
Rapid Development of Modified Vaccinia Virus Ankara (MVA)-Based Vaccine Candidates Against Marburg Virus Suitable for Clinical Use in Humans

 , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Construction of Plasmids pLW73-mCherry and pG06-GFP and Generation of Recombinant MVA Backbone Viruses
2.3. Construction of pIII-MARV-GP and pLW73-MARV-NP and Generation of Recombinant MVA Vaccine Candidates
2.4. In Vitro Characterization of Recombinant MVA Backbone Viruses and MVA Vaccine Candidates
2.5. Western Blot Analysis of Recombinant Proteins
2.6. Immunofluorescence Staining of Recombinant MARV-GP and MARV-NP
2.7. Ethics Statement
2.8. Vaccination Experiments in Mice
2.9. Generation of Peptides, Design of Peptide Pools, and Peptide Prediction
2.10. T Cell Analysis by Enzyme-Linked Immunospot (ELISPOT)
2.11. T Cell Analysis by Intracellular Cytokine Staining (ICS)
2.12. Antigen-Specific IgG Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Statistical Analysis
3. Results
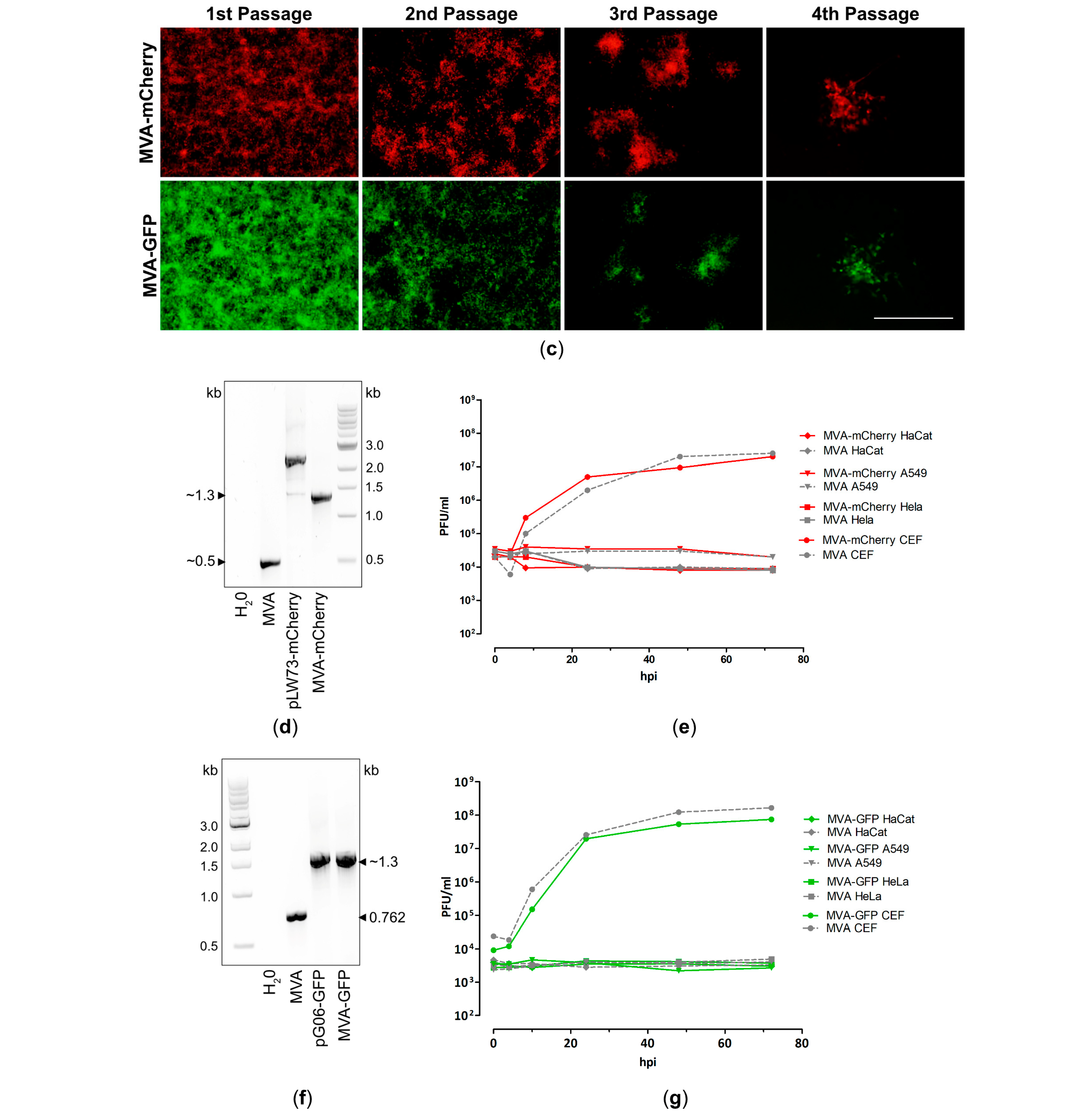
3.1. Generation and In Vitro Characterization of Single Recombinant MVA Backbone Viruses Expressing mCherry or GFP
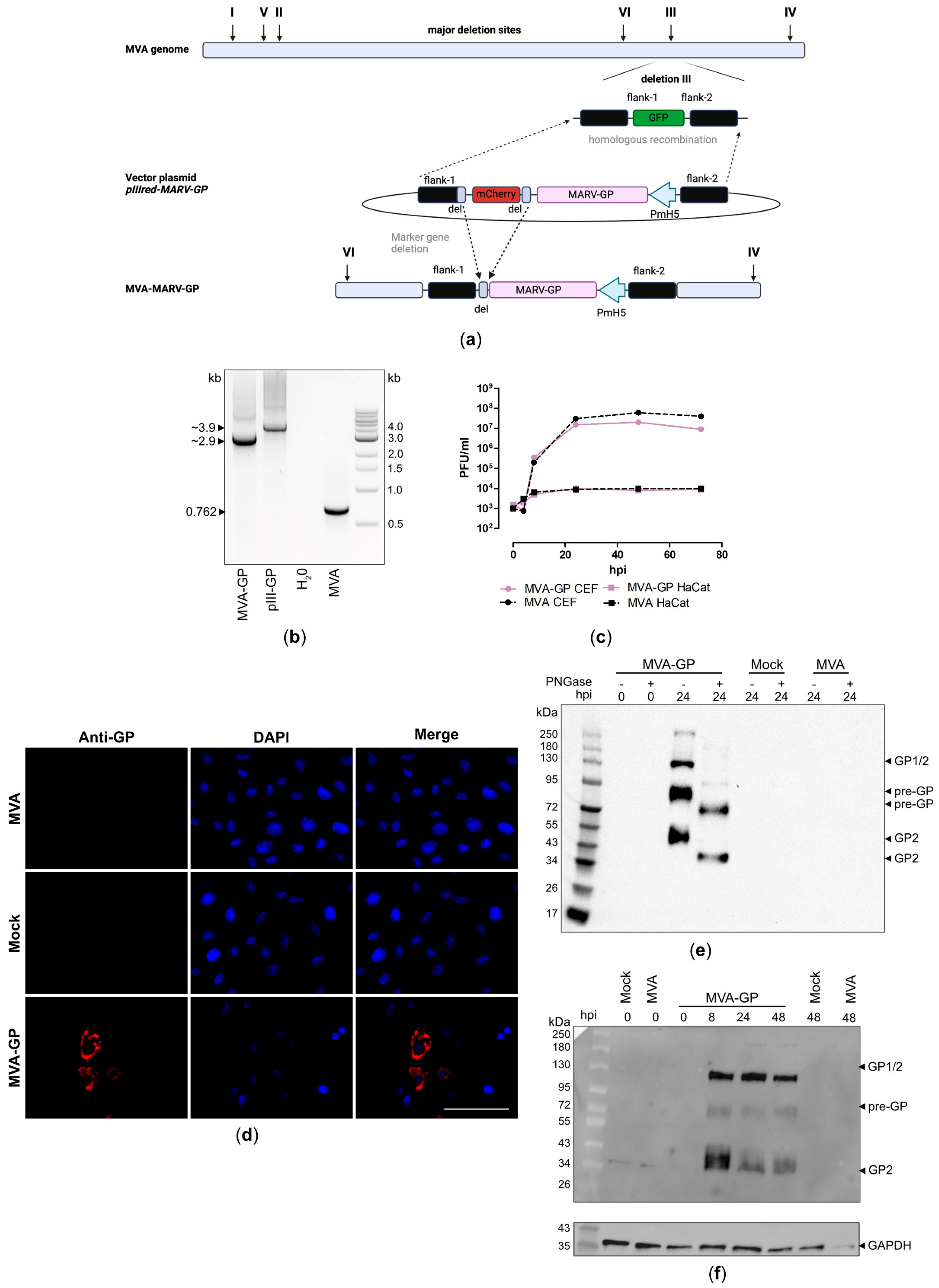
3.2. Design and Generation of a Recombinant MVA-MARV-GP Candidate Vaccine
3.3. Design and Generation of Recombinant MVA-MARV-NP Candidate Vaccine
3.4. Evaluation of MARV-Specific Immune Responses in C57BL/6J Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eneh, S.C.; Okonji, O.C.; Chiburoma, A.G.; Francisca Ogochukwu, O.; Tuwleh, L.; Gideon, I.; Okonji, E.F.; Bushabu, F.N.; Mgbere, O. Marburg virus disease amid COVID-19 in West Africa: An emerging and re-emerging zoonotic epidemic threat, future implications and way forward. Ther. Adv. Infect. Dis. 2023, 10, 20499361231168520. [Google Scholar] [CrossRef] [PubMed]
- Mane Manohar, M.P.; Lee, V.J.; Chinedum Odunukwe, E.U.; Singh, P.K.; Mpofu, B.S.; Oxley, M.C. Advancements in Marburg (MARV) Virus Vaccine Research With Its Recent Reemergence in Equatorial Guinea and Tanzania: A Scoping Review. Cureus 2023, 15, e42014. [Google Scholar] [CrossRef] [PubMed]
- Okonji, O.C.; Okonji, E.F.; Mohanan, P.; Babar, M.S.; Saleem, A.; Khawaja, U.A.; Essar, M.Y.; Hasan, M.M. Marburg virus disease outbreak amidst COVID-19 in the Republic of Guinea: A point of contention for the fragile health system? Clin. Epidemiol. Glob. Health 2021, 13, 100920. [Google Scholar] [CrossRef] [PubMed]
- Kissling, R.E.; Murphy, F.A.; Henderson, B.E. Marburg virus. Ann. N. Y. Acad. Sci. 1970, 174, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Crozier, I.; Kuhn, J.H. A Forgotten Episode of Marburg Virus Disease: Belgrade, Yugoslavia, 1967. Microbiol. Mol. Biol. Rev. 2020, 84, 10–1128. [Google Scholar] [CrossRef]
- Languon, S.; Quaye, O. Filovirus Disease Outbreaks: A Chronological Overview. Virology 2019, 10, 1178122X19849927. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news-room/fact-sheets/detail/marburg-virus-disease (accessed on 13 August 2024).
