
Endemic Human Coronavirus-Specific Nasal Immunoglobulin A and Serum Immunoglobulin G Dynamics in Lower Respiratory Tract Infections
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. The GRACE Study
2.2. Threshold for Serum IgG Responder/Non-Responder Categorization
2.3. Study Participants
2.4. Multiplex Anti-HCoV IgG and IgA Assay
2.5. Data Analysis
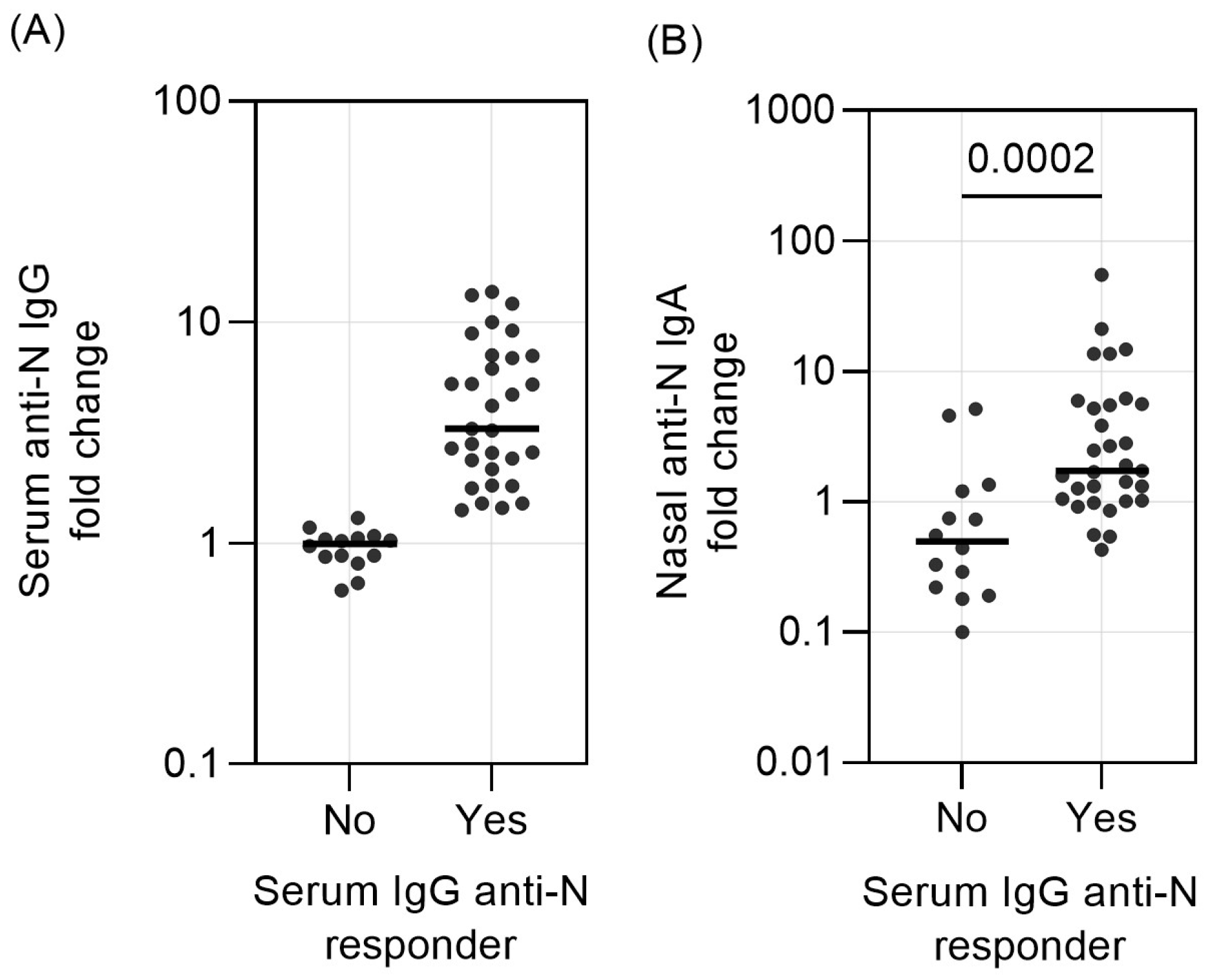
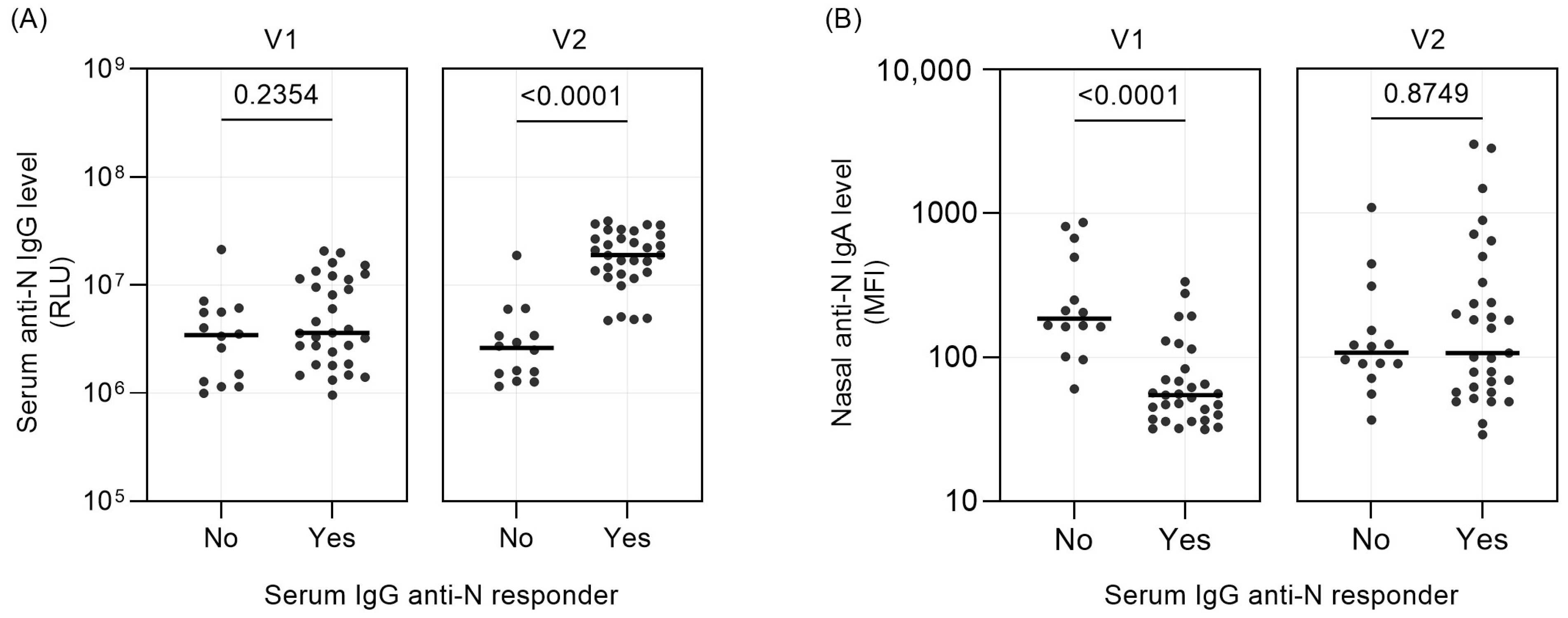
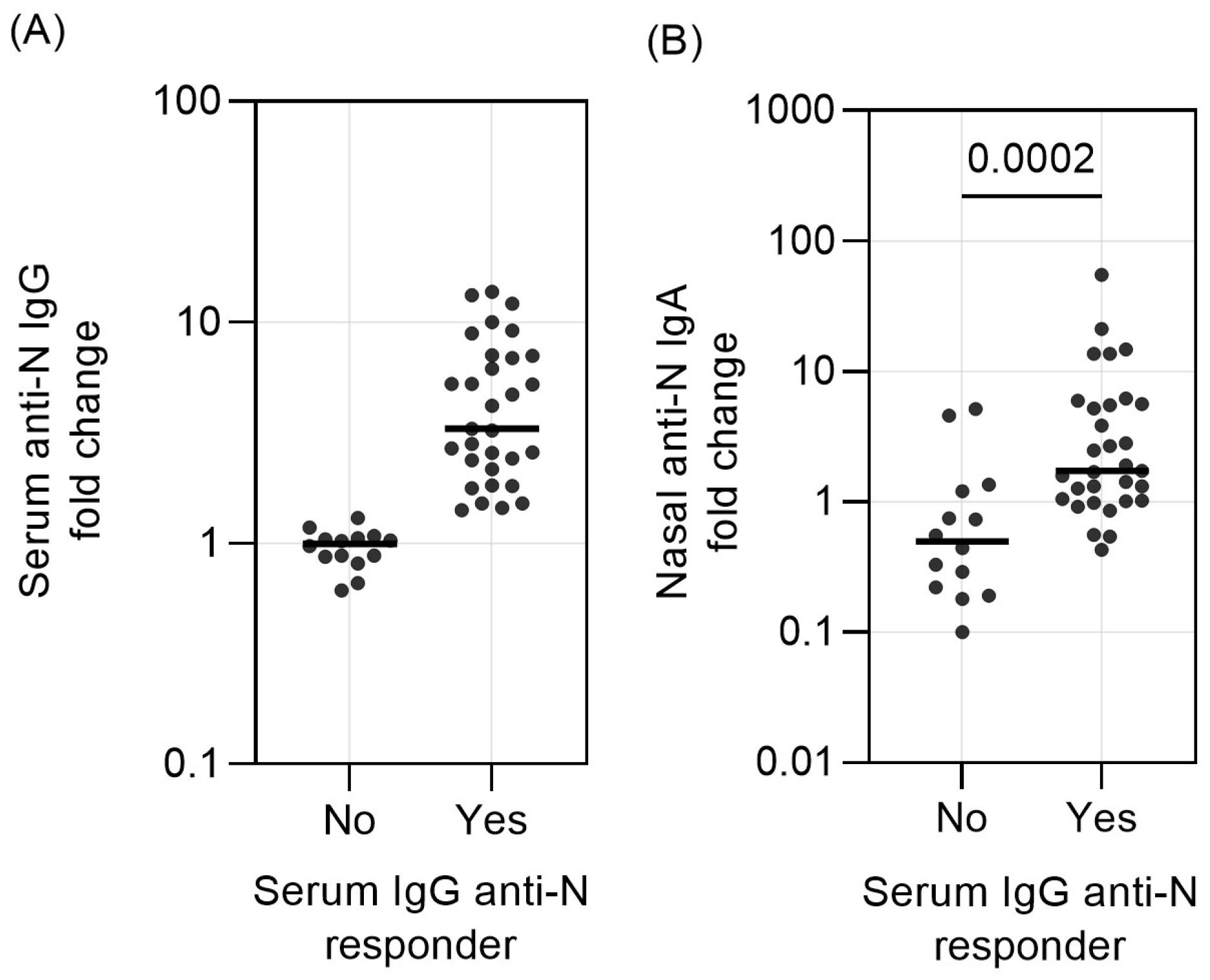
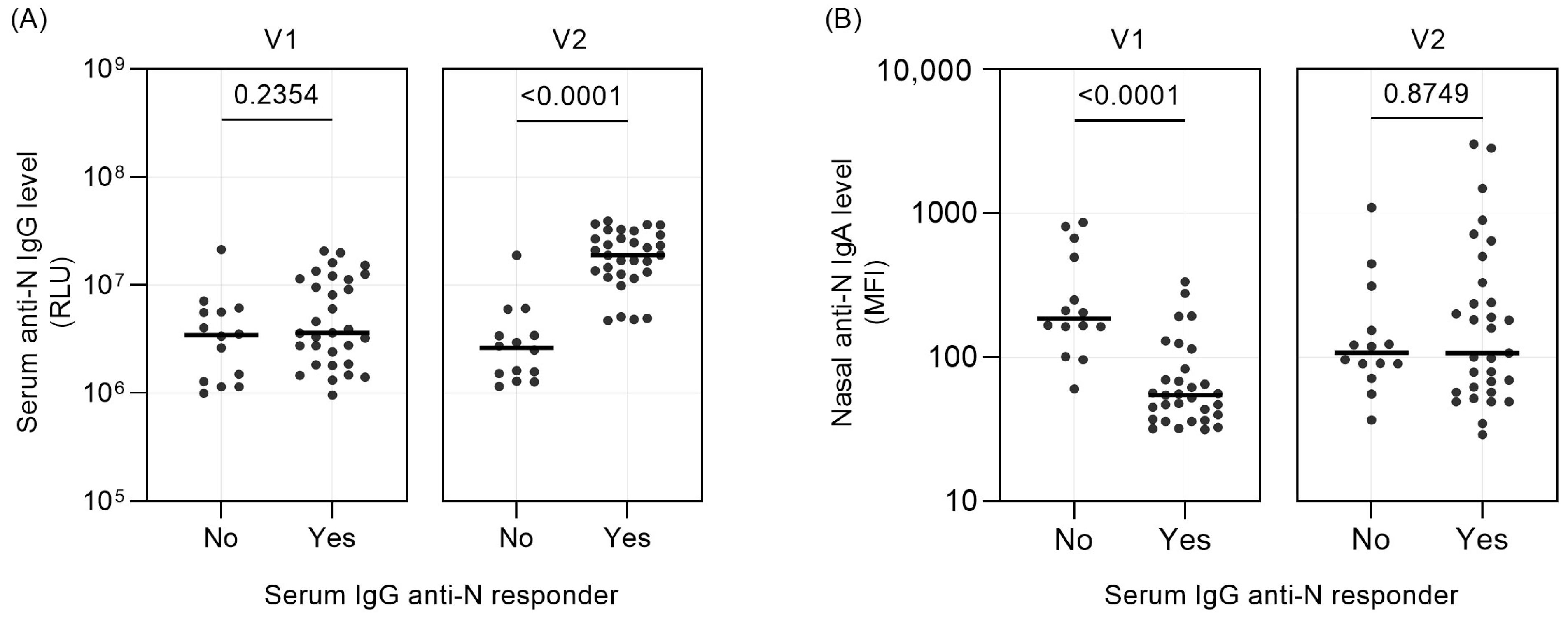
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, Y.; Wunderink, R.G. MERS, SARS and Other Coronaviruses as Causes of Pneumonia. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Sariol, A.; Perlman, S. Lessons for COVID-19 Immunity from Other Coronavirus Infections. Immunity 2020, 53, 248–263. [Google Scholar] [CrossRef]
- Callow, K.A.; Parry, H.F.; Sergeant, M.; Tyrrell, D.A.J. The Time Course of the Immune Response to Experimental Coronavirus Infection of Man. Epidemiol. Infect. 1990, 105, 435–446. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Edridge, A.W.D.; Kaczorowska, J.; Hoste, A.C.R.; Bakker, M.; Klein, M.; Loens, K.; Jebbink, M.F.; Matser, A.; Kinsella, C.M.; Rueda, P.; et al. Seasonal Coronavirus Protective Immunity Is Short-Lasting. Nat. Med. 2020, 26, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Sechan, F.; Grobben, M.; Edridge, A.W.D.; Jebbink, M.F.; Loens, K.; Ieven, M.; Goossens, H.; van Hemert-Glaubitz, S.; van Gils, M.J.; van der Hoek, L. Atypical Antibody Dynamics during Human Coronavirus HKU1 Infections. Front. Microbiol. 2022, 13, 853410. [Google Scholar] [CrossRef]
