VP8*-Specific Antibody-Secreting Cells by ELISpot Assay
2.9. Virus Neutralization Assay
2.10. Statistical Analysis
3. Results
3.1. mRNA Vaccines Induce IFN-α in Serum of Vaccinated Animals 14 h Post Immunization
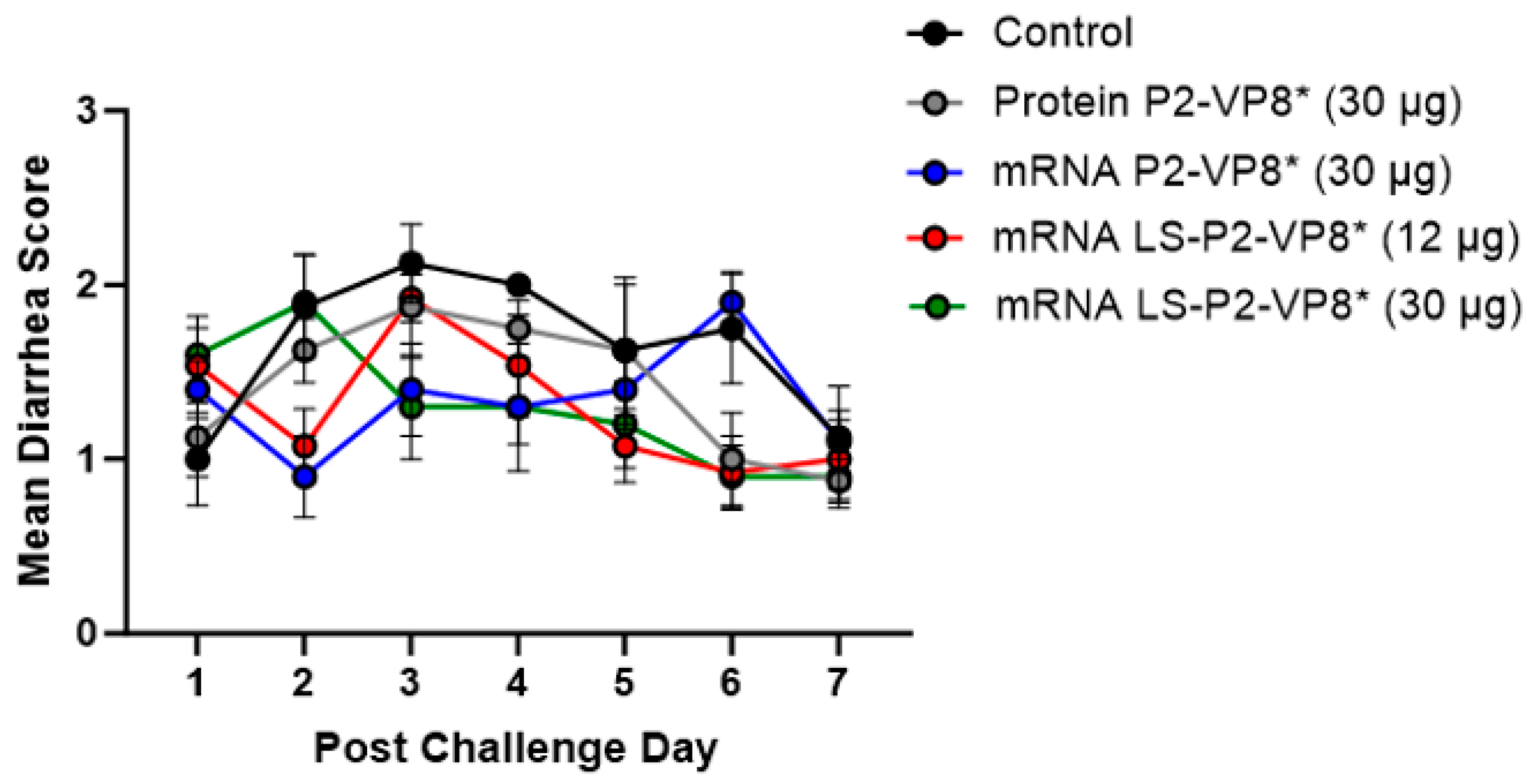
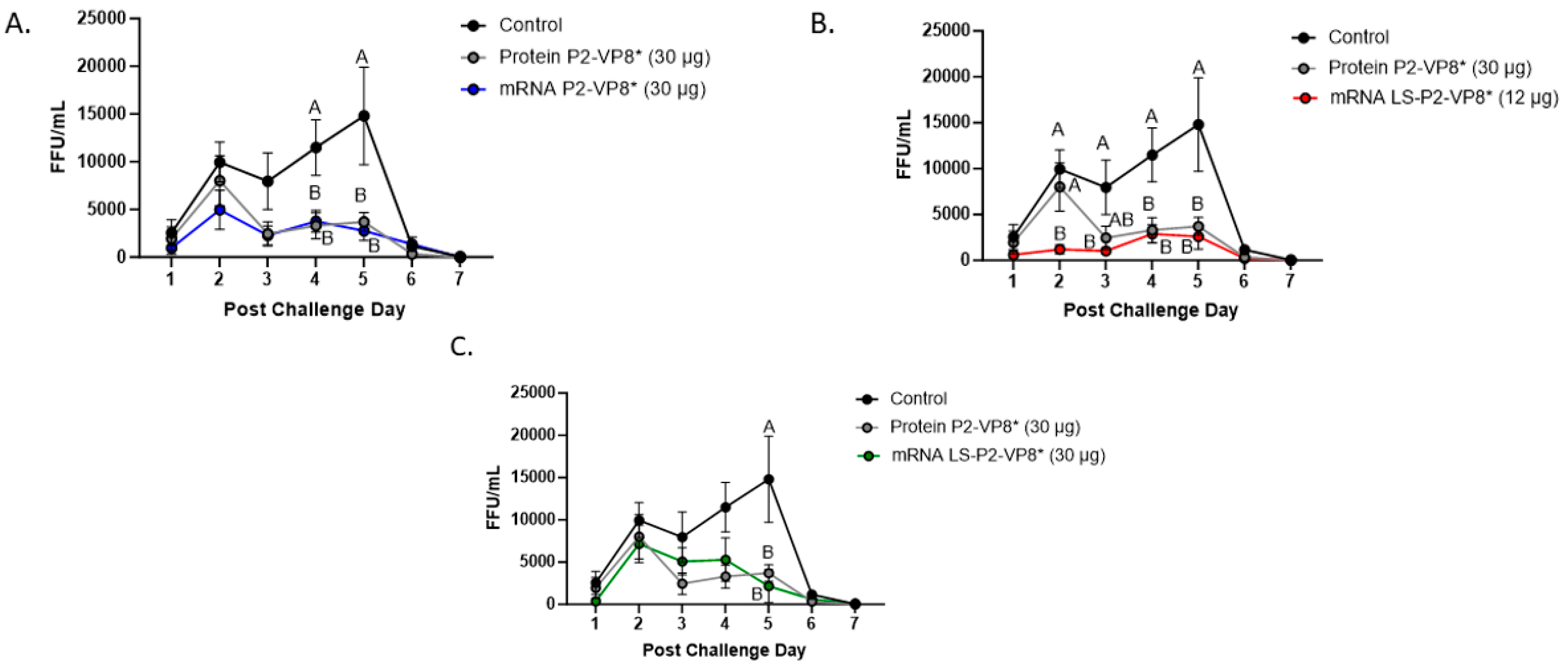
3.2. Both mRNA Vaccine Candidates Conferred Significant Reduction of Severity and Duration of Diarrhea, and Elicited Significant Protection against Infectious Virus Shedding
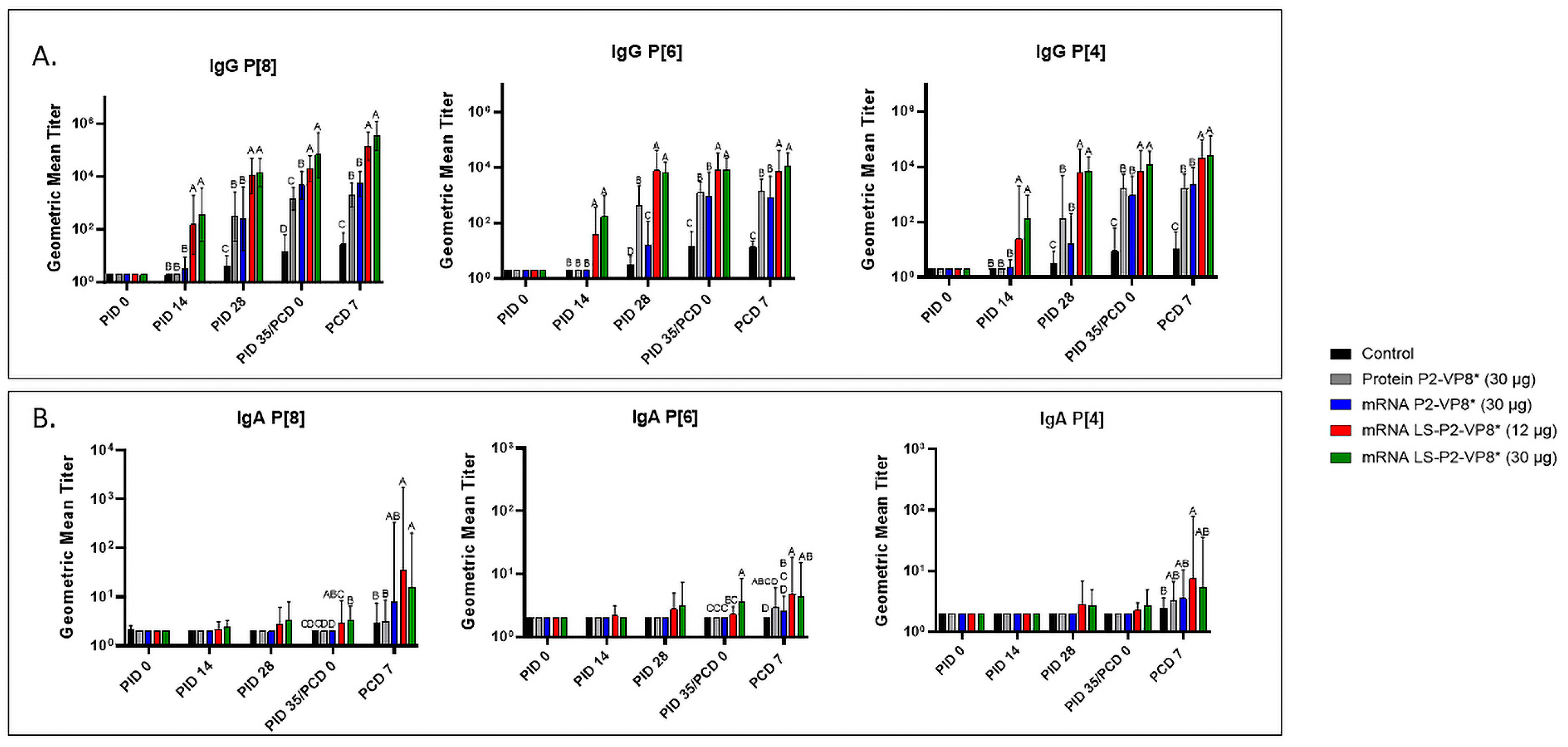
3.3. LS-P2-VP8* Vaccine Induced Strong P-Type-Specific Serum IgG Antibody Responses Pre- and Post-Challenge and Primed for Stronger Serum IgA Antibody Responses Post-Challenge Compared to Other Groups
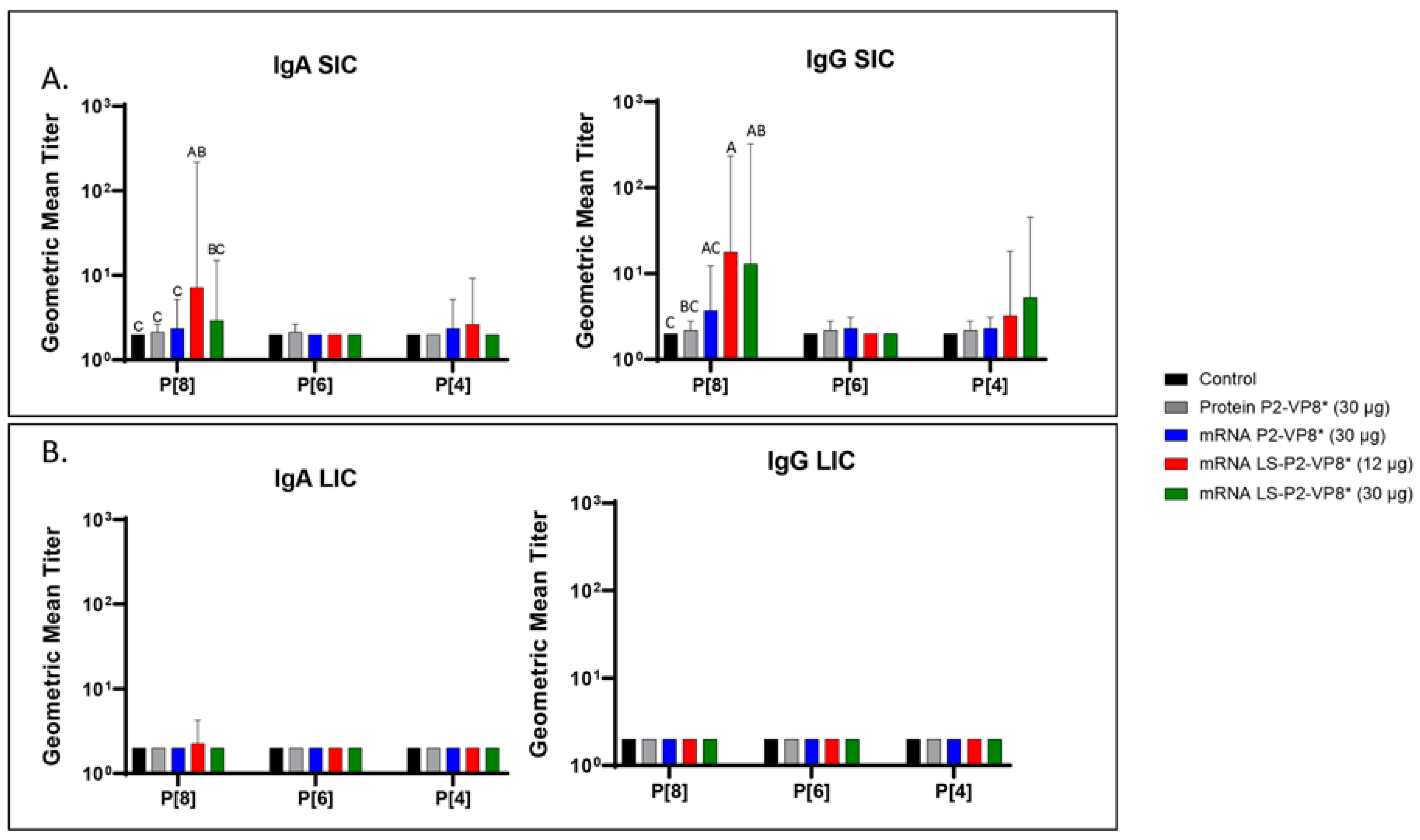
3.4. LS-P2-VP8* Vaccine Primed for Stronger P[8]-Specific Small Intestinal IgA and IgG Antibody Responses Post-Challenge Compared to Other Groups
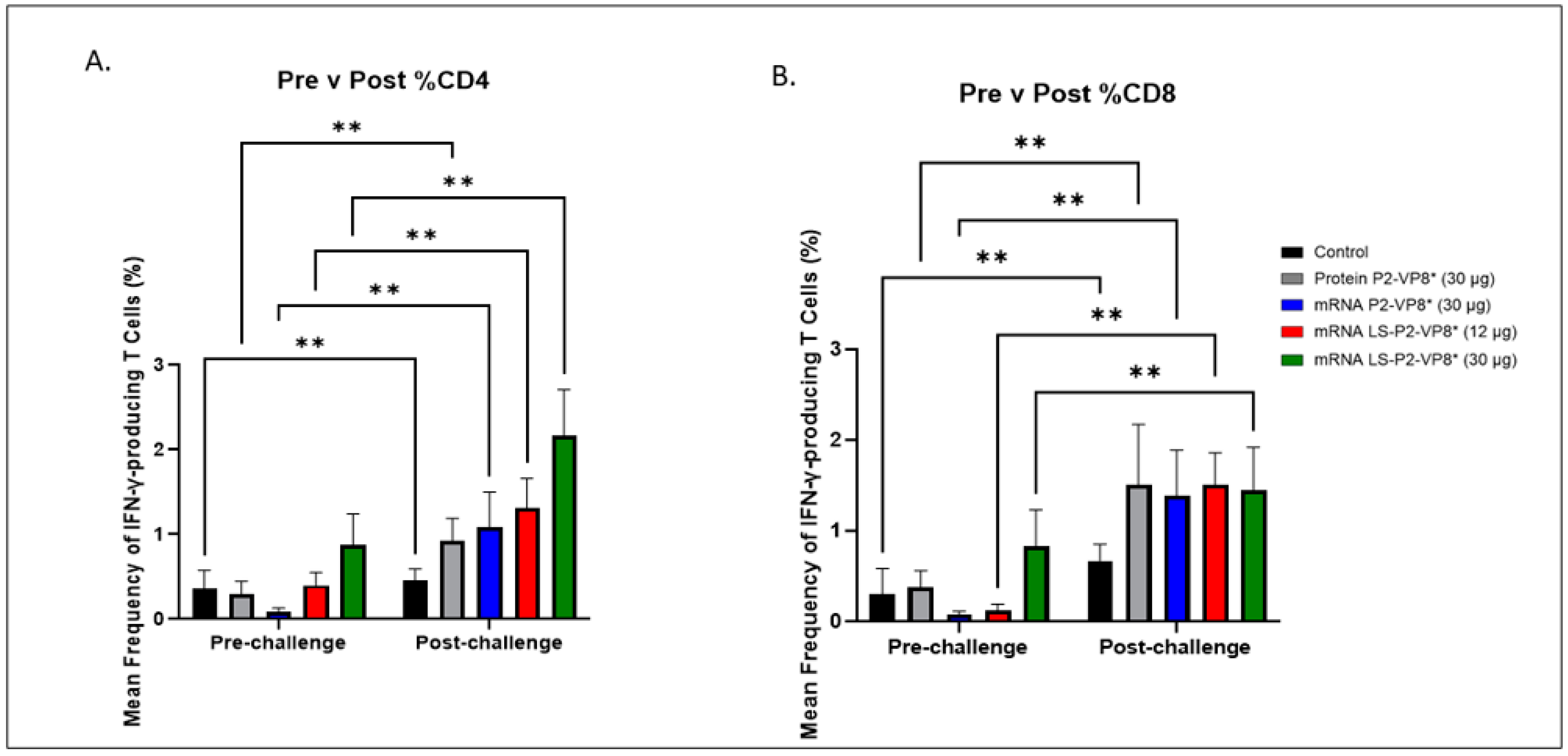
3.5. LS-P2-VP8* (30 µg) Vaccine Induced Stronger HRV-Specific T-Cell Responses in the Blood Pre-Challenge and Primed for Stronger CD4+ Responses Post-Challenge Compared to Other Groups
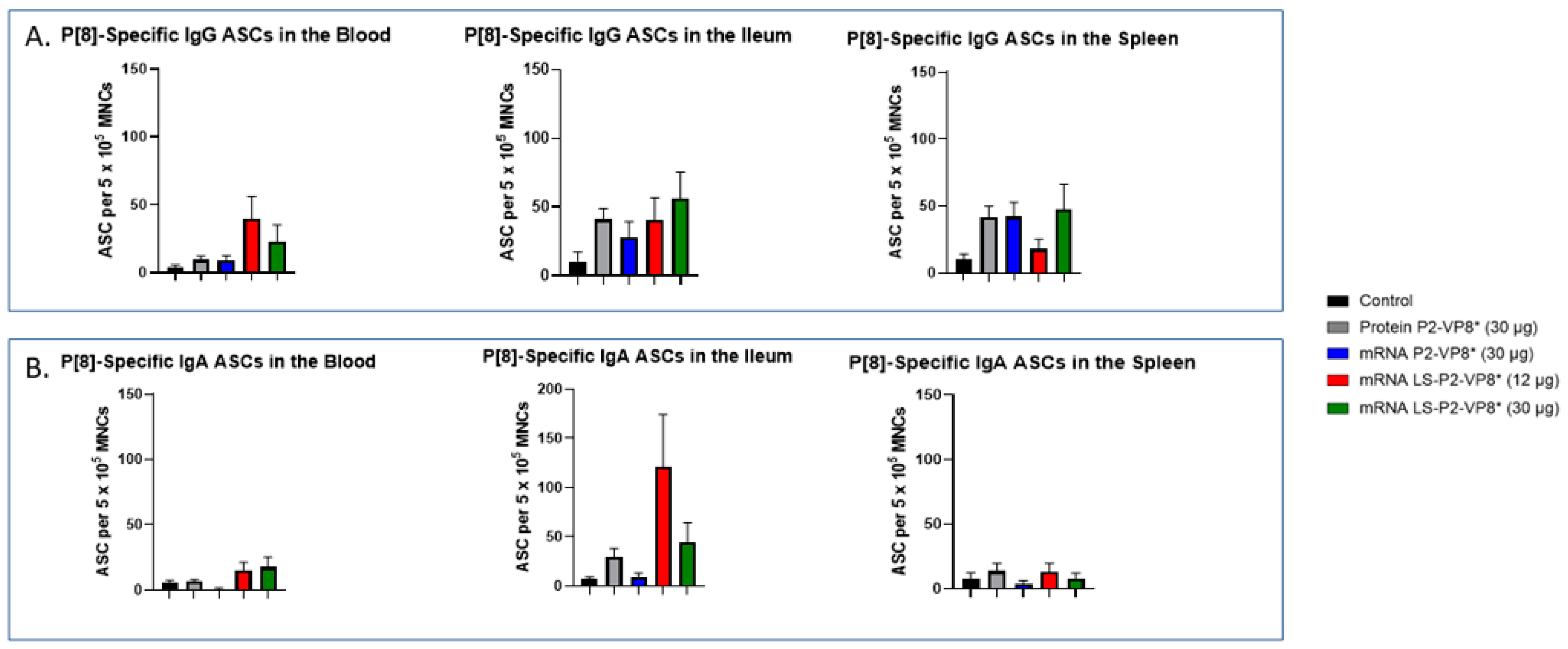
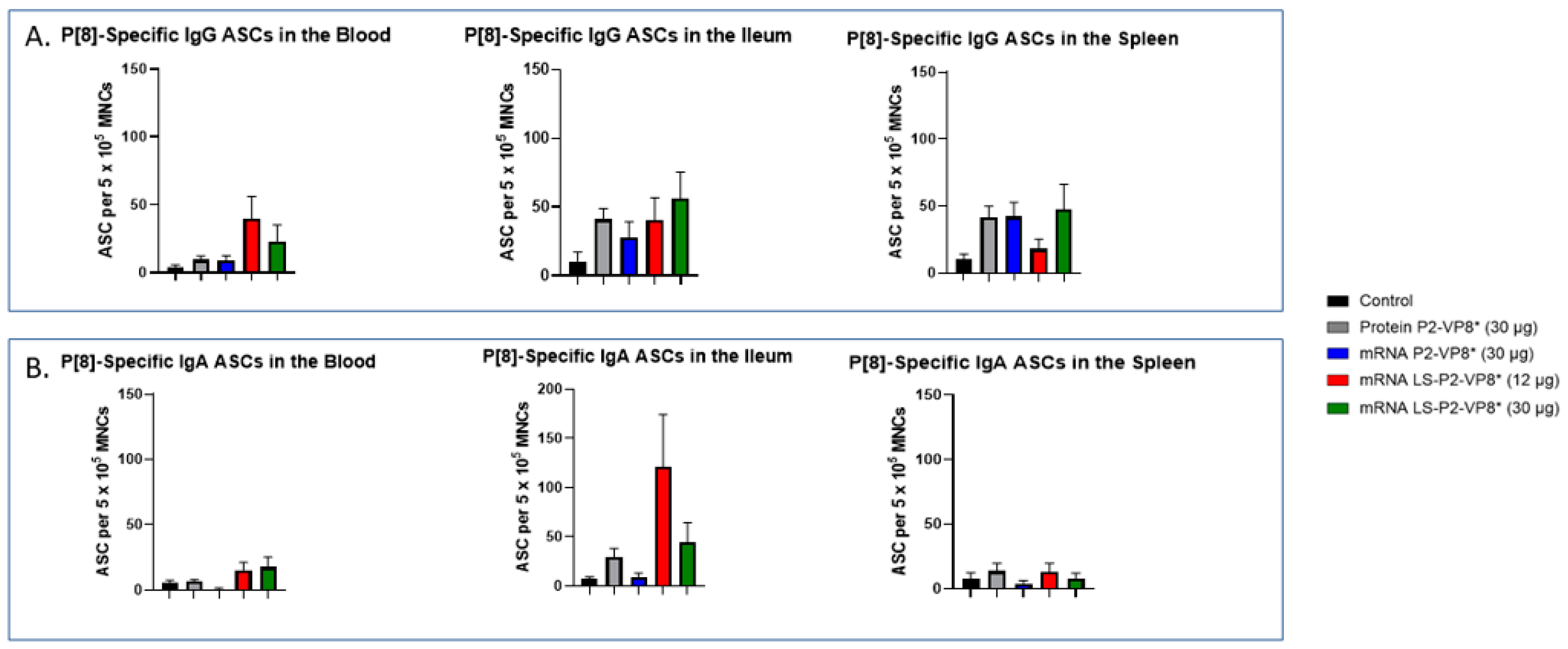
3.6. LS-P2-VP8* Vaccine Primed for Higher Blood and Intestinal ASC Responses Post-Challenge Compared to the Control
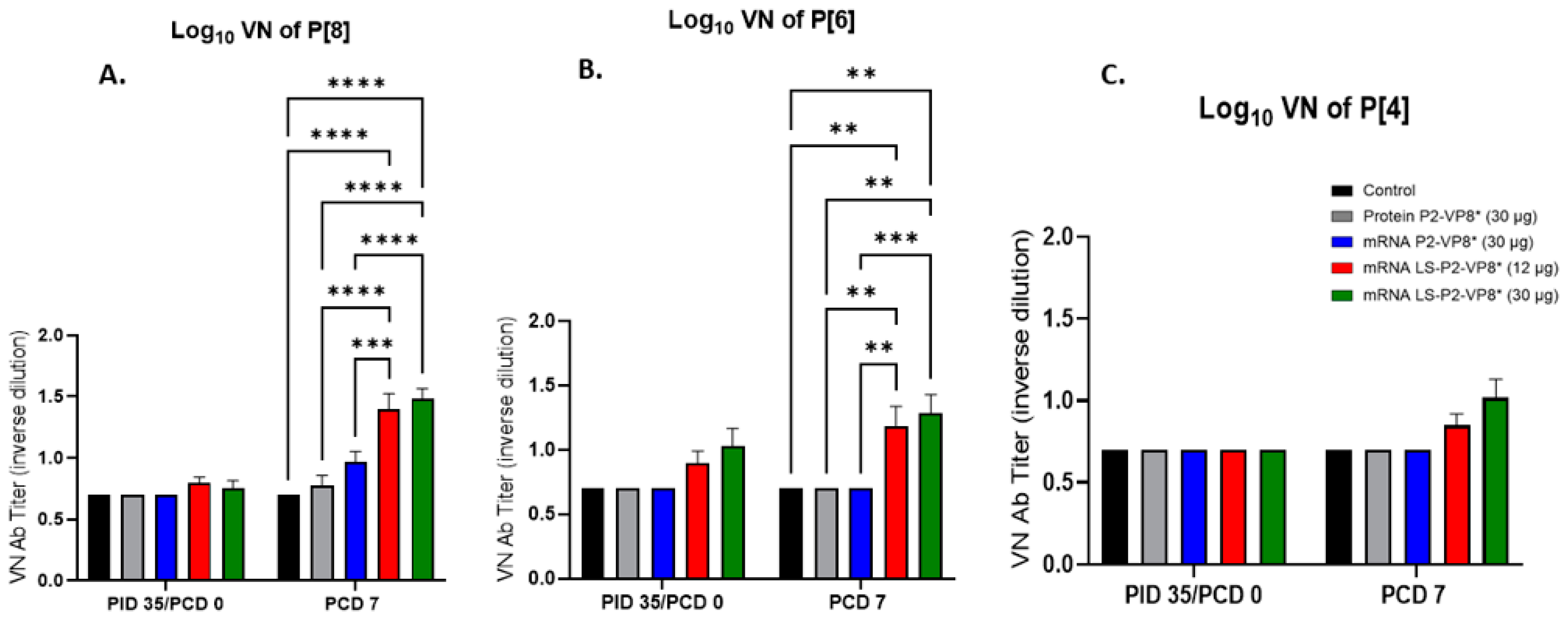
3.7. Pigs Vaccinated with LS-P2-VP8* Were Primed for Significantly Higher P[8]- and P[6]-Specific VN Antibody Titers Post-Challenge Compared to Other Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donato, C.M.; Bines, J.E. Rotaviruses and Rotavirus Vaccines. Pathogens 2021, 10, 959. [Google Scholar] [CrossRef]
- Carcamo-Calvo, R.; Munoz, C.; Buesa, J.; Rodriguez-Diaz, J.; Gozalbo-Rovira, R. The Rotavirus Vaccine Landscape, an Update. Pathogens 2021, 10, 520. [Google Scholar] [CrossRef]
- Chen, J.; Grow, S.; Iturriza-Gomara, M.; Hausdorff, W.P.; Fix, A.; Kirkwood, C.D. The Challenges and Opportunities of Next-Generation Rotavirus Vaccines: Summary of an Expert Meeting with Vaccine Developers. Viruses 2022, 14, 2565. [Google Scholar] [CrossRef]
