
The Protective Effect of IL-17A in Pneumonic Plague Can Be Compensated by Effective Vaccines and Immunization Strategies in Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vivo Treatment with Antibodies
2.3. Bacterial Strains and Vaccines
2.4. Vaccination and Challenge Studies
2.5. Antibody Titers
2.6. Neutralization Antibody Analysis
2.7. Flow Cytometry Analysis
2.8. Statistical Analysis
3. Results
3.1. Vaccinated Animals Are Protected Against Pneumonic Plague
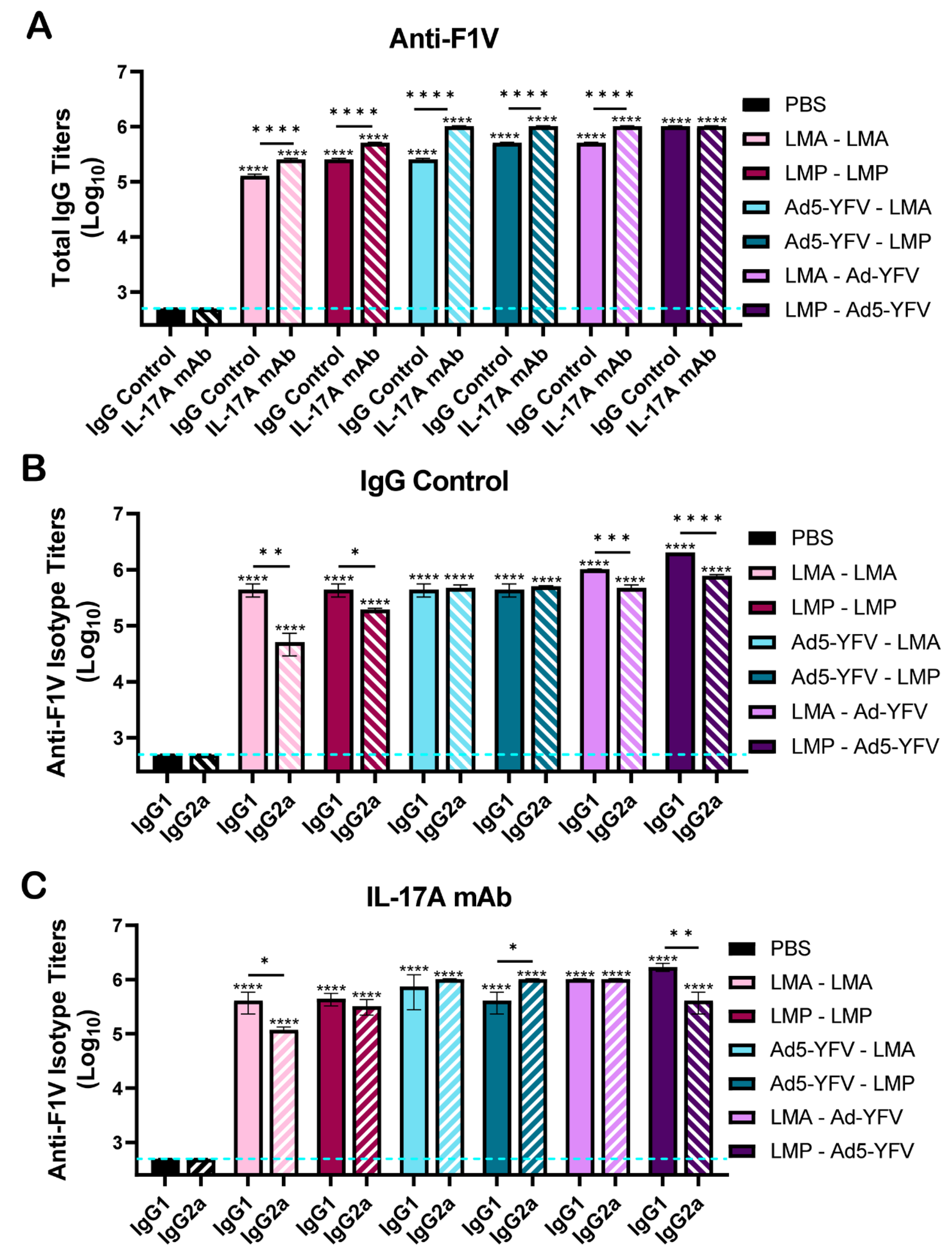
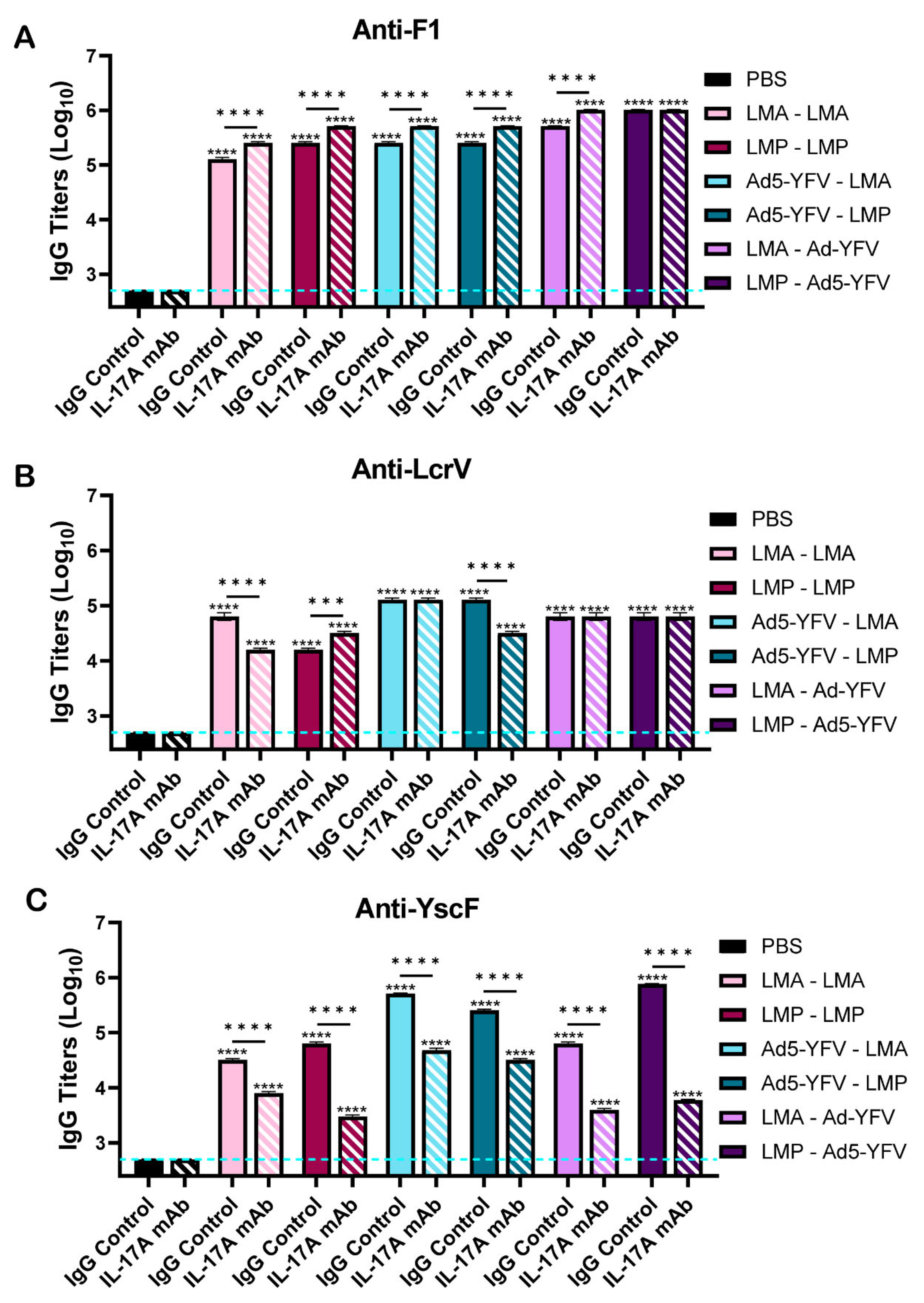
3.2. Vaccinated Mice Exhibit Increased Antibody and B Cell Responses
3.3. T Cell Responses Are Induced by Vaccination
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, R.D.; Fetherston, J.D. Yersinia pestis—Etiologic agent of plague. Clin. Microbiol. Rev. 1997, 10, 35–66. [Google Scholar] [CrossRef]
- Williamson, E.D.; Kilgore, P.B.; Hendrix, E.K.; Neil, B.H.; Sha, J.; Chopra, A.K. Progress on the research and development of plague vaccines with a call to action. NPJ Vaccines 2024, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Kumar, S. Yersinia pestis antibiotic resistance: A systematic review. Osong Public Health Res. Perspect. 2022, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, J.A.; Hendrix, E.K.; Chopra, A.K. Plague vaccines: New developments in an ongoing search. Appl. Microbiol. Biotechnol. 2021, 105, 4931–4941. [Google Scholar] [CrossRef]
- Feodorova, V.A.; Lyapina, A.M.; Khizhnyakova, M.A.; Zaitsev, S.S.; Sayapina, L.V.; Arseneva, T.E.; Trukhachev, A.L.; Lebedeva, S.A.; Telepnev, M.V.; Ulianova, O.V.; et al. Humoral and cellular immune responses to Yersinia pestis Pla antigen in humans immunized with live plague vaccine. PLoS Neglected Trop. Dis. 2018, 12, e0006511. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.K.; Biryukov, S.S.; Klimko, C.P.; Shoe, J.L.; Hunter, M.; Rosario-Acevedo, R.; Fetterer, D.P.; Moody, K.L.; Meyer, J.R.; Rill, N.O.; et al. Protection elicited by attenuated live Yersinia pestis vaccine strains against lethal infection with virulent Y. pestis. Vaccines 2021, 9, 161. [Google Scholar] [CrossRef]