- Strugnell, R.A.; Wijburg, O.L.C. The Role of Secretory Antibodies in Infection Immunity. Nat. Rev. Microbiol. 2010, 8, 656–667. [Google Scholar] [CrossRef]
- Corthésy, B. Multi-Faceted Functions of Secretory IgA at Mucosal Surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef]
- Sato, S.; Kiyono, H. The Mucosal Immune System of the Respiratory Tract. Curr. Opin. Virol. 2012, 2, 225–232. [Google Scholar] [CrossRef]
- Wright, P.F.; Wieland-Alter, W.; Ilyushina, N.A.; Hoen, A.G.; Arita, M.; Boesch, A.W.; Ackerman, M.E.; Van Der Avoort, H.; Oberste, M.S.; Pallansch, M.A.; et al. Intestinal Immunity Is a Determinant of Clearance of Poliovirus After Oral Vaccination. J. Infect. Dis. 2014, 209, 1628–1634. [Google Scholar] [CrossRef]
- Wright, P.F.; Connor, R.I.; Wieland-Alter, W.F.; Hoen, A.G.; Boesch, A.W.; Ackerman, M.E.; Oberste, M.S.; Gast, C.; Brickley, E.B.; Asturias, E.J.; et al. Vaccine-Induced Mucosal Immunity to Poliovirus: Analysis of Cohorts from an Open-Label, Randomised Controlled Trial in Latin American Infants. Lancet Infect. Dis. 2016, 16, 1377–1384. [Google Scholar] [CrossRef]