- Xia, M.; Huang, P.; Tan, M. A Pseudovirus Nanoparticle-Based Trivalent Rotavirus Vaccine Candidate Elicits High and Cross P Type Immune Response. Pharmaceutics 2022, 14, 1597. [Google Scholar] [CrossRef]
- Ramesh, A.; Mao, J.; Lei, S.; Twitchell, E.; Shiraz, A.; Jiang, X.; Tan, M.; Yuan, A.L. Parenterally Administered P24-VP8* Nanoparticle Vaccine Conferred Strong Protection against Rotavirus Diarrhea and Virus Shedding in Gnotobiotic Pigs. Vaccines 2019, 7, 177. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Patel, R.; Kaki, M.; Potluri, V.S.; Kahar, P.; Khanna, D. A comprehensive review of SARS-CoV-2 vaccines: Pfizer, Moderna & Johnson & Johnson. Hum. Vaccin. Immunother. 2022, 18, 2002083. [Google Scholar] [CrossRef] [PubMed]
- Gebre, M.S.; Rauch, S.; Roth, N.; Yu, J.; Chandrashekar, A.; Mercado, N.B.; He, X.; Liu, J.; McMahan, K.; Martinot, A.; et al. Optimization of non-coding regions for a non-modified mRNA COVID-19 vaccine. Nature 2022, 601, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Roth, N.; Schon, J.; Hoffmann, D.; Thran, M.; Thess, A.; Mueller, S.O.; Petsch, B.; Rauch, S. Optimised Non-Coding Regions of mRNA SARS-CoV-2 Vaccine CV2CoV Improves Homologous and Heterologous Neutralising Antibody Responses. Vaccines 2022, 10, 1251. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Primers 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed]
- Roier, S.; Mangala Prasad, V.; McNeal, M.M.; Lee, K.K.; Petsch, B.; Rauch, S. mRNA-based VP8* nanoparticle vaccines against rotavirus are highly immunogenic in rodents. NPJ Vaccines 2023, 8, 190. [Google Scholar] [CrossRef]
- Wen, X.; Wen, K.; Cao, D.; Li, G.; Jones, R.W.; Li, J.; Szu, S.; Hoshino, Y.; Yuan, L. Inclusion of a universal tetanus toxoid CD4(+) T cell epitope P2 significantly enhanced the immunogenicity of recombinant rotavirus DeltaVP8* subunit parenteral vaccines. Vaccine 2014, 32, 4420–4427. [Google Scholar] [CrossRef]