- Tiner, B.L.; Sha, J.; Ponnusamy, D.; Baze, W.B.; Fitts, E.C.; Popov, V.L.; Van Lier, C.J.; Erova, T.E.; Chopra, A.K. Intramuscular immunization of mice with a live-Attenuated triple mutant of yersinia pestis co92 induces robust humoral and cell-mediated immunity to completely protect animals against pneumonic plague. Clin. Vaccine Immunol. 2015, 22, 1255–1268. [Google Scholar] [CrossRef]
- Tiner, B.L.; Sha, J.; Cong, Y.; Kirtley, M.L.; Andersson, J.A.; Chopra, A.K. Immunisation of two rodent species with new live-attenuated mutants of Yersinia pestis CO92 induces protective long-term humoral- and cell-mediated immunity against pneumonic plague. NPJ Vaccines 2016, 1, 16020. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, P.B.; Sha, J.; Andersson, J.A.; Motin, V.L.; Chopra, A.K. A new generation needle- and adjuvant-free trivalent plague vaccine utilizing adenovirus-5 nanoparticle platform. NPJ Vaccines 2021, 6, 21. [Google Scholar] [CrossRef]
- Sha, J.; Kirtley, M.L.; Klages, C.; Erova, T.E.; Telepnev, M.; Ponnusamy, D.; Fitts, E.C.; Baze, W.B.; Sivasubramani, S.K.; Lawrence, W.S.; et al. A Replication-Defective human type 5 adenovirus-based trivalent vaccine confers complete protection against plague in mice and nonhuman primates. Clin. Vaccine Immunol. 2016, 23, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Mahalingam, M.; Zhu, J.; Moayeri, M.; Sha, J.; Lawrence, W.S.; Leppla, S.H.; Chopra, A.K.; Rao, V.B. A Bacteriophage T4 Nanoparticle-Based Dual Vaccine against Anthrax and Plague. mBio 2018, 9, e01926-18. [Google Scholar] [CrossRef]
- Sanapala, S.; Rahav, H.; Patel, H.; Sun, W.; Curtiss, R. Multiple antigens of Yersinia pestis delivered by live recombinant attenuated Salmonella vaccine strains elicit protective immunity against plague. Vaccine 2016, 34, 2410–2416. [Google Scholar] [CrossRef] [PubMed]