- Terauchi, Y.; Sano, K.; Ainai, A.; Saito, S.; Taga, Y.; Ogawa-Goto, K.; Tamura, S.-I.; Odagiri, T.; Tashiro, M.; Fujieda, M.; et al. IgA Polymerization Contributes to Efficient Virus Neutralization on Human Upper Respiratory Mucosa after Intranasal Inactivated Influenza Vaccine Administration. Hum. Vaccines Immunother. 2018, 14, 1351–1361. [Google Scholar] [CrossRef]
- Ainai, A.; van Riet, E.; Ito, R.; Ikeda, K.; Senchi, K.; Suzuki, T.; Tamura, S.; Asanuma, H.; Odagiri, T.; Tashiro, M.; et al. Human Immune Responses Elicited by an Intranasal Inactivated H5 Influenza Vaccine. Microbiol. Immunol. 2020, 64, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Bagga, B.; Cehelsky, J.E.; Vaishnaw, A.; Tomwilkinson, T.; Meyers, R.; Harrison, L.M.; Roddam, P.L.; Walsh, E.E.; DeVincenzo, J.P. Effect of Preexisting Serum and Mucosal Antibody on Experimental Respiratory Syncytial Virus (RSV) Challenge and Infection of Adults. J. Infect. Dis. 2015, 212, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Callow, K.A. Effect of Specific Humoral Immunity and Some Non-Specific Factors on Resistance of Volunteers to Respiratory Coronavirus Infection. Epidemiol. Infect. 1985, 95, 173–189. [Google Scholar] [CrossRef]
- Wright, P.F.; Prevost-Reilly, A.C.; Natarajan, H.; Brickley, E.B.; Connor, R.I.; Wieland-Alter, W.F.; Miele, A.S.; Weiner, J.A.; Nerenz, R.D.; Ackerman, M.E. Longitudinal Systemic and Mucosal Immune Responses to SARS-CoV-2 Infection. J. Infect. Dis. 2022, 226, 1204–1214. [Google Scholar] [CrossRef]