- Groome, M.J.; Fairlie, L.; Morrison, J.; Fix, A.; Koen, A.; Masenya, M.; Jose, L.; Madhi, S.A.; Page, N.; McNeal, M.; et al. Safety and immunogenicity of a parenteral trivalent P2-VP8 subunit rotavirus vaccine: A multisite, randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2020, 20, 851–863. [Google Scholar] [CrossRef]
- Kovacs-Nolan, J.; Mine, Y. Tandem copies of a human rotavirus VP8 epitope can induce specific neutralizing antibodies in BALB/c mice. Biochim. Biophys. Acta 2006, 1760, 1884–1893. [Google Scholar] [CrossRef]
- Wei, Y.; Kumar, P.; Wahome, N.; Mantis, N.J.; Middaugh, C.R. Biomedical Applications of Lumazine Synthase. J. Pharm. Sci. 2018, 107, 2283–2296. [Google Scholar] [CrossRef]
- Jardine, J.; Julien, J.P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef]
- Azuma, Y.; Edwardson, T.G.W.; Hilvert, D. Tailoring lumazine synthase assemblies for bionanotechnology. Chem. Soc. Rev. 2018, 47, 3543–3557. [Google Scholar] [CrossRef]
- Ladenstein, R.; Morgunova, E. Second career of a biosynthetic enzyme: Lumazine synthase as a virus-like nanoparticle in vaccine development. Biotechnol. Rep. 2020, 27, e00494. [Google Scholar] [CrossRef]
- Geng, Q.; Tai, W.; Baxter, V.K.; Shi, J.; Wan, Y.; Zhang, X.; Montgomery, S.A.; Taft-Benz, S.A.; Anderson, E.J.; Knight, A.C.; et al. Novel virus-like nanoparticle vaccine effectively protects animal model from SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009897. [Google Scholar] [CrossRef]
- Yuan, L. Vaccine Efficacy Evaluation: The Gnotobiotic Pig Model, 1st ed.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2022; pp. 1–205. [Google Scholar]
- Desselberger, U.; Huppertz, H.I. Immune responses to rotavirus infection and vaccination and associated correlates of protection. J. Infect. Dis. 2011, 203, 188–195. [Google Scholar] [CrossRef]
- Pabst, R. The pig as a model for immunology research. Cell Tissue Res. 2020, 380, 287–304. [Google Scholar] [CrossRef]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Kim, Y.B. Developmental immunity in the piglet. Birth Defects Orig. Artic. Ser. 1975, 11, 549–557. [Google Scholar]
- Ward, L.A.; Rosen, B.I.; Yuan, L.; Saif, L.J. Pathogenesis of an attenuated and a virulent strain of group A human rotavirus in neonatal gnotobiotic pigs. J. Gen. Virol. 1996, 77 Pt 7, 1431–1441. [Google Scholar] [CrossRef]