- Morton, M.; Garmory, H.S.; Perkins, S.D.; O’Dowd, A.M.; Griffin, K.F.; Turner, A.K.; Bennett, A.M.; Titball, R.W. A Salmonella enterica serovar Typhi vaccine expressing Yersinia pestis F1 antigen on its surface provides protection against plague in mice. Vaccine 2004, 22, 2524–2532. [Google Scholar] [CrossRef]
- Biryukov, S.; Dankmeyer, J.L.; Shamsuddin, Z.; Velez, I.; Rill, N.O.; Rosario-Acevedo, R.; Klimko, C.P.; Shoe, J.L.; Hunter, M.; Ward, M.D.; et al. Impact of Toll-Like Receptor-Specific Agonists on the Host Immune Response to the Yersinia pestis Plague rF1V Vaccine. Front. Immunol. 2021, 12, 726416. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.G.; Anderson, G.W.; Matthew Mauroj, J.; Welkos, S.L.; Andrews, G.P.; Adamovicz, J.; Friedlander, A.M. Protection against experimental bubonic and pneumonic plague by a recombinant capsular Fl-V antigen fusion protein vaccine. Vaccine 1998, 16, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.T.; Eyles, J.E.; Baillie, L.W.; Keane-Myers, A.M. Immunogenicity and efficacy of an anthrax/plague DNA fusion vaccine in a mouse model. FEMS Immunol. Med. Microbiol. 2012, 65, 505–509. [Google Scholar] [CrossRef]
- Shattock, R.J.; Andrianaivoarimanana, V.; McKay, P.F.; Randriantseheno, L.N.; Murugaiah, V.; Samnuan, K.; Rogers, P.; Tregoning, J.S.; Rajerison, M.; Moore, K.M.; et al. A self-amplifying RNA vaccine provides protection in a murine model of bubonic plague. Front. Microbiol. 2023, 14, 1247041. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Levy, Y.; Elia, U.; Cohen, H.; Hazan-Halevy, I.; Aftalion, M.; Ezra, A.; Bar-Haim, E.; Naidu, G.S.; Diesendruck, Y.; et al. A single-dose F1-based mRNA-LNP vaccine provides protection against the lethal plague bacterium. Sci. Adv. 2023, 9, eadg1036. [Google Scholar] [CrossRef] [PubMed]
- Feodorova, V.A.; Motin, V.L. Plague vaccines: Current developments and future perspectives. Emerg. Microbes Infect. 2012, 1, 1–5. [Google Scholar] [CrossRef]
- Sun, W.; Singh, A.K. Plague vaccine: Recent progress and prospects. NPJ Vaccines 2019, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- DynPort Vaccine Company Llc, A.G.C. Safety, Tolerability & Immunogenicity of the Recombinant Plague Vaccine rF1V. 2006. Available online: https://clinicaltrials.gov/study/NCT00097396?term=Safety,%20Tolerability%20%26%20Immunogenicity%20of%20the%20Recombinant%20Plague%20Vaccine%20rF1V&rank=1 (accessed on 29 September 2023).