- ProMed International Society for Infectious Diseases. MARBURG VIRUS DISEASE—RWANDA (07): WHO UPDATE. Available online: https://promedmail.org/promed-post/?id=20241012.8719323 (accessed on 20 October 2024).
- Marzi, A.; Feldmann, H. Filovirus vaccines as a response paradigm for emerging infectious diseases. NPJ Vaccines 2024, 9, 186. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adachi, T.; Adhikari, N.K.J.; Arribas, J.R.; Bah, I.E.; Bausch, D.G.; Bhadelia, N.; Borchert, M.; Brantsæter, A.B.; Brett-Major, D.M.; et al. New filovirus disease classification and nomenclature. Nat. Rev. Microbiol. 2019, 17, 261–263. [Google Scholar] [CrossRef]
- Biedenkopf, N.; Bukreyev, A.; Chandran, K.; Di Paola, N.; Formenty, P.B.H.; Griffiths, A.; Hume, A.J.; Mühlberger, E.; Netesov, S.V.; Palacios, G.; et al. Renaming of genera Ebolavirus and Marburgvirus to Orthoebolavirus and Orthomarburgvirus, respectively, and introduction of binomial species names within family Filoviridae. Arch. Virol. 2023, 168, 220. [Google Scholar] [CrossRef]
- Feldmann, H.; Mühlberger, E.; Randolf, A.; Will, C.; Kiley, M.P.; Sanchez, A.; Klenk, H.D. Marburg virus, a filovirus: Messenger RNAs, gene order, and regulatory elements of the replication cycle. Virus Res. 1992, 24, 1–19. [Google Scholar] [CrossRef]
- Abir, M.H.; Rahman, T.; Das, A.; Etu, S.N.; Nafiz, I.H.; Rakib, A.; Mitra, S.; Emran, T.B.; Dhama, K.; Islam, A.; et al. Pathogenicity and virulence of Marburg virus. Virulence 2022, 13, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Brauburger, K.; Hume, A.J.; Mühlberger, E.; Olejnik, J. Forty-Five Years of Marburg Virus Research. Viruses 2012, 4, 1878–1927. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Fusco, M.L.; Bornholdt, Z.A.; Lee, J.E.; Flyak, A.I.; Matsuoka, R.; Kohda, D.; Yanagi, Y.; Hammel, M.; Crowe, J.E.; et al. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 2015, 160, 904–912. [Google Scholar] [CrossRef]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 1996, 93, 3602–3607. [Google Scholar] [CrossRef]
- Mavrakis, M.; Kolesnikova, L.; Schoehn, G.; Becker, S.; Ruigrok, R.W.H. Morphology of Marburg virus NP-RNA. Virology 2002, 296, 300–307. [Google Scholar] [CrossRef]
- Ruigrok, R.W.H.; Crépin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef]
- Teo, S.P. Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract. 2022, 35, 947–951. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of mRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2016, 97, 187–243. [Google Scholar] [CrossRef] [PubMed]
- Pincus, S.; Tartaglia, J.; Paoletti, E. Poxvirus-based vectors as vaccine candidates. Biologicals 1995, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Pérez, P.; Marcos-Villar, L.; Albericio, G.; Astorgano, D.; Álvarez, E.; Sin, L.; Gómez, C.E.; García-Arriaza, J.; Esteban, M. Highly Attenuated Poxvirus-Based Vaccines Against Emerging Viral Diseases. J. Mol. Biol. 2023, 435, 168173. [Google Scholar] [CrossRef] [PubMed]