- Twitchell, E.L.; Tin, C.; Wen, K.; Zhang, H.; Becker-Dreps, S.; Azcarate-Peril, M.A.; Vilchez, S.; Li, G.; Ramesh, A.; Weiss, M.; et al. Modeling human enteric dysbiosis and rotavirus immunity in gnotobiotic pigs. Gut Pathog. 2016, 8, 51. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as novel technology for passive immunotherapy. Cell Mol. Life Sci. 2019, 76, 301–328. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Melo, M.; Porter, E.; Zhang, Y.; Silva, M.; Li, N.; Dobosh, B.; Liguori, A.; Skog, P.; Landais, E.; Menis, S.; et al. Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol. Ther. 2019, 27, 2080–2090. [Google Scholar] [CrossRef]
- Aldrich, C.; Leroux-Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 2021, 39, 1310–1318. [Google Scholar] [CrossRef]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 29. [Google Scholar] [CrossRef]
- Wen, X.; Cao, D.; Jones, R.W.; Li, J.; Szu, S.; Hoshino, Y. Construction and characterization of human rotavirus recombinant VP8* subunit parenteral vaccine candidates. Vaccine 2012, 30, 6121–6126. [Google Scholar] [CrossRef]
- Fix, A.D.; Harro, C.; McNeal, M.; Dally, L.; Flores, J.; Robertson, G.; Boslego, J.W.; Cryz, S. Safety and immunogenicity of a parenterally administered rotavirus VP8 subunit vaccine in healthy adults. Vaccine 2015, 33, 3766–3772. [Google Scholar] [CrossRef]
- Lakatos, K.; McAdams, D.; White, J.A.; Chen, D. Formulation and preclinical studies with a trivalent rotavirus P2-VP8 subunit vaccine. Human. Vaccines Immunother. 2020, 16, 1957–1968. [Google Scholar] [CrossRef]
- Agarwal, S.; Hickey, J.M.; McAdams, D.; White, J.A.; Sitrin, R.; Khandke, L.; Cryz, S.; Joshi, S.B.; Volkin, D.B. Effect of Aluminum Adjuvant and Preservatives on Structural Integrity and Physicochemical Stability Profiles of Three Recombinant Subunit Rotavirus Vaccine Antigens. J. Pharm. Sci. 2020, 109, 476–487. [Google Scholar] [CrossRef] [PubMed]