- Fröberg, J.; Gillard, J.; Philipsen, R.; Lanke, K.; Rust, J.; van Tuijl, D.; Teelen, K.; Bousema, T.; Simonetti, E.; van der Gaast-de Jongh, C.E.; et al. SARS-CoV-2 Mucosal Antibody Development and Persistence and Their Relation to Viral Load and COVID-19 Symptoms. Nat. Commun. 2021, 12, 5621. [Google Scholar] [CrossRef] [PubMed]
- Hornsby, H.; Nicols, A.R.; Longet, S.; Liu, C.; Tomic, A.; Angyal, A.; Kronsteiner, B.; Tyerman, J.K.; Tipton, T.; Zhang, P.; et al. Omicron Infection Following Vaccination Enhances a Broad Spectrum of Immune Responses Dependent on Infection History. Nat. Commun. 2023, 14, 5065. [Google Scholar] [CrossRef]
- Havervall, S.; Marking, U.; Svensson, J.; Greilert-Norin, N.; Bacchus, P.; Nilsson, P.; Hober, S.; Gordon, M.; Blom, K.; Klingström, J.; et al. Anti-Spike Mucosal IgA Protection against SARS-CoV-2 Omicron Infection. N. Eng. J. Med. 2022, 387, 1333–1336. [Google Scholar] [CrossRef]
- Ieven, M.; Coenen, S.; Loens, K.; Lammens, C.; Coenjaerts, F.; Vanderstraeten, A.; Henriques-Normark, B.; Crook, D.; Huygen, K.; Butler, C.C.; et al. Aetiology of Lower Respiratory Tract Infection in Adults in Primary Care: A Prospective Study in 11 European Countries. Clin. Microbiol. Infect. 2018, 24, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Loens, K.; Van Loon, A.M.; Coenjaerts, F.; Van Aarle, Y.; Goossens, H.; Wallace, P.; Claas, E.J.C.; Ieven, M. Performance of Different Mono- and Multiplex Nucleic Acid Amplification Tests on a Multipathogen External Quality Assessment Panel. J. Clin. Microbiol. 