- Orlova, O.V.; Glazkova, D.V.; Bogoslovskaya, E.V.; Shipulin, G.A.; Yudin, S.M. Development of Modified Vaccinia Virus Ankara-Based Vaccines: Advantages and Applications. Vaccines 2022, 10, 1516. [Google Scholar] [CrossRef]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef]
- Mayr, A.; Stickl, H.; Müller, H.K.; Danner, K.; Singer, H. Der Pockenimpfstamm MVA: Marker, genetische Struktur, Erfahrungen mit der parenteralen Schutzimpfung und Verhalten im abwehrgeschwächten Organismus. Zentralbl. Bakteriol. B 1978, 167, 375–390. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef]
- Tapia, F.; Jordan, I.; Genzel, Y.; Reichl, U. Efficient and stable production of Modified Vaccinia Ankara virus in two-stage semi-continuous and in continuous stirred tank cultivation systems. PLoS ONE 2017, 12, e0182553. [Google Scholar] [CrossRef]
- Jordan, I.; Lohr, V.; Genzel, Y.; Reichl, U.; Sandig, V. Elements in the Development of a Production Process for Modified Vaccinia Virus Ankara. Microorganisms 2013, 1, 100–121. [Google Scholar] [CrossRef]
- Su, J.; Brunner, L.; Ates Oz, E.; Sacherl, J.; Frank, G.; Kerth, H.A.; Thiele, F.; Wiegand, M.; Mogler, C.; Aguilar, J.C.; et al. Activation of CD4 T cells during prime immunization determines the success of a therapeutic hepatitis B vaccine in HBV-carrier mouse models. J. Hepatol. 2023, 78, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Kupke, A.; Volz, A.; Dietzel, E.; Freudenstein, A.; Schmidt, J.; Shams-Eldin, H.; Jany, S.; Sauerhering, L.; Krähling, V.; Gellhorn Serra, M.; et al. Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein. Vaccines 2022, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Meyer Zu Natrup, C.; Tscherne, A.; Dahlke, C.; Ciurkiewicz, M.; Shin, D.-L.; Fathi, A.; Rohde, C.; Kalodimou, G.; Halwe, S.; Limpinsel, L.; et al. Stabilized recombinant SARS-CoV-2 spike antigen enhances vaccine immunogenicity and protective capacity. J. Clin. Investig. 2022, 132, e159895. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, A.; Schwarz, J.H.; Rohde, C.; Kupke, A.; Kalodimou, G.; Limpinsel, L.; Okba, N.M.A.; Bošnjak, B.; Sandrock, I.; Odak, I.; et al. Immunogenicity and efficacy of the COVID-19 candidate vector vaccine MVA-SARS-2-S in preclinical vaccination. Proc. Natl. Acad. Sci. USA 2021, 118, e2026207118. [Google Scholar] [CrossRef] [PubMed]
- Mooij, P.; García-Arriaza, J.; Pérez, P.; Lázaro-Frías, A.; Verstrepen, B.E.; Böszörményi, K.P.; Mortier, D.; Fagrouch, Z.; Kiemenyi-Kayere, G.; Niphuis, H.; et al. Poxvirus MVA Expressing SARS-CoV-2 S Protein Induces Robust Immunity and Protects Rhesus Macaques From SARS-CoV-2. Front. Immunol. 2022, 13, 845887. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Pérez, P.; Astorgano, D.; Albericio, G.; Kerstens, W.; Thibaut, H.J.; Coelmont, L.; Weynand, B.; Labiod, N.; Delgado, R.; et al. Optimized vaccine candidate MVA-S(3P) fully protects against SARS-CoV-2 infection in hamsters. Front. Immunol. 2023, 14, 1163159. [Google Scholar] [CrossRef]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.S.; et al. A T Cell-Inducing Influenza Vaccine for the Elderly: Safety and Immunogenicity of MVA-NP+M1 in Adults Aged over 50 Years. PLoS ONE 2012, 7, e48322. [Google Scholar] [CrossRef]