- DynPort Vaccine Company Llc, A.G.C. Recombinant Plague Vaccine rF1V in Healthy Volunteers. In ClinicalTrials.gov; 2008. Available online: https://clinicaltrials.gov/study/NCT00332956?term=Recombinant%20Plague%20Vaccine%20rF1V%20in%20Healthy%20Volunteers.%20&rank=1 (accessed on 29 September 2023).
- DynPort Vaccine Company Llc, A.G.C. Randomized Single-Blinded Study to Evaluate Safety and Immunogenicity of Recombinant Plague Vaccine With and Without Adjuvant. In ClinicalTrials.gov; 2012. Available online: https://clinicaltrials.gov/study/NCT01122784?term=Randomized%20Single-Blinded%20Study%20to%20Evaluate%20Safety%20and%20Immunogenicity%20of%20Recombinant%20Plague%20Vaccine%20With%20and%20Without%20Adjuvant.%20&rank=1 (accessed on 29 September 2023).
- PharmAthene, U.K.L. One Year Study to Evaluate Three Different Adjuvanted Doses of the Recombinant Plague Vaccine (rF1 and rV Antigens). In ClinicalTrials.gov; 2007. Available online: https://clinicaltrials.gov/study/NCT00246467?term=One%20Year%20Study%20to%20Evaluate%20Three%20Different%20Adjuvanted%20Doses%20of%20the%20Recombinant%20Plague%20Vaccine%20&rank=1 (accessed on 29 September 2023).
- Diseases, N.I.o.A.a.I. Dose Escalation Trial of a Plague Vaccine, Flagellin/F1/V, in Healthy Adult Volunteers. In ClinicalTrials.gov; 2014. Available online: https://clinicaltrials.gov/study/NCT01381744?term=.%20Dose%20Escalation%20Trial%20of%20a%20Plague%20Vaccine,%20Flagellin%2FF1%2FV,%20in%20Healthy%20Adult%20Volunteers.%20&rank=1 (accessed on 29 September 2023).
- Jiangsu Province Centers for Disease Control and, P. Immunogenicity and Safety of Subunit Plague Vaccine. In ClinicalTrials.gov; 2015. Available online: https://clinicaltrials.gov/study/NCT02596308?term=.%20Immunogenicity%20and%20Safety%20of%20Subunit%20Plague%20Vaccine&rank=1 (accessed on 29 September 2023).