2012, 50, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Kiyono, H.; Pabst, R.; Russell, M.W. Terminology: Nomenclature of Mucosa-Associated Lymphoid Tissue. Mucosal Immunol. 2008, 1, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Fröberg, J.; Koomen, V.J.C.H.; van der Gaast-de Jongh, C.E.; Philipsen, R.; GeurtsvanKessel, C.H.; de Vries, R.D.; Baas, M.C.; van der Molen, R.G.; de Jonge, M.I.; Hilbrands, L.B.; et al. Primary Exposure to SARS-CoV-2 via Infection or Vaccination Determines Mucosal Antibody-Dependent ACE2 Binding Inhibition. J. Infect. Dis. 2023, 229, 137–146. [Google Scholar] [CrossRef]
- Ravichandran, S.; Grubbs, G.; Tang, J.; Lee, Y.; Huang, C.; Golding, H.; Khurana, S. Systemic and Mucosal Immune Profiling in Asymptomatic and Symptomatic SARS-CoV-2-Infected Individuals Reveal Unlinked Immune Signatures. Sci. Adv. 2021, 7, 6533–6545. [Google Scholar] [CrossRef]
- Cohen, J.I.; Dropulic, L.; Wang, K.; Gangler, K.; Morgan, K.; Liepshutz, K.; Krogmann, T.; Ali, M.A.; Qin, J.; Wang, J.; et al. Comparison of Levels of Nasal, Salivary, and Plasma Antibody to Severe Acute Respiratory Syndrome Coronavirus 2 during Natural Infection and after Vaccination. Clin. Infect. Dis. 2022, 76, 1391–1399. [Google Scholar] [CrossRef]