- Choi, E.M.-L.; Lacarra, B.; Afolabi, M.O.; Ale, B.M.; Baiden, F.; Bétard, C.; Foster, J.; Hamzé, B.; Schwimmer, C.; Manno, D.; et al. Safety and immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in infants: A phase 2, randomised, double-blind, active-controlled trial in Guinea and Sierra Leone. Lancet Glob. Health 2023, 11, e1743–e1752. [Google Scholar] [CrossRef]
- Happe, M.; Hofstetter, A.R.; Wang, J.; Yamshchikov, G.V.; Holman, L.A.; Novik, L.; Strom, L.; Kiweewa, F.; Wakabi, S.; Millard, M.; et al. Heterologous cAd3-Ebola and MVA-EbolaZ vaccines are safe and immunogenic in US and Uganda phase 1/1b trials. NPJ Vaccines 2024, 9, 67. [Google Scholar] [CrossRef]
- Bockstal, V.; Shukarev, G.; McLean, C.; Goldstein, N.; Bart, S.; Gaddah, A.; Anumenden, D.; Stoop, J.N.; Marit de Groot, A.; Pau, M.G.; et al. First-in-human study to evaluate safety, tolerability, and immunogenicity of heterologous regimens using the multivalent filovirus vaccines Ad26.Filo and MVA-BN-Filo administered in different sequences and schedules: A randomized, controlled study. PLoS ONE 2022, 17, e0274906. [Google Scholar] [CrossRef]
- Wussow, F.; Kha, M.; Kim, T.; Ly, M.; Yll-Pico, M.; Kar, S.; Lewis, M.G.; Chiuppesi, F.; Diamond, D.J. Synthetic multiantigen MVA vaccine COH04S1 and variant-specific derivatives protect Syrian hamsters from SARS-CoV-2 Omicron subvariants. NPJ Vaccines 2023, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Americo, J.L.; Cotter, C.A.; Earl, P.L.; Liu, R.; Moss, B. Intranasal inoculation of an MVA-based vaccine induces IgA and protects the respiratory tract of hACE2 mice from SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2202069119. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Americo, J.L.; Cotter, C.A.; Earl, P.L.; Erez, N.; Peng, C.; Moss, B. One or two injections of MVA-vectored vaccine shields hACE2 transgenic mice from SARS-CoV-2 upper and lower respiratory tract infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2026785118. [Google Scholar] [CrossRef] [PubMed]
- Chiuppesi, F.; Nguyen, V.H.; Park, Y.; Contreras, H.; Karpinski, V.; Faircloth, K.; Nguyen, J.; Kha, M.; Johnson, D.; Martinez, J.; et al. Synthetic multiantigen MVA vaccine COH04S1 protects against SARS-CoV-2 in Syrian hamsters and non-human primates. NPJ Vaccines 2022, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.M.; Marín-López, A.; Chiem, K.; Jimenez-Cabello, L.; Ullah, I.; Utrilla-Trigo, S.; Calvo-Pinilla, E.; Lorenzo, G.; Moreno, S.; Ye, C.; et al. Vaccinia Virus Strain MVA Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Glycoprotein Induces Robust Protection and Prevents Brain Infection in Mouse and Hamster Models. Vaccines 2023, 11, 1006. [Google Scholar] [CrossRef]
- Pérez, P.; Albericio, G.; Astorgano, D.; Flores, S.; Sánchez-Corzo, C.; Sánchez-Cordón, P.J.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; et al. Preclinical immune efficacy against SARS-CoV-2 beta B.1.351 variant by MVA-based vaccine candidates. Front. Immunol. 2023, 14, 1264323. [Google Scholar] [CrossRef]