- McAdams, D.; Lakatos, K.; Estrada, M.; Chen, D.; Plikaytis, B.; Sitrin, R.; White, J.A. Quantification of trivalent non-replicating rotavirus vaccine antigens in the presence of aluminum adjuvant. J. Immunol. Methods 2021, 494, 113056. [Google Scholar] [CrossRef]
- McAdams, D.; Estrada, M.; Holland, D.; Singh, J.; Sawant, N.; Hickey, J.M.; Kumar, P.; Plikaytis, B.; Joshi, S.B.; Volkin, D.B.; et al. Concordance of in vitro and in vivo measures of non-replicating rotavirus vaccine potency. Vaccine 2022, 40, 5069–5078. [Google Scholar] [CrossRef]
- Yuan, L.; Jobst, P.M.; Weiss, M. Gnotobiotic Pigs: From Establishing Facility to Modeling Human Infectious Diseases. In Gnotobiotics; Trenton, R., Schoeb, K.A.E., Eds.; Sarah Tenney: Westerville, OH, USA, 2017; pp. 349–364. [Google Scholar]
- Yuan, L.; Wen, K.; Azevedo, M.S.; Gonzalez, A.M.; Zhang, W.; Saif, L.J. Virus-specific intestinal IFN-gamma producing T cell responses induced by human rotavirus infection and vaccines are correlated with protection against rotavirus diarrhea in gnotobiotic pigs. Vaccine 2008, 26, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.J.; Ward, L.A.; Rosen, B.I.; To, T.L.; Saif, L.J. Systemic and intestinal antibody-secreting cell responses and correlates of protective immunity to human rotavirus in a gnotobiotic pig model of disease. J. Virol. 1996, 70, 3075–3083. [Google Scholar] [CrossRef]
- Vega, C.G.; Garaicoechea, L.L.; Degiuseppe, J.I.; Bok, M.; Rivolta, A.A.; Piantanida, A.P.; Asenzo, G.; Aduriz Guerrero, M.; Wigdorovitz, A.; Stupka, J.A.; et al. ROTADIAL: The first nanobody-based immunoassay to detect Group A Rotavirus. J. Virol. Methods 2021, 298, 114279. [Google Scholar] [CrossRef]
- Knowlton, D.R.; Spector, D.M.; Ward, R.L. Development of an improved method for measuring neutralizing antibody to rotavirus. J. Virol. Methods 1991, 33, 127–134. [Google Scholar] [CrossRef]
- Kenward, M.G.; Roger, J.H. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 1997, 53, 983–997. [Google Scholar] [CrossRef]
- Otero, C.E.; Langel, S.N.; Blasi, M.; Permar, S.R. Maternal antibody interference contributes to reduced rotavirus vaccine efficacy in developing countries. PLoS Pathog. 2020, 16, e1009010. [Google Scholar] [CrossRef]