- Keuning, M.W.; Grobben, M.; Bijlsma, M.W.; Anker, B.; Berman-de Jong, E.P.; Cohen, S.; Felderhof, M.; de Groen, A.E.; de Groof, F.; Rijpert, M.; et al. Differences in Systemic and Mucosal SARS-CoV-2 Antibody Prevalence in a Prospective Cohort of Dutch Children. Front. Immunol. 2022, 13, 976382. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, Y.; Omata, K.; Shimizu, Y.; Kinoshita-Iwamoto, N.; Terada, M.; Suzuki, T.; Morioka, S.; Uemura, Y.; Ohmagari, N.; Maeda, K.; et al. SARS-CoV-2-Neutralizing Humoral IgA Response Occurs Earlier but Is Modest and Diminishes Faster than IgG Response. Microbiol. Spectr. 2022, 10, e0271622. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Demographics 1 | Non-Responder (n = 14) | Responder (n = 31) | p Value 2 |
|---|---|---|---|
| Age in years | 41 (34–56) | 51 (36–57) | 0.470 |
| Male sex | 5 (36%) | 12 (39%) | 0.848 |
| Smoking past or present | 5 (36%) | 13 (42%) | 0.753 |
| Comorbidities | |||
| Lung 3 | 1 (7%) | 4 (13%) | 0.569 |
| Heart 4 | 2 (14%) | 2 (6%) | 0.578 |
| Diabetes | 0 (0%) | 1 (3%) | 0.999 |
| Symptoms at enrollment (V1) | |||
| Cough | 14 (100%) | 31 (100%) | NA 5 |
| Phlegm | 13 (93%) | 23 (74%) | 0.236 |
| Breathlessness | 7 (50%) | 18 (72%) | 0.749 |
| Wheezing | 7 (50%) | 8 (26%) | 0.172 |
| Runny nose | 13 (93%) | 23 (74%) | 0.236 |
| Fever | 4 (29%) | 12 (39%) | 0.738 |
| Chest pain | 5 (36%) | 15 (48%) | 0.526 |
| Muscle pain | 7 (50%) | 14 (45%) | 0.999 |
| Disturbed sleep | 6 (43%) | 2 (68%) | 0.188 |
| Headache | 10 (71%) | 22 (71%) | 0.999 |
| Generally feeling unwell | 12 (86%) | 28 (90%) | 0.639 |