- Jiangsu Province Centers for Disease Control and, P. Immunogenicity and Safety of Subunit Vaccine of Plague Vaccine With Two Immunization Regimens. In ClinicalTrials.gov; 2022. Available online: https://clinicaltrials.gov/study/NCT05330624?term=.%20Immunogenicity%20and%20Safety%20of%20Subunit%20Vaccine%20of%20Plague%20Vaccine%20With%20Two%20Immunization%20Regimens&rank=1 (accessed on 29 September 2023).
- OV Group. Investigating a Vaccine Against Plague (PlaVac); Centre for Clinical Vaccinology and Tropical Medicine (CCVTM): Oxford, UK, 2021; pp. 1–21. [Google Scholar]
- Dynavax Technologies, C. Trial of the Immunogenicity, Safety, and Tolerability of rF1V Vaccine With CpG 1018® Adjuvant Compared With rF1V Vaccine in Adults 18 to 55 Years of Age. In ClinicalTrials.gov; 2024. Available online: https://clinicaltrials.gov/study/NCT05506969?term=Trial%20of%20the%20Immunogenicity,%20Safety,%20and%20Tolerability%20of%20rF1V%20Vaccine%20With%20CpG%201018%20Adjuvant%20Compared%20With%20rF1V%20Vaccine&rank=1 (accessed on 29 September 2023).
- Sha, J.; Endsley, J.J.; Kirtley, M.L.; Foltz, S.M.; Huante, M.B.; Erova, T.E.; Kozlova, E.V.; Popov, V.L.; Yeager, L.A.; Zudina, I.V.; et al. Characterization of an F1 deletion mutant of Yersinia pestis CO92, pathogenic role of F1 antigen in bubonic and pneumonic plague, and evaluation of sensitivity and specificity of F1 antigen capture-based dipsticks. J. Clin. Microbiol. 2011, 49, 1708–1715. [Google Scholar] [CrossRef]
- Meka-Mechenko, T.V. F1-negative natural Y. pestis Strains. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1967. [Google Scholar]
- Tiner, B.L.; Sha, J.; Kirtley, M.L.; Erova, T.E.; Popov, V.L.; Baze, W.B.; van Lier, C.J.; Ponnusamy, D.; Andersson, J.A.; Motin, V.L.; et al. Combinational deletion of three membrane protein-encoding genes highly attenuates Yersinia pestis while retaining immunogenicity in a mouse model of pneumonic plague. Infect. Immun. 2015, 83, 1318–1338. [Google Scholar] [CrossRef]
- Sha, J.; Rosenzweig, J.A.; Kirtley, M.L.; van Lier, C.J.; Fitts, E.C.; Kozlova, E.V.; Erova, T.E.; Tiner, B.L.; Chopra, A.K. A non-invasive in vivo imaging system to study dissemination of bioluminescent Yersinia pestis CO92 in a mouse model of pneumonic plague. Microb. Pathog. 2013, 55, 39–50. [Google Scholar] [CrossRef]
- van Lier, C.J.; Tiner, B.L.; Chauhan, S.; Motin, V.L.; Fitts, E.C.; Huante, M.B.; Endsley, J.J.; Ponnusamy, D.; Sha, J.; Chopra, A.K. Further characterization of a highly attenuated Yersinia pestis CO92 mutant deleted for the genes encoding Braun lipoprotein and plasminogen activator protease in murine alveolar and primary human macrophages. Microb. Pathog. 2015, 80, 27–38. [Google Scholar] [CrossRef]
- Matson, J.S.; Durick, K.A.; Bradley, D.S.; Nilles, M.L. Immunization of mice with YscF provides protection from Yersinia pestis infections. BMC Microbiol. 2005, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Powell, B.S.; Goodin, J. Yersinia pestis Yop secretion protein F: Purification, characterization, and protective efficacy against bubonic plague. Protein Expr. Purif. 2005, 42, 166–172. [Google Scholar] [CrossRef]
- Lathem, W.W.; Price, P.A.; Miller, V.L.; Goldman, W.E. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 2007, 315, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Zhou, J.; Yang, H.; Wang, X.; Zhang, X.; Wang, Q.; Wu, X.; Han, Y.; Song, Y.; Tan, Y.; et al. IL-17A Produced by Neutrophils Protects against Pneumonic Plague through Orchestrating IFN-γ–Activated Macrophage Programming. J. Immunol. 2014, 192, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-S.; Kummer, L.W.; Szaba, F.M.; Smiley, S.T. IL-17 Contributes to Cell-Mediated Defense against Pulmonary Yersinia pestis Infection. J. Immunol. 2011, 186, 1675–1684. [Google Scholar] [CrossRef]
- Kilgore, P.B.; Sha, J.; Hendrix, E.K.; Motin, V.L.; Chopra, A.K. Combinatorial Viral Vector-Based and Live Attenuated Vaccines without an Adjuvant to Generate Broader Immune Responses to Effectively Combat Pneumonic Plague. mBio 2021, 12, e03223-21. [Google Scholar] [CrossRef] [PubMed]
- ThermoScientific. Instructions EZ-Link Sulfo-NHS-Biotin. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0016134_2161850_EZLinkSulfoNHS_Biotin_UG.pdf (accessed on 27 November 2024).