- Kremer, M.; Volz, A.; Kreijtz, J.H.C.M.; Fux, R.; Lehmann, M.H.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. 2012, 890, 59–92. [Google Scholar] [CrossRef]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Agresti, A.; Comunian, C.; Milani, A.; Capua, I.; Erfle, V.; Siccardi, A.G. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J. Virol. Methods 2010, 163, 195–204. [Google Scholar] [CrossRef]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Erfle, V.; Staib, C.; Sutter, G.; Siccardi, A.G. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. J. Virol. Methods 2009, 156, 37–43. [Google Scholar] [CrossRef]
- Barbieri, A.; Panigada, M.; Soprana, E.; Di Mario, G.; Gubinelli, F.; Bernasconi, V.; Recagni, M.; Donatelli, I.; Castrucci, M.R.; Siccardi, A.G. Strategies to obtain multiple recombinant modified vaccinia Ankara vectors. Applications to influenza vaccines. J. Virol. Methods 2018, 251, 7–14. [Google Scholar] [CrossRef]
- Carroll, M.W.; Moss, B.E. coli beta-glucuronidase (GUS) as a marker for recombinant vaccinia viruses. Biotechniques 1995, 19, 352–354, 356. [Google Scholar] [PubMed]
- Chakrabarti, S.; Brechling, K.; Moss, B. Vaccinia virus expression vector: Coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol. Cell. Biol. 1985, 5, 3403–3409. [Google Scholar] [CrossRef] [PubMed]
- Falkner, F.G.; Moss, B. Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J. Virol. 1988, 62, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Mackett, M.; Smith, G.L.; Moss, B. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J. Virol. 1984, 49, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Moss, B.; Wyatt, L.S. Generation of Recombinant Vaccinia Viruses. Curr. Protoc. Protein Sci. 2017, 89, 5.13.1–5.13.18. [Google Scholar] [CrossRef]
- Sánchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. Isolation of recombinant MVA using F13L selection. Methods Mol. Biol. 2012, 890, 93–111. [Google Scholar] [CrossRef]
- Tscharke, D.C.; Smith, G.L. Notes on transient host range selection for engineering vaccinia virus strain MVA. Biotechniques 2002, 33, 186–188. [Google Scholar] [CrossRef]
- Antoshkina, I.V.; Glazkova, D.V.; Urusov, F.A.; Bogoslovskaya, E.V.; Shipulin, G.A. Comparison of Recombinant MVA Selection Methods Based on F13L, D4R and K1L Genes. Viruses 2022, 14, 528. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Shors, S.T.; Murphy, B.R.; Moss, B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996, 14, 1451–1458. [Google Scholar] [CrossRef]