| Interference with daily activity | 9 (64%) | 28 (90%) | 0.085 |
| Confusion and disorientation | 1 (7%) | 2 (6%) | 0.999 |
| Diarrhea | 3 (21%) | 1 (3%) | 0.082 |
| Duration of illness/cough prior to V1-sampling in days | |||
| Illness | 7 (3–7.25) | 4 (3–5) | 0.146 |
| Cough | 6 (3–8.25) | 4 (2–7) | 0.227 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sechan, F.; Loens, K.; Goossens, H.; Ieven, M.; van der Hoek, L. Endemic Human Coronavirus-Specific Nasal Immunoglobulin A and Serum Immunoglobulin G Dynamics in Lower Respiratory Tract Infections. Vaccines 2024, 12, 90. https://doi.org/10.3390/vaccines12010090
Sechan F, Loens K, Goossens H, Ieven M, van der Hoek L. Endemic Human Coronavirus-Specific Nasal Immunoglobulin A and Serum Immunoglobulin G Dynamics in Lower Respiratory Tract Infections. Vaccines. 2024; 12(1):90. https://doi.org/10.3390/vaccines12010090
Chicago/Turabian StyleSechan, Ferdyansyah, Katherine Loens, Herman Goossens, Margareta Ieven, and Lia van der Hoek. 2024. "Endemic Human Coronavirus-Specific Nasal Immunoglobulin A and Serum Immunoglobulin G Dynamics in Lower Respiratory Tract Infections" Vaccines 12, no. 1: 90. https://doi.org/10.3390/vaccines12010090
APA StyleSechan, F., Loens, K., Goossens, H., Ieven, M., & van der Hoek, L. (2024). Endemic Human Coronavirus-Specific Nasal Immunoglobulin A and Serum Immunoglobulin G Dynamics in Lower Respiratory Tract Infections. Vaccines, 12(1), 90. https://doi.org/10.3390/vaccines12010090