- Williamson, E.D.; Flick-Smith, H.C.; Waters, E.; Miller, J.; Hodgson, I.; Le Butt, C.S.; Hill, J. Immunogenicity of the rF1+rV vaccine for plague with identification of potential immune correlates. Microb. Pathog. 2007, 42, 11–21. [Google Scholar] [CrossRef]
- Sha, J.; Agar, S.L.; Baze, W.B.; Olano, J.P.; Fadl, A.A.; Erova, T.E.; Wang, S.; Foltz, S.M.; Suarez, G.; Motin, V.L.; et al. Braun lipoprotein (Lpp) contributes to virulence of yersiniae: Potential role of Lpp in inducing bubonic and pneumonic plague. Infect. Immun. 2008, 76, 1390–1409. [Google Scholar] [CrossRef] [PubMed]
- Andrianaivoarimanana, V.; Randriantseheno, L.N.; Moore, K.M.; Walker, N.J.; Lonsdale, S.G.; Kempster, S.; Almond, N.A.; Rajerison, M.; Williamson, E.D. Potential human immunotherapeutics for plague. Immunother. Adv. 2021, 1, ltab020. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, G.; Rampling, T.; Powlson, J.; Morter, R.; Wright, D.; Hill, A.V.S.; Ewer, K.J. Activation-induce markers detect vaccine-specific CD4+ T cell responses not measured by assays conventionally used in clinical trials. Vaccines 2018, 6, 50. [Google Scholar] [CrossRef]
- Gray, J.I.; Westerhof, L.M.; MacLeod, M.K.L. The roles of resident, central and effector memory CD4 T-cells in protective immunity following infection or vaccination. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, L.; Moore, B.B.; Zhang, S.; Xiao, P.; Decker, A.M.; Wang, H.L. IL-17: Balancing Protective Immunity and Pathogenesis. J. Immunol. Res. 2023, 2023, 3360310. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, L.; Li, R.; Huang, Y.; Wang, S. The IL-17 family in diseases: From bench to bedside. Signal Transduct. Target. Ther. 2023, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Li, N.; Gade, P.; Kalvakolanu, D.V.; Weibley, T.; Doble, B.; Woodgett, J.R.; Wood, T.D.; Gaffen, S.L. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci. Signal 2009, 2, ra8. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Compán, V.; Puig, L.; Vidal, S.; Ramírez, J.; Llamas-Velasco, M.; Fernández-Carballido, C.; Almodóvar, R.; Pinto, J.A.; Galíndez-Aguirregoikoa, E.; Zarco, P.; et al. The paradigm of IL-23-independent production of IL-17F and IL-17A and their role in chronic inflammatory diseases. Front. Immunol. 2023, 14, 1191782. [Google Scholar] [CrossRef]
- Lin, Y.; Slight, S.R.; Khader, S.A. Th17 cytokines and vaccine-induced immunity. Semin. Immunopathol. 2010, 32, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Gaffen, S.L.; Kolls, J.K. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009, 2, 403–411. [Google Scholar] [CrossRef]
- Khader, S.A.; Bell, G.K.; Pearl, J.E.; Fountain, J.J.; Rangel-Moreno, J.; Cilley, G.E.; Shen, F.; Eaton, S.M.; Gaffen, S.L.; Swain, S.L.; et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 2007, 8, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; D’Souza, S.; Adnet, P.Y.; Laali, R.; Jurion, F.; Palfliet, K.; Huygen, K. Priming but not boosting with plasmid DNA encoding mycolyl-transferase Ag85A from Mycobacterium tuberculosis increases the survival time of Mycobacterium bovis BCG vaccinated mice against low dose intravenous challenge with M. tuberculosis H37Rv. Vaccine 2006, 24, 3353–3364. [Google Scholar] [CrossRef]