- von Creytz, I.; Gerresheim, G.K.; Lier, C.; Schneider, J.; Schauflinger, M.; Benz, M.; Kämper, L.; Rohde, C.; Eickmann, M.; Biedenkopf, N. Rescue and characterization of the first West African Marburg virus 2021 from Guinea. Heliyon 2023, 9, e19613. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Aksular, M.; Calvo-Pinilla, E.; Marín-López, A.; Ortego, J.; Chambers, A.C.; King, L.A.; Castillo-Olivares, J. A single dose of African horse sickness virus (AHSV) VP2 based vaccines provides complete clinical protection in a mouse model. Vaccine 2018, 36, 7003–7010. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; Lázaro-Frías, A.; Zamora, C.; Sánchez-Cordón, P.J.; Astorgano, D.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; García-Arriaza, J. A Single Dose of an MVA Vaccine Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Protein Neutralizes Variants of Concern and Protects Mice From a Lethal SARS-CoV-2 Infection. Front. Immunol. 2022, 12, 824728. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, N.K.; Aljamaan, F.; Aljami, H.A.; Alenazi, M.W.; Albalawi, H.; Almasoud, A.; Alharthi, F.J.; Azhar, E.I.; Barhoumi, T.; Bosaeed, M.; et al. Immunogenicity of High-Dose MVA-Based MERS Vaccine Candidate in Mice and Camels. Vaccines 2022, 10, 1330. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Reche, P.A.; Glutting, J.-P.; Reinherz, E.L. Prediction of MHC class I binding peptides using profile motifs. Hum. Immunol. 2002, 63, 701–709. [Google Scholar] [CrossRef]
- Malm, M.; Tamminen, K.; Vesikari, T.; Blazevic, V. Norovirus-Specific Memory T Cell Responses in Adult Human Donors. Front. Microbiol. 2016, 7, 1570. [Google Scholar] [CrossRef]
- Fiore-Gartland, A.; Manso, B.A.; Friedrich, D.P.; Gabriel, E.E.; Finak, G.; Moodie, Z.; Hertz, T.; de Rosa, S.C.; Frahm, N.; Gilbert, P.B.; et al. Pooled-Peptide Epitope Mapping Strategies Are Efficient and Highly Sensitive: An Evaluation of Methods for Identifying Human T Cell Epitope Specificities in Large-Scale HIV Vaccine Efficacy Trials. PLoS ONE 2016, 11, e0147812. [Google Scholar] [CrossRef]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72 Pt 5, 1031–1038. [Google Scholar] [CrossRef]
- Volchkov, V.; Klenk, H.D. Proteolytic Processing of Filovirus Glycoproteins. In Activation of Viruses by Host Proteases; Springer: Berlin/Heidelberg, Germany, 2018; pp. 99–108. [Google Scholar] [CrossRef]
- Feldmann, H.; Volchkov, V.E.; Volchkova, V.A.; Ströher, U.; Klenk, H.-D. Biosynthesis and role of filoviral glycoproteins. J. Gen. Virol. 2001, 82, 2839–2848. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent Modifications of the Ebola Virus Glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.L.; King, T.H.; Alfson, K.J.; Albanese, K.A.; Smith, J.N.P.; Smock, P.; Jakubik, J.; Goez-Gazi, Y.; Gazi, M.; Dutton, J.W.; et al. Single-Shot ChAd3-MARV Vaccine in Modified Formulation Buffer Shows 100% Protection of NHPs. Vaccines 2022, 10, 1935. [Google Scholar] [CrossRef] [PubMed]
- Hunegnaw, R.; Honko, A.N.; Wang, L.; Carr, D.; Murray, T.; Shi, W.; Nguyen, L.; Storm, N.; Dulan, C.N.M.; Foulds, K.E.; et al. A single-shot ChAd3-MARV vaccine confers rapid and durable protection against Marburg virus in nonhuman primates. Sci. Transl. Med. 2022, 14, eabq6364. [Google Scholar] [CrossRef] [PubMed]
- ProMed International Society for Infectious Diseases. MARBURG VIRUS DISEASE—RWANDA (04): UPDATE, VACCINATION TRIAL. Available online: https://promedmail.org/promed-post/?id=20241008.8719234 (accessed on 16 October 2024).