- Kaul, D.; Ogra, P.L. Mucosal responses to parenteral and mucosal vaccines. Dev. Biol. Stand. 1998, 95, 141–146. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gomara, M.; Maes, P.; Patton, J.T.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Engevik, M.A.; Engevik, K.A.; Scribano, F.J.; Gebert, J.T.; Danhof, H.A.; Nelson, J.C.; Kellen, J.S.; Strtak, A.C.; et al. Rotavirus induces intercellular calcium waves through ADP signaling. Science 2020, 370, 3621. [Google Scholar] [CrossRef]
- Angel, J.; Steele, A.D.; Franco, M.A. Correlates of protection for rotavirus vaccines: Possible alternative trial endpoints, opportunities, and challenges. Hum. Vaccin. Immunother. 2014, 10, 3659–3671. [Google Scholar] [CrossRef]
- Corthesy, B.; Benureau, Y.; Perrier, C.; Fourgeux, C.; Parez, N.; Greenberg, H.; Schwartz-Cornil, I. Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J. Virol. 2006, 80, 10692–10699. [Google Scholar] [CrossRef]
- Pollock, L.; Bennett, A.; Jere, K.C.; Mandolo, J.; Dube, Q.; Bar-Zeev, N.; Heyderman, R.S.; Cunliffe, N.A.; Iturriza-Gomara, M. Plasma Rotavirus-specific IgA and Risk of Rotavirus Vaccine Failure in Infants in Malawi. Clin. Infect. Dis. 2022, 75, 41–46. [Google Scholar] [CrossRef]
- Baker, J.M.; Tate, J.E.; Leon, J.; Haber, M.J.; Pitzer, V.E.; Lopman, B.A. Postvaccination Serum Antirotavirus Immunoglobulin A as a Correlate of Protection Against Rotavirus Gastroenteritis Across Settings. J. Infect. Dis. 2020, 222, 309–318. [Google Scholar] [CrossRef]
- Westerman, L.E.; McClure, H.M.; Jiang, B.; Almond, J.W.; Glass, R.I. Serum IgG mediates mucosal immunity against rotavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 7268–7273. [Google Scholar] [CrossRef]
- Wen, K.; Bui, T.; Weiss, M.; Li, G.; Kocher, J.; Yang, X.; Jobst, P.M.; Vaught, T.; Ramsoondar, J.; Ball, S.; et al. B-Cell-Deficient and CD8 T-Cell-Depleted Gnotobiotic Pigs for the Study of Human Rotavirus Vaccine-Induced Protective Immune Responses. Viral Immunol. 2016, 29, 112–127. [Google Scholar] [CrossRef]