- Vordermeier, H.M.; Villarreal-Ramos, B.; Cockle, P.J.; McAulay, M.; Rhodes, S.G.; Thacker, T.; Gilbert, S.C.; McShane, H.; Hill, A.V.; Xing, Z.; et al. Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect. Immun. 2009, 77, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Mitsdoerffer, M.; Lee, Y.; Jäger, A.; Kim, H.J.; Korn, T.; Kolls, J.K.; Cantor, H.; Bettelli, E.; Kuchroo, V.K. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 2010, 107, 14292–14297. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Østergaard, M.; Wei, J.C.; Sieper, J.; Järvinen, P.; Tam, L.S.; Salvarani, C.; Kim, T.H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: Results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; Van den Bosch, F.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet 2018, 392, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.P.; Mattapallil, M.J.; Raychaudhuri, K.; Bing, S.J.; Wu, S.; Zhong, Y.; Wang, W.; Chen, Z.; Silver, P.B.; Jittayasothorn, Y.; et al. The Cytokine IL-17A Limits Th17 Pathogenicity via a Negative Feedback Loop Driven by Autocrine Induction of IL-24. Immunity 2020, 53, 384–397.e385. [Google Scholar] [CrossRef] [PubMed]
- Novatchkova, M.; Leibbrandt, A.; Werzowa, J.; Neubüser, A.; Eisenhaber, F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem. Sci. 2003, 28, 226–229. [Google Scholar] [CrossRef]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Forthal, D.N. Functions of Antibodies. Microbiol. Spectr. 2014, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Campodónico, V.L.; Llosa, N.J.; Grout, M.; Döring, G.; Maira-Litrán, T.; Pier, G.B. Evaluation of flagella and flagellin of Pseudomonas aeruginosa as vaccines. Infect. Immun. 2010, 78, 746–755. [Google Scholar] [CrossRef]
- Bishop, A.L.; Schild, S.; Patimalla, B.; Klein, B.; Camilli, A. Mucosal immunization with Vibrio cholerae outer membrane vesicles provides maternal protection mediated by antilipopolysaccharide antibodies that inhibit bacterial motility. Infect. Immun. 2010, 78, 4402–4420. [Google Scholar] [CrossRef]
- Richards, A.F.; Baranova, D.E.; Pizzuto, M.S.; Jaconi, S.; Willsey, G.G.; Torres-Velez, F.J.; Doering, J.E.; Benigni, F.; Corti, D.; Mantis, N.J. Recombinant Human Secretory IgA Induces Salmonella Typhimurium Agglutination and Limits Bacterial Invasion into Gut-Associated Lymphoid Tissues. ACS Infect. Dis. 2021, 7, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zheng, B.; Lu, J.; Wu, H.; Zhang, Q.; Jiao, L.; Pan, H.; Zhou, J. Evaluation of human antibodies from vaccinated volunteers for protection against Yersinia pestis infection. Microbiol. Spectr. 2024, 12, e0105424. [Google Scholar] [CrossRef] [PubMed]
- Velappan, N.; Biryukov, S.S.; Rill, N.O.; Klimko, C.P.; Rosario-Acevedo, R.; Shoe, J.L.; Hunter, M.; Dankmeyer, J.L.; Fetterer, D.P.; Bedinger, D.; et al. Characterization of two affinity matured Anti-Yersinia pestis F1 human antibodies with medical countermeasure potential. PLoS ONE 2024, 19, e0305034. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Yahagi, A.; Hamada, S.; Begum, M.D.; Watanabe, H.; Kawakami, K.; Suda, T.; Sudo, K.; Nakae, S.; Iwakura, Y.; et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 2007, 178, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ritchea, S.; Logar, A.; Slight, S.; Messmer, M.; Rangel-Moreno, J.; Guglani, L.; Alcorn, J.F.; Strawbridge, H.; Park, S.M.; et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 2009, 31, 799–810. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mukasa, R.; Hatton, R.D.; Weaver, C.T. Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 2009, 21, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Cooney, L.A.; Towery, K.; Endres, J.; Fox, D.A. Sensitivity and resistance to regulation by IL-4 during Th17 maturation. J. Immunol. 2011, 187, 4440–4450. [Google Scholar] [CrossRef] [PubMed]