- Suschak, J.J.; Schmaljohn, C.S. Vaccines against Ebola virus and Marburg virus: Recent advances and promising candidates. Hum. Vaccin. Immunother. 2019, 15, 2359–2377. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Lim, S.; Kaserer, M.; Lülf, A.; Marr, L.; Jany, S.; Deeg, C.A.; Pijlman, G.P.; Koraka, P.; Osterhaus, A.D.M.E.; et al. Immunogenicity and protective efficacy of recombinant Modified Vaccinia virus Ankara candidate vaccines delivering West Nile virus envelope antigens. Vaccine 2016, 34, 1915–1926. [Google Scholar] [CrossRef]
- Koch, T.; Dahlke, C.; Fathi, A.; Kupke, A.; Krähling, V.; Okba, N.M.A.; Halwe, S.; Rohde, C.; Eickmann, M.; Volz, A.; et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: An open-label, phase 1 trial. Lancet Infect. Dis. 2020, 20, 827–838. [Google Scholar] [CrossRef]
- Mittler, E.; Kolesnikova, L.; Strecker, T.; Garten, W.; Becker, S. Role of the Transmembrane Domain of Marburg Virus Surface Protein GP in Assembly of the Viral Envelope. J. Virol. 2007, 81, 3942–3948. [Google Scholar] [CrossRef]
- Sänger, C.; Mühlberger, E.; Lötfering, B.; Klenk, H.-D.; Becker, S. The Marburg virus surface protein GP is phosphorylated at its ectodomain. Virology 2002, 295, 20–29. [Google Scholar] [CrossRef]
- Meyer, M.; Gunn, B.M.; Malherbe, D.C.; Gangavarapu, K.; Yoshida, A.; Pietzsch, C.; Kuzmina, N.A.; Saphire, E.O.; Collins, P.L.; Crowe, J.E.; et al. Ebola vaccine–induced protection in nonhuman primates correlates with antibody specificity and Fc-mediated effects. Sci. Transl. Med. 2021, 13, eabg6128. [Google Scholar] [CrossRef]
- Cross, R.W.; Mire, C.E.; Feldmann, H.; Geisbert, T.W. Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discov. 2018, 17, 413–434. [Google Scholar] [CrossRef]
- Stonier, S.W.; Herbert, A.S.; Kuehne, A.I.; Sobarzo, A.; Habibulin, P.; Dahan, C.V.A.; James, R.M.; Egesa, M.; Cose, S.; Lutwama, J.J.; et al. Marburg virus survivor immune responses are Th1 skewed with limited neutralizing antibody responses. J. Exp. Med. 2017, 214, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.F.; Rubin, S.; Krause, P.R. Approaches to demonstrating the effectiveness of filovirus vaccines: Lessons from Ebola and COVID-19. Front. Immunol. 2023, 14, 1109486. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, D.C.; Domi, A.; Hauser, M.J.; Meyer, M.; Gunn, B.M.; Alter, G.; Bukreyev, A.; Guirakhoo, F. Modified vaccinia Ankara vaccine expressing Marburg virus-like particles protects guinea pigs from lethal Marburg virus infection. NPJ Vaccines 2020, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.S.; Mohamadzadeh, M. Status and challenges of filovirus vaccines. Vaccine 2007, 25, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Changula, K.; Yoshida, R.; Noyori, O.; Marzi, A.; Miyamoto, H.; Ishijima, M.; Yokoyama, A.; Kajihara, M.; Feldmann, H.; Mweene, A.S.; et al. Mapping of conserved and species-specific antibody epitopes on the Ebola virus nucleoprotein. Virus Res. 2013, 176, 83–90. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Y.; Dong, C.; Gonzalez, G.X.; Song, Y.; Zhu, W.; Kim, J.; Wei, L.; Wang, B.-Z. Influenza NP core and HA or M2e shell double-layered protein nanoparticles induce broad protection against divergent influenza A viruses. Nanomedicine 2021, 40, 102479. [Google Scholar] [CrossRef]