- Fleming, S.B. Viral Inhibition of the IFN-Induced JAK/STAT Signalling Pathway: Development of Live Attenuated Vaccines by Mutation of Viral-Encoded IFN-Antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8(+) T Cell Immunity to mRNA Vaccines. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Groome, M.J.; Koen, A.; Fix, A.; Page, N.; Jose, L.; Madhi, S.A.; McNeal, M.; Dally, L.; Cho, I.; Power, M.; et al. Safety and immunogenicity of a parenteral P2-VP8-P[8] subunit rotavirus vaccine in toddlers and infants in South Africa: A randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2017, 17, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Fellows, T.; Page, N.; Fix, A.; Flores, J.; Cryz, S.; McNeal, M.; Iturriza-Gomara, M.; Groome, M.J. Association between Immunogenicity of a Monovalent Parenteral P2-VP8 Subunit Rotavirus Vaccine and Fecal Shedding of Rotavirus following Rotarix Challenge during a Randomized, Double-Blind, Placebo-Controlled Trial. Viruses 2023, 15, 1809. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | Vaccine | Challenge |
|---|---|---|---|
| 1 | 8 | Irrelevant LNP-formulated mRNA vaccine (Control) | Wa HRV (G1P[8]) |
| 2 | 8 | Trivalent alum-adjuvanted P2-VP8* protein vaccine (Protein P2-VP8*) | |
| 3 | 10 | Trivalent LNP-formulated P2-VP8* mRNA vaccine (P2-VP8*) | |
| 4 | 13 | Trivalent LNP-formulated LS-P2-VP8* mRNA vaccine (LS-P2-VP8* [12 µg]) | |
| 5 | 10 | Trivalent LNP-formulated LS-P2-VP8* mRNA vaccine (LS-P2-VP8* [30 µg]) |
| Clinical Signs of Diarrhea b | Virus Shedding (CCIF) c | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatments (Vaccine Dose µg) a | n | Percentage with Diarrhea | Mean Days to Onset | Mean Duration Days | Mean Cumulative Fecal Score | AUC of Diarrhea | Percentage of Shedding Virus | Mean Days to Onset | Mean Duration Days | Mean Peak Titer (FFU/mL of Feces) f | AUC of Virus Shedding |
| Control | 8 | 8/8 (100%) | 2 | 4.6 (0.53) Ade | 11.5 (0.78) A | 10.4 (0.65) A | 8/8 (100%) | 1.5 | 5.4 (0.18) A | 2.1 × 104 (3645) A | 4.7 × 104 (6881) A |