- DiPiazza, A.T.; Leist, S.R.; Abiona, O.M.; Moliva, J.I.; Werner, A.; Minai, M.; Nagata, B.M.; Bock, K.W.; Phung, E.; Schäfer, A.; et al. COVID-19 vaccine mRNA-1273 elicits a protective immune profile in mice that is not associated with vaccine-enhanced disease upon SARS-CoV-2 challenge. Immunity 2021, 54, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Monin, L.; Slight, S.; Uche, U.; Blanchard, E.; Fallert Junecko, B.A.; Ramos-Payan, R.; Stallings, C.L.; Reinhart, T.A.; Kolls, J.K.; et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 2014, 10, e1004099. [Google Scholar] [CrossRef] [PubMed]
- Borkner, L.; Curham, L.M.; Wilk, M.M.; Moran, B.; Mills, K.H.G. IL-17 mediates protective immunity against nasal infection with Bordetella pertussis by mobilizing neutrophils, especially Siglec-F. Mucosal Immunol. 2021, 14, 1183–1202. [Google Scholar] [CrossRef]
- Bowen, W.; Batra, L.; Pulsifer, A.R.; Yolcu, E.S.; Lawrenz, M.B.; Shirwan, H. Robust Th1 cellular and humoral responses generated by the Yersinia pestis rF1-V subunit vaccine formulated to contain an agonist of the CD137 pathway do not translate into increased protection against pneumonic plague. Vaccine 2019, 37, 5708–5716. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.L.; Biryukov, S.S.; Rill, N.O.; Klimko, C.P.; Hunter, M.; Dankmeyer, J.L.; Miller, J.A.; Shoe, J.L.; Mlynek, K.D.; Talyansky, Y.; et al. Sex differences in immune protection in mice conferred by heterologous vaccines for pneumonic plague. Front. Immunol. 2024, 15, 1397579. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, P.B.; Sha, J.; Hendrix, E.K.; Neil, B.H.; Lawrence, W.S.; Peel, J.E.; Hittle, L.; Woolston, J.; Sulakvelidze, A.; Schwartz, J.A.; et al. A bacteriophage cocktail targeting Yersinia pestis provides strong post-exposure protection in a rat pneumonic plague model. Microbiol. Spectr. 2024, 12, e0094224. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S.; Maggi, E.; Liotta, F.; Cosmi, L.; Annunziato, F. Properties and origin of human Th17 cells. Mol. Immunol. 2009, 47, 3–7. [Google Scholar] [CrossRef]
- Toy, D.; Kugler, D.; Wolfson, M.; Vanden Bos, T.; Gurgel, J.; Derry, J.; Tocker, J.; Peschon, J. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 2006, 177, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Romagnani, S. Human and murine Th17. Curr. Opin. HIV AIDS 2010, 5, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Deng, Z.; Cao, C.; Li, L. IL-17 polymorphisms and asthma risk: A meta-analysis of 11 single nucleotide polymorphisms. J. Asthma 2015, 52, 981–988. [Google Scholar] [CrossRef]
- Niu, Y.M.; Yuan, H.; Zhou, Y. Interleukin-17 gene polymorphisms contribute to cancer risk. Mediat. Inflamm. 2014, 2014, 128490. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrix, E.K.; Sha, J.; Kilgore, P.B.; Neil, B.H.; Verma, A.K.; Chopra, A.K. The Protective Effect of IL-17A in Pneumonic Plague Can Be Compensated by Effective Vaccines and Immunization Strategies in Mice. Vaccines 2024, 12, 1361. https://doi.org/10.3390/vaccines12121361
Hendrix EK, Sha J, Kilgore PB, Neil BH, Verma AK, Chopra AK. The Protective Effect of IL-17A in Pneumonic Plague Can Be Compensated by Effective Vaccines and Immunization Strategies in Mice. Vaccines. 2024; 12(12):1361. https://doi.org/10.3390/vaccines12121361
Chicago/Turabian StyleHendrix, Emily K., Jian Sha, Paul B. Kilgore, Blake H. Neil, Atul K. Verma, and Ashok K. Chopra. 2024. "The Protective Effect of IL-17A in Pneumonic Plague Can Be Compensated by Effective Vaccines and Immunization Strategies in Mice" Vaccines 12, no. 12: 1361. https://doi.org/10.3390/vaccines12121361
APA StyleHendrix, E. K., Sha, J., Kilgore, P. B., Neil, B. H., Verma, A. K., & Chopra, A. K. (2024). The Protective Effect of IL-17A in Pneumonic Plague Can Be Compensated by Effective Vaccines and Immunization Strategies in Mice. Vaccines, 12(12), 1361. https://doi.org/10.3390/vaccines12121361