- Sayedahmed, E.E.; Elshafie, N.O.; dos Santos, A.P.; Jagannath, C.; Sambhara, S.; Mittal, S.K. Development of NP-Based Universal Vaccine for Influenza A Viruses. Vaccines 2024, 12, 157. [Google Scholar] [CrossRef]
- He, B.; Hu, T.; Yan, X.; Pa, Y.; Liu, Y.; Liu, Y.; Li, N.; Yu, J.; Zhang, H.; Liu, Y.; et al. Isolation, characterization, and circulation sphere of a filovirus in fruit bats. Proc. Natl. Acad. Sci. USA 2024, 121, e2313789121. [Google Scholar] [CrossRef]
- Natesan, M.; Jensen, S.M.; Keasey, S.L.; Kamata, T.; Kuehne, A.I.; Stonier, S.W.; Lutwama, J.J.; Lobel, L.; Dye, J.M.; Ulrich, R.G. Human Survivors of Disease Outbreaks Caused by Ebola or Marburg Virus Exhibit Cross-Reactive and Long-Lived Antibody Responses. Clin. Vaccine Immunol. 2016, 23, 717–724. [Google Scholar] [CrossRef]
- Backes, S.; Sperling, K.M.; Zwilling, J.; Gasteiger, G.; Ludwig, H.; Kremmer, E.; Schwantes, A.; Staib, C.; Sutter, G. Viral host-range factor C7 or K1 is essential for modified vaccinia virus Ankara late gene expression in human and murine cells, irrespective of their capacity to inhibit protein kinase R-mediated phosphorylation of eukaryotic translation initiation factor 2alpha. J. Gen. Virol. 2010, 91, 470–482. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tscherne, A.; Kalodimou, G.; Kupke, A.; Rohde, C.; Freudenstein, A.; Jany, S.; Kumar, S.; Sutter, G.; Krähling, V.; Becker, S.; et al. Rapid Development of Modified Vaccinia Virus Ankara (MVA)-Based Vaccine Candidates Against Marburg Virus Suitable for Clinical Use in Humans. Vaccines 2024, 12, 1316. https://doi.org/10.3390/vaccines12121316
Tscherne A, Kalodimou G, Kupke A, Rohde C, Freudenstein A, Jany S, Kumar S, Sutter G, Krähling V, Becker S, et al. Rapid Development of Modified Vaccinia Virus Ankara (MVA)-Based Vaccine Candidates Against Marburg Virus Suitable for Clinical Use in Humans. Vaccines. 2024; 12(12):1316. https://doi.org/10.3390/vaccines12121316
Chicago/Turabian StyleTscherne, Alina, Georgia Kalodimou, Alexandra Kupke, Cornelius Rohde, Astrid Freudenstein, Sylvia Jany, Satendra Kumar, Gerd Sutter, Verena Krähling, Stephan Becker, and et al. 2024. "Rapid Development of Modified Vaccinia Virus Ankara (MVA)-Based Vaccine Candidates Against Marburg Virus Suitable for Clinical Use in Humans" Vaccines 12, no. 12: 1316. https://doi.org/10.3390/vaccines12121316
APA StyleTscherne, A., Kalodimou, G., Kupke, A., Rohde, C., Freudenstein, A., Jany, S., Kumar, S., Sutter, G., Krähling, V., Becker, S., & Volz, A. (2024). Rapid Development of Modified Vaccinia Virus Ankara (MVA)-Based Vaccine Candidates Against Marburg Virus Suitable for Clinical Use in Humans. Vaccines, 12(12), 1316. https://doi.org/10.3390/vaccines12121316