| Protein P2-VP8* (30 µg) | 8 | 8/8 (100%) | 1.9 | 3.1 (0.35) AB | 9.9 (0.74) AB | 8.9 (0.78) AB | 8/8 (100%) | 1.3 | 4.9 (0.30) AB | 1.0 × 104 (2182) ABC | 1.9 × 104 (4030) AB |
| mRNA P2-VP8* (30 µg) | 10 | 10/10 (100%) | 2.4 | 3.1 (0.10) B | 9.4 (0.48) AB | 8.2 (0.48) B | 10/10 (100%) | 1.8 | 4.0 (0.33) BC | 6.9 × 103 (1849) CD | 1.6 × 104 (4932) BC |
| mRNA LS-P2-VP8* (12 µg) | 13 | 12/13 (92%) | 1.9 | 3.3 (0.51) AB | 9.1 (0.85) B | 7.8 (0.72) B | 13/13 (100%) | 1.7 | 3.6 (0.37) C | 4.5 × 103 (1182) D | 8.3 × 103 (2619) C |
| mRNA LS-P2-VP8* (30 µg) | 10 | 9/10 (90%) | 2.4 | 2.5 (0.48) B | 9.1 (0.91) B | 7.9 (0.83) B | 10/10 (100%) | 1.7 | 4.1 (0.43) BC | 1.2 × 104 (2335) ABC | 2.1 × 104 (5103) B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hensley, C.; Roier, S.; Zhou, P.; Schnur, S.; Nyblade, C.; Parreno, V.; Frazier, A.; Frazier, M.; Kiley, K.; O’Brien, S.; et al. mRNA-Based Vaccines Are Highly Immunogenic and Confer Protection in the Gnotobiotic Pig Model of Human Rotavirus Diarrhea. Vaccines 2024, 12, 260. https://doi.org/10.3390/vaccines12030260
Hensley C, Roier S, Zhou P, Schnur S, Nyblade C, Parreno V, Frazier A, Frazier M, Kiley K, O’Brien S, et al. mRNA-Based Vaccines Are Highly Immunogenic and Confer Protection in the Gnotobiotic Pig Model of Human Rotavirus Diarrhea. Vaccines. 2024; 12(3):260. https://doi.org/10.3390/vaccines12030260
Chicago/Turabian StyleHensley, Casey, Sandro Roier, Peng Zhou, Sofia Schnur, Charlotte Nyblade, Viviana Parreno, Annie Frazier, Maggie Frazier, Kelsey Kiley, Samantha O’Brien, and et al. 2024. "mRNA-Based Vaccines Are Highly Immunogenic and Confer Protection in the Gnotobiotic Pig Model of Human Rotavirus Diarrhea" Vaccines 12, no. 3: 260. https://doi.org/10.3390/vaccines12030260
APA StyleHensley, C., Roier, S., Zhou, P., Schnur, S., Nyblade, C., Parreno, V., Frazier, A., Frazier, M., Kiley, K., O’Brien, S., Liang, Y., Mayer, B. T., Wu, R., Mahoney, C., McNeal, M. M., Petsch, B., Rauch, S., & Yuan, L. (2024). mRNA-Based Vaccines Are Highly Immunogenic and Confer Protection in the Gnotobiotic Pig Model of Human Rotavirus Diarrhea. Vaccines, 12(3), 260. https://doi.org/10.3390/vaccines12030260